

Abstract
Background: MiR-29c, a member of the miR-29 family, has been recognized to play an important role in hepatitis C virus (HCV) infection. However, the underlying molecular mechanism of miR-29c involved in HCV replication is not fully understood. Methods: RT-qPCR assay was used to detect the expression pattern of miR-29c and signal transducer and activator of transcription 3 (STAT3) mRNA in JFH-1-infected Huh7 cells. HCV replication was evaluated by the expression of HCV RNA, non-structural protein 5A (NS5A) and non-structural protein 3 (NS3). Dual-Luciferase Reporter assay was applied to search for the candidate target mRNAs of miR-29c. Western blot assay was performed to detect the protein level of double-stranded RNA-dependent protein kinase R (PKR), (2′-5′)-oligoadenylate synthetase (OAS) and interferon regulatory transcription factor 1 (IRF1). Results: miR-29c expression was down-regulated, and STAT3 mRNA and protein expressions were up-regulated in JFH-1-infected Huh7 cells. MiR-29c overexpression or STAT3 knockdown repressed HCV replication, while miR-29c depletion or STAT3 upregulation promoted HCV replication. Additionally, STAT3 was a direct target of miR-29c, and miR-29c suppressed STAT3 protein expression in Huh7 cells. Moreover, STAT3 overexpression reversed miR-29c-mediated suppression on HCV replication. Furthermore, the anti-miR-29c-mediated inhibitory effect on type I IFN response was abated following STAT3 knockdown. Conclusions: miR-29c might repress HCV infection via promoting type I IFN response by targeting STAT3 in JFH-1-infected Huh7 cells, offering a promising avenue for HCV treatment.
MiR-29c, a member of the miR-29 family, has been recognized to play an important role in hepatitis C virus (HCV) infection.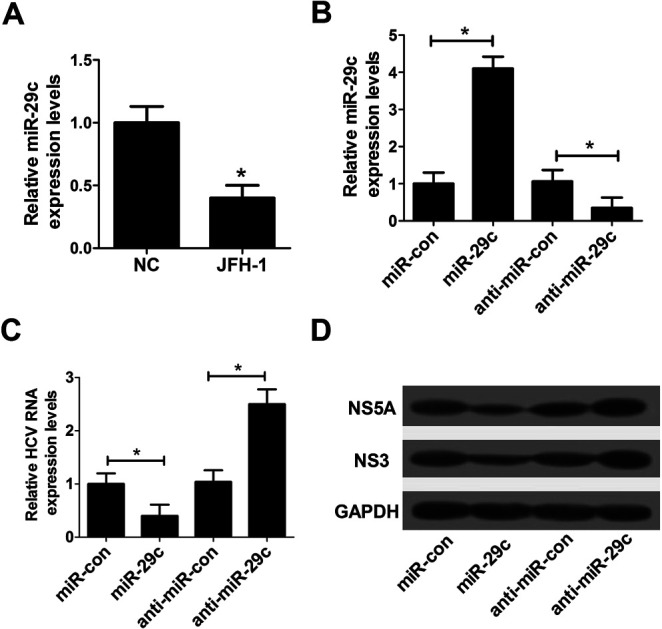
1. Introduction
Hepatitis C virus (HCV), a hepatotropic RNA virus with a prevalence of about 3% worldwide, causes serious liver damage, including liver fibrosis, liver cirrhosis and hepatocellular carcinoma.1 Generally, HCV is divided into six major genotypes (types 1–6), and the JFH-1 strain of HCV belongs to genotype 2a and is the first HCV strain that can produce HCV particles in Huh7 cells.2 At present, the main factors of HCV infection are the unsterile medical procedures and unsafe injection drugs used in countries with high HCV prevalence.3 Despite great advances in treatment options for HCV infection, such as sofosbuvir/velpatasvir, sofosbuvir/simeprevir and sofosbuvir/ledipasvir combinations, the common adverse events and expensive costs are still a great challenge.4,5 Therefore, it's very important to explore the pathogenesis of HCV infection in order to identify more effective therapy strategies for HCV.
