

Abstract
Brain tumors result in significant morbidity and mortality in both children and adults. Recent data indicates that immunotherapies may offer a survival benefit after standard of care has failed for malignant brain tumors. Modest results from several late phase clinical trials, however, underscore the need for more refined, comprehensive strategies that incorporate new mechanistic and pharmacologic knowledge. Recently, oncometabolism has emerged as an adjunct modality for combinatorial treatment approaches necessitated by the aggressive, refractory nature of high-grade glioma and other progressive malignant brain tumors. Manipulation of metabolic processes in cancer and immune cells that comprise the tumor microenvironment through controlled targeting of oncogenic pathways may be utilized to maximize the efficacy of immunotherapy and improve patient outcomes. Herein, we summarize preclinical and early phase clinical trial research of oncometabolism-based therapeutics that may augment immunotherapy by exploiting the biochemical and genetic underpinnings of brain tumors. We also examine metabolic pathways related to immune cells that target tumor cells, termed ‘tumor immunometabolism’. Specifically, we focus on glycolysis and altered glucose metabolism, including glucose transporters, hexokinase, pyruvate dehydrogenase, and lactate dehydrogenase, glutamine, and we discuss targeting arginase, adenosine, and indoleamine 2,3-dioxygenase (IDO), and toll-like receptors. Lastly, we summarize future directions targeting metabolism in combination with emerging therapies such as oncolytic virotherapy, vaccines, and chimeric antigen receptor T cells.
Keywords: brain tumor, oncometabolism, metabolism, immunotherapy, cancer, glioma
Introduction
Primary central nervous system (CNS) tumors are the most common solid malignancy in children and have an average annual age-adjusted incidence rate of 23.4 and 6.1 per 100,000 adults and children, respectively [1]. Gliomas, the most common primary CNS tumor across all ages, are categorized by the World Health Organization (WHO) as grade 1 and 2 gliomas (generally regarded as low-grade gliomas, LGGs) and grade 3 and 4 (regarded as high-grade gliomas, HGGs) [2, 3]. Glioblastoma (GBM, grade 4) is the most aggressive subtype, and carries a uniformly fatal prognosis despite an intensive multimodal treatment regimen comprised of surgical resection, chemotherapy, and radiation. Median overall survival of GBM patients is approximately 18 months, and the 5-year survival rate has remained below 10% for the past several decades [4–6]. Therefore, there is a pressing need for novel, mechanistically targeted therapies to improve outcomes of patients with HGGs.
Recent advances have renewed interest in immunotherapies (a broad spectrum of modalities including checkpoint inhibitors, vaccines, cellular therapy and oncolytic virotherapy) for the treatment of brain tumors. However, major barriers still exist to the development of safe, effective immunotherapies for brain tumors. These include the immunologically exclusive (‘cold’) or immunosuppressive tumor microenvironment, the immune privilege afforded by the blood brain barrier, the low somatic mutational burden that limits the likelihood of neoantigen presentation (commonly seen with pediatric brain tumors), and the metabolism of the tumor (i.e., oncometabolism).
Oncometabolism, defined as the metabolic reprogramming that accompanies oncogenesis, tumor progression, and central aspects of malignant transformation, is a critical modulator of response to immunotherapy [7]. Altered tumor metabolism is a biological hallmark of gliomas, where biochemical pathways are often disrupted by mutations in genes encoding metabolic enzymes. Metabolic intermediates, known as oncometabolites, can accumulate abnormally either upstream (e.g. L-2-hydroxyglutarate [2-HG], succinate, fumarate) or downstream (e.g., D-2-HG) of these enzymatic perturbations and can also be excreted from the cancer cell into the local environment [8, 9]. Cancer-associated alterations in several key metabolic pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, glutaminolysis, and fatty acid oxidation [10] often cause constitutive upregulation of metabolic pathways. This range of integrated pathways served as the molecular basis of the metabolic plasticity exhibited by cancer cells in response to treatment and which can lead to resistance phenotypes. Together these pathways ensure effective production of ATP and other metabolites required for the generation of biomass for tumor growth as well as inhibition of the antitumor immune response [11, 12]. Tumor cells and activated immune cells both require a continuous supply of nutrients, including glucose and amino acids such as arginine, glutamine, and tryptophan to carry out anabolic macromolecule synthesis. Alterations in tumor cell metabolism that favor tumor growth may also dampen antitumor immune responses either directly or by contributing to an immunosuppressive tumor microenvironment via metabolic competition and signaling.