MicroRNAs (miRNAs), a class of non-coding single-stranded RNA molecules containing 19–23 nucleotides (nt), could bind to the 3′-untranslated regions (3′-UTR) of target mRNAs to make gene silence by the stimulation of mRNA degradation or translational repression.6 Increasing evidence presents that most miRNAs play important roles in a series of biological processes.7 Moreover, several miRNAs are reported to be related to virus infection diseases, including HCV infection.8 In a recent document, Zhang et al.9 found that miR-155 expression induced by HCV facilitated hepatocyte proliferation and tumorigenesis via regulating Wnt signaling. Moreover, miR-122 expression was remarkably decreased in patients with HCV infection and miR-122 was confirmed to be associated with a poor response to INF-based treatment.10 The miR-29 family, including miR-29a, miR-29b and miR-29c, have been recognized as HCV replication suppressor that were down-regulated in most HCV-infected patients.11 A recent report also revealed that miR-29c and its target gene PPARGC1A might be used as candidate biomarkers of JFH-1-infected Huh7 cells via bioinformatics analysis.12 However, the underlying molecular mechanisms of miR-29c in HCV replication are not fully understood.
The present study aimed to explore the basic principles of miR-29c in regulating HCV replication in JFH-1-infected Huh7 cells. Function analysis found that overexpression of miR-29c repressed HCV replication and signal transducer and activator of transcription 3 (STAT3) up-regulation promoted HCV replication. Further mechanistic analysis demonstrated that miR-29c targeted the 3′UTR of STAT3 mRNA to suppress HCV replication via type I IFN response.
2. Materials and methods
2.1. Cell culture and treatment
Human hepatoma cells (Huh7) were purchased from American Tissue Culture Collection (ATCC, Manassas, VA, USA), and cultured in Dulbecco's modified Eagle's medium (11995065, DMEM, Gibco, Rockville, MD, USA) supplemented with 10% fetal bovine serum (10099141, FBS, Gibco), 100 U ml−1 penicillin (15140122, Gibco) and 100 U ml−1 streptomycin (15140122, Gibco). Cells were maintained at 37 °C in a humidified atmosphere with 5% CO2.
For leukemia inhibitory factor (LIF) treatment, Huh7 cells were cultured in growth medium for 24 h. Then, cells were pretreated with various concentrations (0, 10, 20 μM) of LIF (ab57665, Abcam, Cambridge, UK) for 1 h before infection of JFH-1 (0.01 MOI) for 24 h.
For AG490 treatment, Huh7 cells were pretreated with various concentrations (0, 10, 20 μM) of AG490 (ab120950, Abcam) for 1 h prior to infection with JFH-1 (0.01 MOI) for 24 h.
2.2. Virus preparation
JFH-1 strain, belonging to genotype 2a of HCV, was produced and propagated as previously reported.13 Briefly, pJFH-1 plasmid was assembled containing the full-length JFH-1 cDNA downstream of the T7 RNA promoter by Sangon Biotech (Shanghai, China). Then, the JFH-1 RNA was synthesized by transcription with pJFH-1 plasmid and MEGAscript RNAi Kit (AM1626, Thermo Fisher Scientific, Waltham, MA, USA). 20 μg of JFH-1 RNA was transfected into 5.0 × 106 Huh7 cells by electroporation and the culture medium with JFH-1 was harvested by a 0.22 μm filter (slgv033rb, Millipore, Billerica, MA, USA) at 5 days after transfection. To detect the HCV RNA titers, total RNA was extracted from 100 μl samples with GenEluteTM Total RNA Purification Kit (RTN9602, Sigma-Aldrich, St. Louis, MO, USA), and expression of HCV RNA was measured by real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR). HCV RNA was measured by using the PCR primers 5′-TCTGCGGAACCGGTGAGTA-3′ (sense) and 5′-TCAGGCAGTACCACAAGGC-3′ (antisense), based on the JFH-1 sequence (GenBank accession no. AB047639).
2.3. Cell transfection
The full-length sequences of STAT3 were amplified by PCR and then cloned into pcDNA3.1 vector (V87020, Invitrogen, Waltham, MA, USA) to construct pcDNA-STAT3 overexpression plasmid (pcDNA-STAT3). MiR-29c mimics, miR-29c inhibitor (anti-miR-29c), siRNA targeting STAT3 (si-STAT3) and all corresponding negative control oligonucleotides (miR-con, anti-miR-con, si-con) were obtained from Sangon Biotech. Huh7 cells (3 × 105 per well) were inoculated into 6-well plates and cultured for 24 h. Then, cells were transfected with 20 nM miRNA mimics, 50 nM anti-miRs, 40 nM si-RNAs or 50 ng pcDNAs using Lipofectamine™ 3000 transfection reagent (L3000001, Invitrogen) referring to the instructions of manufacturer.
2.4. Dual-Luciferase Reporter assay
Putative miR-29c targets were predicted using the online software algorithms, including TargetScan (http://www.targetscan.org/) and MiRanda (http://microrna.sanger.ac.uk/). The partial sequences of STAT3 3′-UTR containing the putative binding sites of miR-29c were synthesized by PCR and cloned into the pGL3 Luciferase Reporter Vector (E1751, Promega, Madison, WI) to generate the wild-type pGL3-STAT3 luciferase reporter plasmid (WT-STAT3). Site-directed mutagenesis of miR-29c complementary bases was generated using Q5 Site-Directed Mutagenesis Kit (E0554S, New England Biolabs, Ipswich, MA, USA) and then cloned into the pGL3-control vector to construct mutant-type pGL3-STAT3 luciferase reporter plasmid (MUT-STAT3). Huh7 cells were transfected with WT-STAT3 or MUT-STAT3 together with miR-con, miR-29c mimics, anti-miR-con, or anti-miR-29c. After 24 h, cells were collected for the measurement of luciferase activity using Dual-Luciferase Reporter assay (E1910, Promega).
2.5. Total RNA extraction and RT-qPCR
The TRIzol Plus RNA Purification Kit (12183555, Invitrogen) was used for total RNA extraction of treated Huh7 cells according to the manufacturer's instructions. Equal amount of RNA (500 ng) was reversely transcribed into cDNA using M-MLV reverse transcriptase (AM2043, Invitrogen). The qPCR reactions were performed using a preheated 7900HT fast real-time PCR detection system (4329001, Thermo Fisher Scientific) with the EXPRESS One-Step SYBR™ GreenER™ Kit (1179001K, Invitrogen). Gene expression levels were measured by the means of the 2−ΔΔCt method with β-actin or U6 as an internal control. For RT-qPCR analysis, the following primers were used: miR-29c: 5′-CGATTTCTCCTGGTGTTCA-3′ (forward) and 5′-ACCGATTTCAAATGGTGC-3′ (reverse); STAT3: 5′-CCCGTTTTGCCTTCATTT-3′ (forward) and 5′-CAGGGGCTTCCAACCTTT-3′ (reverse); β-actin: 5′-ACACTGTGCCCATCTACGAGGGG-3′ (forward) and 5′-ATGATGGAGTTGAAGGTAGTTTCGTGGAT-3′ (reverse); U6: 5′-GCTTCGGCAGCACATATACTAAAAT-3′ (forward) and 5′-CGCTTCACGAATTTGCGTGTCAT-3′ (reverse).
2.6. Western blot
At 24 h after infection, Huh7 cells were lysed using RIPA buffer (R0278, Sigma-Aldrich) with Complete™ Protease Inhibitor Cocktail (Roche, South San Francisco, CA, USA). 50 μg of protein per sample was separated by 10% SDS-PAGE and transferred to the PVDF membranes (IBFP0812C, Millipore, Billerica, MA, USA), followed by blocking with 5% nonfat milk in TBST (10 mM Tris–HCl, pH = 8.0, 150 mM NaCl containing 0.1% v/v Tween 20) for at least 1 h. Then, the PVDF membranes were incubated overnight at 4 °C with primary monoclonal antibody against non-structural protein 5A (NS5A) (ab20342, 1 : 1000, Abcam), non-structural protein 3 (NS3) (ab65407, 1 : 3000, Abcam), STAT3 (ab68153, 1 : 1000, Abcam), STAT3-Y705 (ab76315, 1 : 3000, Abcam), double-stranded RNA-dependent protein kinase R (PKR) (3072S, 1 : 1000, Cell Signaling Technology, Danvers, MA, USA), (2′-5′)-oligoadenylate synthetase (OAS) (14498S, 1 : 1000, Cell Signaling Technology), interferon regulatory transcription factor 1 (IRF1) (8478S, 1 : 1000, Cell Signaling Technology), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (5174T, 1 : 1000, Cell Signaling Technology). After being washed three times with TBST, the PVDF membranes were further incubated with HRP-conjugated secondary antibodies (SC2357, 1 : 1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 30 min. Then, protein expression was detected using VersaDoc 4000 MP imaging system (Bio-Rad Laboratories, Hercules, CA, USA).
2.7. Statistical analysis
All experiments were repeated independently at least three times. The data were analyzed using the software of SPSS 16.0 (SPSS Inc., IL, USA) and GraphPad Prism v5.0 (GraphPad Software Inc., CA, USA). A Student's t test and one-way analysis variance (ANOVA) were used to assess significant differences between groups. All data were represented as mean ± standard deviation (mean ± SD) and P < 0.05 was considered to be statistically significant.