In this review, we examine the major pathways involved in oncometabolism and in the metabolic pathways related to immune cells that target tumor cells (i.e., tumor immunometabolism) focusing on GBM as an example. By doing so, we discuss novel metabolic oriented therapies currently in preclinical studies and early phase clinical trials, which can be exploited to augment the efficacy of immunotherapies in HGGs.
Oncometabolism and tumor immunometabolism
Nearly a century ago, Otto Warburg first reported that rapidly-growing cancer cells exhibit an enhanced metabolic plasticity to meet their substantial energy and growth requirements. Specifically, this is accomplished by significantly engaging glycolysis in conjunction with mitochondrial oxidative phosphorylation in the absence of underlying hypoxia, a phenomena termed “aerobic glycolysis” [13]. Since these initial discoveries, it has now become apparent that metabolic switching to aerobic glycolysis is a widespread adaptation to changing cellular phenotype, particularly in cells in the immune system (discussed below) [14]. Beyond aerobic glycolysis, other metabolic pathways such as amino acid metabolism, lipid synthesis, and nucleotide synthesis are also altered in HGGs [15–17]. Many of these modulations are the consequence of aberrant gene expression, epigenetic remodeling, or dysregulation of upstream growth signaling pathways. In the past decades, the identification of specific oncogenic mutations in metabolic enzymes has garnered much excitement as drug targets, and a number of genomic studies are underway investigating critical events in metabolic reprogramming and disease initiation of HGGs [18]. Most importantly, recent advances in the field of immunometabolism have uncovered pathways specific to immune function and signaling that are key for the host anti-tumorigenic response (Figure 1). This has led to novel therapies being tested in clinical trials targeting oncometabolism and tumor immunometabolism (Table 1 and Figure 2).
Figure 1.
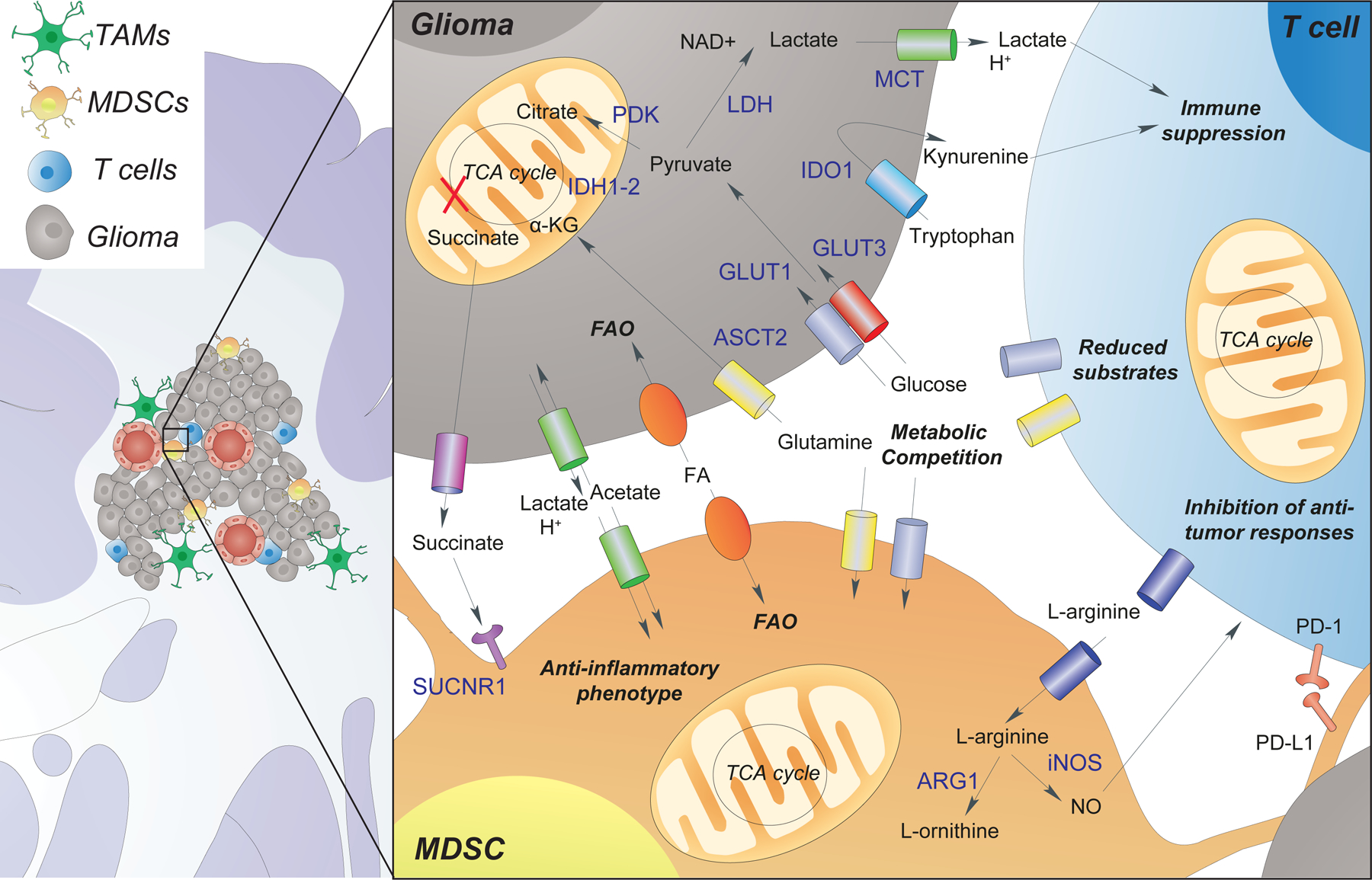
Metabolic interaction between glioma cells, T cells, and myeloid-derived suppressor cells (MDSCs). Glioma cells uptake nutrients from the extracellular space, which include glucose, glutamine, amino acids, acetate, and fatty acids (FAs) and use these nutrients to compensate for increased energy demands (e.g., high glycolytic rate). This leads to a metabolic competition with other cell types within