3. Results
3.1. Overexpression of miR-29c repressed HCV replication in JFH-1-infected Huh7 cells
In order to investigate the effect of miR-29c on HCV infection, RT-qPCR assay was performed to detect the expression pattern of miR-29c in JFH-1-infected Huh7 cells. The data revealed that the expression of miR-29c was evidently decreased in JFH-1-infected Huh7 cells compared with the negative control (Fig. 1A). Further, to explore the function of miR-29c on HCV replication, miR-29c mimics and anti-miR-29c were synthesized and transfected into Huh7 cells, respectively. As displayed in Fig. 1B, the expression level of miR-29c was strikingly promoted by the introduction of miR-29c mimics, while miR-29c expression was greatly inhibited after transfection with anti-miR-29c compared with the respective control. RT-qPCR assay showed that miR-29c up-regulation markedly hindered HCV RNA expression in JFH-1-infected Huh7 cells, while miR-29c knockdown facilitated HCV RNA expression (Fig. 1C). A previous document has verified that NS3 and NS5A proteins are the central catalytic enzymes of HCV replicase.14 Consistently, western blot assay also showed that miR-29c overexpression led to a suppression of HCV replication, revealed by the decrease of NS5A and NS3 protein levels, while miR-29c depletion exhibited an opposite effect (Fig. 1D). These data suggested that miR-29c might play an important role in the development and progression of HCV infection.
Fig. 1. Overexpression of miR-29c repressed HCV replication in JFH-1-infected Huh7 cells. (A) RT-qPCR assay was performed to detect the expression pattern of miR-29c in JFH-1-infected Huh7 cells. (B) The expression level of miR-29c was detected in Huh7 cells transfected with miR-29c mimics or anti-miR-29c. (C and D) Huh7 cells transfected with miR-29c mimics, anti-miR-29c or respective control were infected with JFH-1 (0.01 MOI) for 24 h, followed by the detection of HCV RNA expression by RT-qPCR (C), NS3 and NS5A proteins expression by western blot assay (D). GAPDH was used as the internal reference. *P < 0.05 vs. respective control.

3.2. STAT3 was a direct target gene of miR-29c
To further explore the underlying molecular mechanism of miR-29c on HCV infection, the software algorithms were used to search for the endogenous target genes of miR-29c. Interestingly, the predicted data revealed that there existed some complementary sites between miR-29c and 3′-UTR of STAT3 mRNA, indicating STAT3 might be a direct target of miR-29c (Fig. 2A). Then, Dual-Luciferase Reporter assay was employed to validate the assumption by transfecting constructed wild-type or mutant-type pGL3-STAT3 luciferase reporter vectors (WT-STAT3 or MUT-STAT3) into Huh7 cells together with miR-con, miR-29c mimics, anti-miR-con or anti-miR-29c. These results demonstrated that miR-29c overexpression remarkably inhibited luciferase activity in Huh7 cells transfected with WT-STAT3 vector, but no evident effect was observed in MUT-STAT3 group (Fig. 2B). Conversely, introduction of anti-miR-29c led to an increase of luciferase activity of WT-STAT3 vector, and little change was displayed in MUT-STAT3 group (Fig. 2C). Further, we investigated the effect of miR-29c on STAT3 expression in Huh7 cells by the introduction of miR-29c mimics and anti-miR-29c. The experiment data presented that up-regulated miR-29c significantly lowered the expression of STAT3 protein, while miR-29c knockdown enhanced the protein level of STAT3 (Fig. 2D) compared with their counterparts respectively. Taken together, these results indicated that STAT3 was a direct target gene of miR-29c and miR-29c suppressed the expression of STAT3 protein.
Fig. 2. STAT3 was a direct target of miR-29c. (A) The wild and mutant 3′-UTR of STAT3 mRNA with the putative binding sites of miR-29c. (B and C) Dual-Luciferase Reporter assay was performed to explore whether STAT3 was a direct target of miR-29c. Wild-type or mutant-type pGL3-STAT3 luciferase vectors (WT-STAT3 or MUT-STAT3) were constructed and transfected into Huh7 cells together with miR-con or miR-29c mimics (B), anti-miR-con or anti-miR-29c (C). (D) Huh7 cells were transfected with miR-con, miR-29c mimics, anti-miR-con or anti-miR-29c for 48 h, followed by the measurement of STAT3 protein expression. *P < 0.05 vs. corresponding control.

3.3. Up-regulation of STAT3 promoted HCV replication in JFH-1-infected Huh7 cells
To further investigate the effect of STAT3 on HCV replication, STAT3 expression pattern was detected by RT-qPCR and western blot assay in JFH-1-infected Huh7 cells. The results demonstrated that the expression of STAT3 mRNA (Fig. 3A) and STAT3 protein (Fig. 3B) were significantly increased in JFH-1-infected Huh7 cells. Then, STAT3 overexpression plasmid (pcDNA-STAT3) was constructed and transfected into Huh7 cells prior to infection with JFH-1. As displayed in Fig. 3C, STAT3 expression was strikingly enhanced by the introduction of pcDNA-STAT3. Moreover, the results displayed that STAT3 overexpression led to a promotion of HCV replication, revealed by the increase of HCV RNA expression (Fig. 3D), as well as the enhancement of NS5A and NS3 proteins levels (Fig. 3E).
Fig. 3. STAT3 overexpression promoted HCV replication in JFH-1-infected Huh7 cells. RT-qPCR assay of STAT3 mRNA expression (A) and western blot assay of STAT3 protein expression (B) in JFH-1-infected Huh7 cells. (C–E) At 48 h after transfection with pDNA-STAT3, Huh7 cells were infected with JFH-1 (0.01 MOI) for 24 h, followed by the evaluation of STAT3 protein expression (C), HCV RNA expression (D), NS3 and NS5A protein expressions (E). (F–H) Huh7 cells were pretreated with 0, 10, and 20 μM of LIF for 1 h before infection with JFH-1 (0.01 MOI) for 24 h, followed by the assessment of STAT3-Y705 phosphorylation (F), HCV RNA expression (G), NS3 and NS5A proteins expression (H). *P < 0.05 vs. corresponding control.

It was previously reported that the active form of STAT3 could result in a marked increase in HCV replication.15 LIF, a known activator of STAT3, was confirmed to induce phosphorylation-Y705 of STAT3 protein.16 In this research, Huh7 cells were pre-treated with various concentrations (0, 10, and 20 μM) of LIF for 1 h before infection of JFH-1. The data showed that STAT3-Y705 phosphorylation was evidently increased by LIF treatment (Fig. 3F). Then, we further explored the effect of STAT3-Y705 phosphorylation on HCV replication. These results displayed that STAT3-Y705 phosphorylation triggered by LIF treatment remarkably facilitated HCV replication, presented by the increase of HCV RNA expression (Fig. 3G), as well as the elevation of NS5A and NS3 proteins levels (Fig. 3H). All these results implied that STAT3 overexpression promoted HCV replication possibly via STAT3-Y705 phosphorylation in JFH-1-infected Huh7 cells.
3.4. STAT3 silence repressed HCV replication in JFH-1-infected Huh7 cells
Subsequently, siRNA of STAT3 (si-STAT3) was synthesized and transfected into Huh7 cells to examine its knockdown efficiency. The data presented that the expression of STAT3 mRNA (Fig. 4A) and protein (Fig. 4B) were significantly attenuated by the introduction with si-STAT3, in comparison to their counterparts, respectively. Hence, si-STAT3 was used to further explore the effect of STAT3 knockdown on HCV replication. The results elucidated that STAT3 depletion resulted in a decline of HCV replication, embodied by the decrease of HCV RNA expression (Fig. 4C), as well as the down-regulation of NS5A and NS3 proteins (Fig. 4D).
Fig. 4. STAT3 silence repressed HCV replication in JFH-1-infected Huh7 cells. RT-qPCR assay of STAT3 mRNA expression (A) and western blot assay of STAT3 expression (B) in Huh7 cells transfected with si-STAT3 or si-con. (C and D) Huh7 cells transfected with si-STAT3 or si-con were infected with JFH-1 for 24 h, followed by the analysis of HCV RNA expression (C), NS5A and NS3 protein expressions (D). (E–G) Huh7 cells were pretreated with various concentrations (0, 10, and 20 μM) of AG490 for 1 h and then infected with JFH-1 for 24 h, followed by the detection of STAT3-Y705 phosphorylation (E), HCV RNA expression (F), NS3 and NS5A proteins expression (G). *P < 0.05 vs. respective control.

AG490, a JAK2 inhibitor, was verified to suppress the phosphorylation level of STAT3.17 Thus, AG490 was employed to investigate the effect of STAT3 phosphorylation inhibition on HCV replication. As shown in Fig. 4E, STAT3-Y705 phosphorylation was effectively depressed by AG490 treatment. Moreover, AG490 treatment led to a dramatic suppression of HCV replication, demonstrated by the decrease of HCV RNA expression (Fig. 4F), as well as the reduction of NS5A and NS3 proteins levels (Fig. 4G). All these data indicated that STAT3 down-regulation blocked HCV replication partly through the decrease of phosphorylation at Y705 in JFH-1-infected Huh7 cells.