the microenvironment (e.g., T cells), which become significantly affected by the lack of key nutrients (e.g., glucose and glutamine). Glioma cells metabolize tryptophan to the immunosuppressive molecule kynurenine via (Indoleamine 2,3-Dioxygenase 1 (IDO1). Furthermore, the increased release of lactate and succinate from the glioma, leads to both T cell suppression and MDSC polarization towards an anti-inflammatory phenotype. Polarized MDSCs reduce the extracellular levels of L-arginine, release nitric oxide (NO), and express the programmed death-ligand 1 (PD-L1), further inhibiting T cell anti-tumor responses. Other abbreviations: α-KG (α -ketoglutarate), programmed cell death protein 1 (PD-1), Nicotinamide adenine dinucleotide + (NAD+), lactate dehydrogenase (LDH), alanine, serine, cysteine transporter 2 (ASCT2), succinate receptor 1 (SUCNR1), arginase 1 (ARG1), monocarboxylate transporter (MCT), glucose transporter (GLUT) and fatty acid oxidation (FAO).
Table 1.
Summary of ongoing and completed clinical trials targeting metabolism
| Target | Drug name | Phase | Cancer type | NCTa Number |
|---|---|---|---|---|
| Tumor metabolism | ||||
| IDH1b | Ivosidenib (AG-120) |
I | Advanced solid tumors including Glioma | NCT02073994 |
| IDH2 | Enasidenib (AG-221) |
Approved | Acute myeloid leukemia | |
| IDH1/2 | Vorasidenib (AG-881) |
I | Low grade glioma | NCT03343197 |
| FASNc | TVB-2640 | I | Solid tumors | NCT02223247 |
| GLS1d | CB-839 | I/II | IDH-Mutated Diffuse Astrocytoma | NCT03528642 |
| MCT1e | AZD3965 | I | Solid tumors | NCT01791595 |
| PDKf | Dichloroacetate | II | Malignant gliomas | NCT00540176 |
| Immuno-metabolism | ||||
| Arginase | CB-1158 | I/II | Solid tumor | NCT03314935 |
| IDO1g | Indoximod (1-MT) |
I | Malignant Brain Tumors | NCT02502708 |
| II | Pediatric brain Tumors | NCT04049669 | ||
| I/II | Malignant Brain Tumors | NCT02052648 | ||
| Epacadostat | II | Recurrent Glioblastoma | NCT03532295 | |
| I/II | Multiple solid malignancies | NCT02178722, NCT02327078 | ||
| Adenosine | TTX-030 | NCT03884556 | ||
| IPH5201 | I | Solid tumors | NCT04261075 | |
| SRF-617 | NCT04336098 | |||
| TLR3h | Poly-ICLC | I/II | Glioblastoma | NCT00262730 |
| TLR7/8 | MEDI9197 | I | Solid tumor | NCT02556463 |
National Clinical Trial,
Isocitrate dehydrogenase 1,
Fatty acid synthase,
Glutaminase,
Monocarboxylate transporter 1,
Pyruvate dehydrogenase kinase,
Indoleamine 2, 3-dioxygenase 1,
Toll-like receptor 3
Figure 2.