3.5. Restoration of STAT3 expression reversed miR-29c-mediated inhibitory effect on HCV replication in JFH-1-infected Huh7 cells
To further explore whether the suppressive effect of miR-29c on HCV replication was mediated by STAT3, Huh7 cells were transfected with miR-29c mimics, anti-miR-29c, miR-29c mimics + pcDNA-STAT3 or anti-miR-29c + si-STAT3 prior to infection with JFH-1. The results displayed that miR-29c-triggered decline of HCV replication was strikingly reversed by the introduction of pcDNA-STAT3, presented by higher HCV RNA expression (Fig. 5A), as well as enhanced NS5A and NS3 protein levels (Fig. 5B). On the contrary, STAT3 knockdown evidently attenuated anti-miR-29c-mediated promotive effect on HCV replication, manifested by lower HCV RNA expression (Fig. 5C), as well as decreased NS5A and NS3 protein levels (Fig. 5D). Taken together, these results suggested that miR-29c repressed HCV replication by down-regulating STAT3 expression in JFH-1-infected Huh7 cells.
Fig. 5. Restoration of STAT3 expression reversed miR-29c-mediated inhibitory effect on HCV replication in JFH-1-infected Huh7 cells. (A and B) Huh7 cells transfected with miR-29c mimics alone, or together with STAT3 were infected with JFH-1 (0.01 MOI) for 24 h, followed by the assay of HCV RNA expression (A), NS3 and NS5A proteins expression (B). (C and D) Huh7 cells were transfected with anti-miR-29c alone, or together with si-STAT3 prior to infection with JFH-1 (0.01 MOI) for 24 h, followed by the determination of HCV RNA expression (C), NS3 and NS5A proteins expression (D). *P < 0.05 vs. respective control.

3.6. Anti-miR-29c-mediated suppression on type I IFN response was abated following STAT3 knockdown
Type I IFN response has been confirmed to be the first line of host defense controlling viral infection via regulation of IFN-stimulated genes (ISGs), including PKR, OAS and IRF1.18 As a result, we further investigated the effect of STAT3 on type I IFN response in JFH-1-infected Huh7 cells. si-STAT3 was employed to detected the effect on the expression of PKR, OAS and IRF1. The data displayed that the expressions of PKR, OAS and IRF1 at both mRNA (Fig. 6A–C) and protein (Fig. 6D) levels were drastically promoted by the introduction of si-STAT3 compared with negative control. The results suggested that STAT3 knockdown might stimulate the activation of type I IFN response. Subsequently, to explore the effect of miR-29c on type I IFN response, anti-miR-29c was transfected into Huh7 cells. These data delineated that down-regulation of miR-29c resulted in a substantial drop in the expression levels of PKR, OAS, IRF1 at both mRNA (Fig. 6E–G) and protein (Fig. 6H) levels. These data suggested that miR-29c might play a vital role in the regulation of type I IFN response. Furthermore, the inhibitory effect of anti-miR-29c on type I IFN response was prominently abrogated after transfection with si-STAT3 (Fig. 6E–H). All these results implied that miR-29c depletion repressed the activation of type I IFN response by up-regulating STAT3 expression.
Fig. 6. Anti-miR-29c-mediated suppression on type I IFN response was abated following STAT3 knockdown. RT-qPCR assay of PKR mRNA (A), OAS mRNA (B), IRF1 mRNA (C) expression and western bolt assay of PKR/OAS/IRF1 proteins expression (D) in Huh7 cells transfected with si-STAT3 or si-con. RT-qPCR assay of PKR mRNA (E), OAS mRNA (F), IRF1 mRNA (G) expression and western bolt assay of PKR/OAS/IRF1 proteins expression (H) in Huh7 cells transfected with anti-miR-29c alone, or together with si-STAT3. *P < 0.05 vs. corresponding control.

4. Discussion
MiR-29c, a member of miR-29 family, located on chromosome 1q32 and was found by small RNA library sequencing.19 A previous report revealed that miR-29c might function as a tumor suppressor through targeting endogenous gene WIP1 in liver carcinoma which was mainly caused by the infection of HBV and HCV.20 Also, it was demonstrated that miR-29c inhibited proliferation and induced apoptosis in the development and progression of HBV-related hepatocellular carcinoma via targeting gene TNFAIP3.21 Additionally, miR-29c down-regulation repressed the expression of extracellular matrix proteins and miR-29c up-regulation reduced HCV RNA abundance in hepatic stellate cells.11 In the present study, we found that the expression of miR-29c was evidently decreased in JFH-1-infected Huh7 cells. Moreover, miR-29c overexpression led to a suppression of HCV replication. These findings suggested that miR-29c might play an important role in the development and progression of HCV infection.
Compared with the classical transcription factors, miRNAs exerted their functions mainly through regulating the expression of endogenous target genes. As a result, the software algorithms were used to search for the candidate target genes of miR-29c. Of interest, the data revealed that there existed some complementary sites between miR-29c and STAT3 mRNA, indicating that STAT3 might interact with miR-29c. Subsequently, dual luciferase assay verified that STAT3 was a direct target gene of miR-29c, and miR-29c hindered STAT3 protein expression by binding to the 3′UTR of STAT3 mRNA.
STAT3, a transcription factor expressed in a series of metabolic tissues, is activated via phosphorylation of Tyr705 and Tyr727 to exert its function.22 McCartney et al.15 proved that HCV replication could promote STAT3 activation, in turn, the activation of STAT3 could enhance HCV replication. A recent report showed that STAT3 knockdown obviously inhibited HCV replication, implied that targeting STAT3 might be a promising strategy for preventing HCV.23 Tacke et al.24 demonstrated that extracellular HCV core activated STAT3 on human macrophages and HCV core-induced STAT3 activation depended on cytokine IL-6 production. In addition, Xiong et al.25 found that phosphorylated STAT3 enhanced the expression of lncRNA Lethe and facilitated HCV replication via type I IFN response. In this study, we discovered that STAT3 enhanced HCV replication and STAT3 down-regulation decreased HCV replication. Furthermore, restoration of STAT3 expression counteracted miR-29c-mediated inhibitory effect on HCV replication in JFH-1-infected Huh7 cells. Taken together, these results provided further evidence that STAT3 might be a critical mediator in HCV infection.
Type I IFN response is recognized to be the first line of host defense and thus plays a critical role to protect hepatocytes against HCV infection.26 As demonstrated by a previous study, STAT3 is a negative regulator of type I IFN response.27 In the present study, we also verified that STAT3 depletion induced the activation of type I IFN response by increasing the expression of PKR, OAS and IRF1. Moreover, miR-29c knockdown led to a suppression of type I IFN response. Based on the present finding that STAT3 was a direct target of miR-29c, we further investigated whether miR-29c depletion repressed the activation of type I IFN response by targeting STAT3. As expected, STAT3 knockdown reversed anti-miR-29c-mediated inhibition on type I IFN response. All the data suggested that miR-29c down-regulation repressed the activation of type I IFN response by targeting STAT3. Contrary with our findings, Wu et al.28 proved that miR-146a was a potent negative regulator of type I IFN response by targeting STAT3, resulting in the enhancement of human cytomegalovirus in MRC-5 cells. Zhu et al.29 found that miR-19a depletion enhanced HBV replication via repressing type I IFN response by targeting SOCS1 in pregnant women.
5. Conclusion
Our study demonstrated that miR-29c repressed HCV infection via enhancing type I IFN response by targeting STAT3 in JFH-1-infected Huh7 cells, indicating that miR-29c might be a useful marker and potential therapeutic target of HCV infection.
Authors' contribution
This work was conceived and designed by Yanjing Wang. The experiment procedures and data analysis were performed by Yuanyuan Li. The manuscript was prepared and reviewed by Yanjing Wang.
Conflicts of interest
The authors have no conflict of interest to declare.
Funding
This research received no specific grant from any funding agency in the public.
Supplementary Material
Acknowledgments
Thanks for all participants involved in this research.