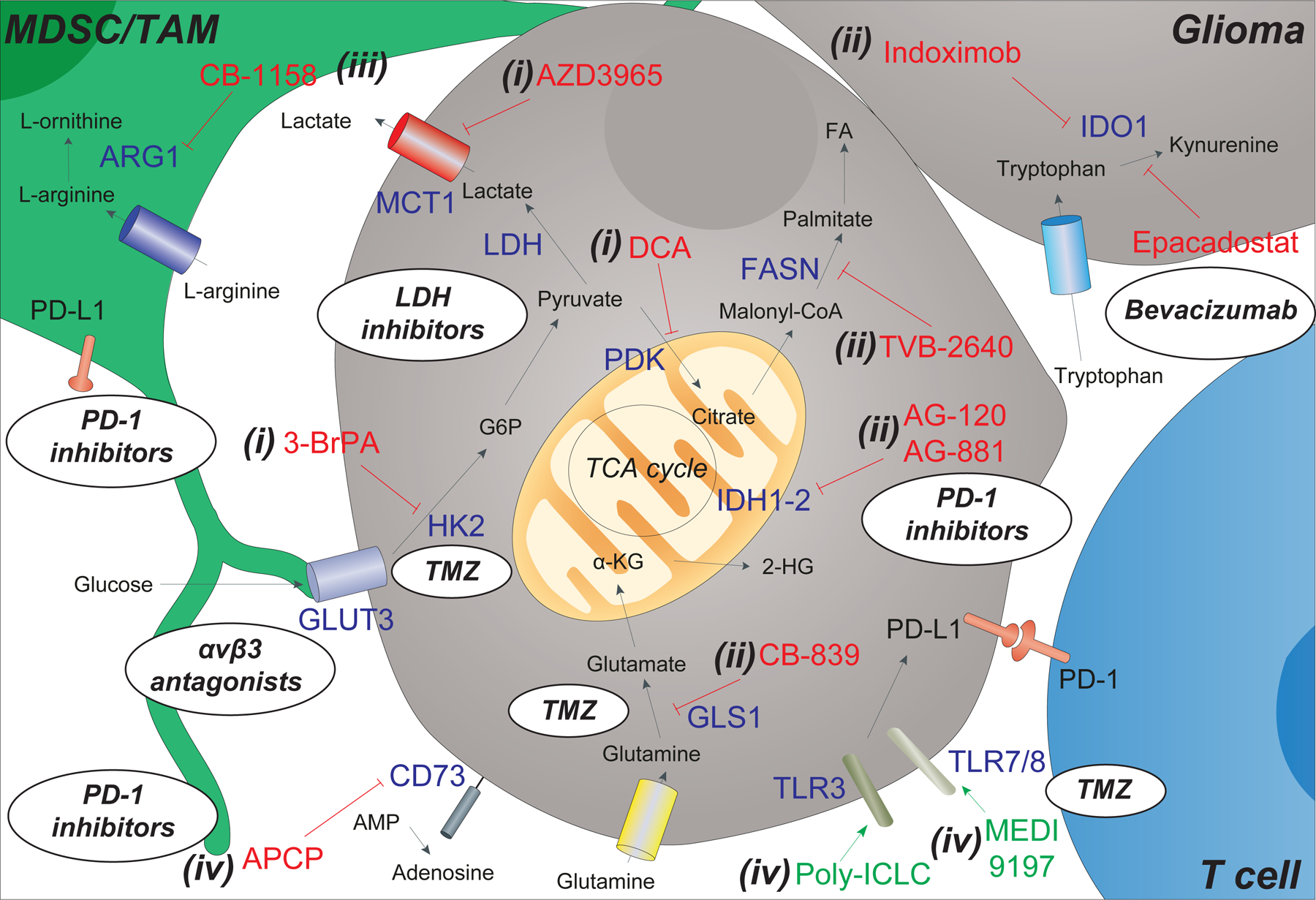
Targeting tumor metabolism in gliomas with biologics and small molecule inhibitors. Cell metabolism and the availability of metabolic substrates in the glioma microenvironment is dependent on the activity of multiple cell types, including myeloid-derived suppressor cells (MDSCs), tumor associate macrophages (TAMs), glioma cells, and T cells. Inhibitors (in red) and agonists (in green) aimed at interfering with specific targets (blue) of this complex metabolic network have been developed and are under clinical testing. These include (i) inhibitors that act on altered glucose and lactate metabolism in gliomas [such as 3-brompyruvate (3-BrPA), dichloroacetate (DCA) and AZD3965]; (ii) inhibitors of other metabolic enzymes in glioma [such as isocitrate dehydrogenases (IDH) 1–2 via ivosidenib (AG-120) and vorasidenib (AG-881), glutaminase (GLS1) via CB-839, fatty acid synthase (FASN) via TVB-2640, and indoleamine 2,3-dioxygenase (IDO)1 via indoximod (1-MT) and epacadostat (INCB024360)]; (iii) inhibitors of arginase (ARG) 1 in TAM via molecules such as CB-1158; and (iv) modulators of membrane bound proteins [such as the 5′-nucleotidase CD73 via α, β-methylene ADP (APCP), and the toll like receptors (TLRs) 3–7/8 via the agonists poly (I:C) stabilized by lysine (poly-ICLC) and MEDI9197]. Oval inserts/bubbles show how current and/or future combination of immunotherapies and chemotherapies can be used to overcome certain metabolic alterations of the glioma. These include the use of αvβ3 antagonists to target glioma cells overexpressing GLUT3, LDH inhibitors (e.g., NCI-006) to target the overexpression of lactate dehydrogenase (LDH), bevacizumab (VEGF inhibitor) in combination with epacadostat (IDO inhibitor), PD-1 inhibitors that are used in synergy with APCP, AB-881, and CB-1158 treatments, and finally the combination of temozolomide (TMZ) with TLR inhibitors and HK2 inhibitors (the latter being able to significantly enhance the efficacy of TMZ treatment).
Other abbreviations: FA (fatty acids), α-KG (α -ketoglutarate), 2-HG (2-hydroxyglutarate), GSH (glutathione), adenosine monophosphate (AMP), Programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1).