References
- Wang S. H., Yeh S. H. and Chen P. J., Hepatitis C Virus II, 2016, pp. 109–136 [Google Scholar]
- Wakita T. Methods Mol. Biol. 2009;510:305–327. doi: 10.1007/978-1-59745-394-3_23. [DOI] [PubMed] [Google Scholar]
- Welzel T. Petersen J. Herzer K. Ferenci P. Gschwantler M. Cornberg M. Ingiliz P. Berg T. Spengler U. Weiland O. J. Hepatol. 2016;64:S781–S782. [Google Scholar]
- Horsleysilva J. L. Vargas H. E. Gastroenterol. Hepatol. 2017;48:22–31. [Google Scholar]
- Maan R. van der Meer A. J. F1000Research. 2016;5:367. doi: 10.12688/f1000research.7399.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R. C. Burge C. B. Methods Mol. Biol. 2014;1097:457–476. doi: 10.1007/978-1-62703-709-9_21. [DOI] [PubMed] [Google Scholar]
- Deiuliis J. A. Int. J. Obes. 2016;40:88–101. doi: 10.1038/ijo.2015.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appourchaux K. Dokmak S. Rescherigon M. Treton X. Lapalus M. Gattolliat C. H. Porchet E. Martinotpeignoux M. Boyer N. Vidaud M. Bedossa P. Marcellin P. Bièche I. Estrabaud E. Asselah T. Sci. Rep. 2016;6:34935. doi: 10.1038/srep34935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Wei W. Cheng N. Wang K. Li B. Jiang X. Sun S. Hepatology. 2012;56:1631–1640. doi: 10.1002/hep.25849. [DOI] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M. Krol J. Markiewicz I. Heim M. H. Filipowicz W. Nat. Med. 2009;15:31–33. doi: 10.1038/nm.1902. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S. Friedman R. C. Marquez R. T. Keck K. Kong B. Icardi M. S. Brown K. E. Burge C. B. Schmidt W. N. Wang Y. McCaffrey A. P. J. Infect. Dis. 2011;203:1753–1762. doi: 10.1093/infdis/jir186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P. Wu M. Chen H. Xu J. Wu M. Li M. Qian F. Xu J. OncoTargets Ther. 2016;9:191–202. doi: 10.2147/OTT.S91748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T. Choi Y. Elmowalid G. Sapp R. K. Barth H. Furusaka A. Mishiro S. Wakita T. Krawczynski K. Liang T. J. Hepatology. 2008;48:732–740. doi: 10.1002/hep.22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q. Xu C. C. Virus Res. 2009;145:63–73. doi: 10.1016/j.virusres.2009.06.005. [DOI] [PubMed] [Google Scholar]
- McCartney E. M. Helbig K. J. Narayana S. K. Eyre N. S. Aloia A. L. Beard M. R. Hepatology. 2013;58:1558–1568. doi: 10.1002/hep.26496. [DOI] [PubMed] [Google Scholar]
- Cheng J. G. Stewart C. L. Proc. Natl. Acad. Sci. U. S. A. 2001;98:8680–8685. doi: 10.1073/pnas.151180898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. J. Chen R. F. Deng X. G. Zhou Y. Ye X. Yu M. Tang J. He X. Y. Cheng D. Zeng B. FEBS Lett. 2014;588:566–573. doi: 10.1016/j.febslet.2013.11.041. [DOI] [PubMed] [Google Scholar]
- Gonzáleznavajas J. M. Lee J. David M. Raz E. Nat. Rev. Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P. Rusu M. Sheridan R. Sewer A. Iovino N. Aravin A. Pfeffer S. Rice A. Kamphorst A. O. Landthaler M. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. Li D. Sidler C. Rodriguezjuarez R. Singh N. Heyns M. Ilnytskyy Y. Bronson R. T. Kovalchuk O. Oncotarget. 2015;6:9937–9950. doi: 10.18632/oncotarget.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. M. Wang Y. Fan C. G. Xu F. F. Sun W. S. Liu Y. G. Jia J. H. Biochem. Biophys. Res. Commun. 2011;411:586–592. doi: 10.1016/j.bbrc.2011.06.191. [DOI] [PubMed] [Google Scholar]
- Mashili F. Chibalin A. V. Krook A. Zierath J. R. Diabetes. 2013;62:457–465. doi: 10.2337/db12-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C. Y. Huang C. F. Yeh M. L. Huang J. F. Yu M. L. Chuang W. L. J. Hepatol. 2016;64:S413–S414. doi: 10.1016/S0168-8278(16)00662-0. [DOI] [Google Scholar]
- Tacke R. S. Tosellotrampont A. Nguyen V. Mullins D. W. Hahn Y. S. J. Biol. Chem. 2011;286:10847–10855. doi: 10.1074/jbc.M110.217653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y. Yuan J. Zhang C. Zhu Y. Kuang X. Lan L. Wang X. Biomed. Pharmacother. 2015;72:165–171. doi: 10.1016/j.biopha.2015.04.019. [DOI] [PubMed] [Google Scholar]
- Liu J. P. Ye L. Wang X. Li J. L. Ho W. Z. Transplant Infect. Dis. 2011;13:24–32. doi: 10.1111/j.1399-3062.2010.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. B. Levy D. E. Lee C. K. J. Immunol. 2011;187:2578–2585. doi: 10.4049/jimmunol.1004128. [DOI] [PubMed] [Google Scholar]
- Wu J. Wei B. Wang L. Int. J. Clin. Exp. Med. 2016;9:10044–10051. [Google Scholar]
- Zhu Y. X. Wang M. Bian Q. Ma X. P. Zhao X. P. Int. J. Clin. Exp. Pathol. 2017;10:4348–4355. [Google Scholar]