Targeting tumor preference for glycolysis
Considerable work has been dedicated to investigating differences in glycolysis that may exist between tumor cells and immune cells in order to create metabolically-targeted antitumor therapies. Chief among them is the different metabolic regulation of T cells vs cancer cells. Effective T cell activation requires a transition to a state of rapid growth and differentiation into effector subtypes (T helper 1 [Th1], Th2, and Th17) that is marked by increased energetic and biosynthetic demands [19]. These demands are met with increased nutrient uptake and increased metabolism, resulting in a significant upregulation of glucose and amino acid transporters [20, 21]. However, many tumors, particularly HGGs, also exhibit aerobic glycolysis, which creates a competition for nutrients between tumor cells and T cells in the tumor microenvironment (TME) [22, 23]. A recent study suggests intrinsic cell programming, not nutrient competition, dictates immune cell metabolism in the TME [24]. Therefore, differences in glycolytic oncometabolism and tumor immunometabolism may be important for future development of effective immunotherapies, such as those seen with specific glucose transporters (Figures 1 and 2).
GBM increases glucose metabolism via aerobic glycolysis and increased expression of glucose transporters 1 and 3 (GLUT1 and GLUT3) for energy production and tumor growth [25]. While GLUT1 is also known to be essential for T cell activation, preferential expression of GLUT3 has been shown to be critical to tumorigenesis and cancer stem cell maintenance, and correlates with poor survival in GBM patients [26, 27]. As such, GLUT3-specific inhibitors that obstruct glucose uptake, decrease glycolytic capacity, and inhibit the growth of patient-derived GBM xenografts have been successfully tested in vitro [28]. Antiangiogenic therapies such as vascular endothelial growth factor (VEGF) inhibitors (e.g., bevacizumab) work by binding circulating VEGF and preventing its interaction with cell surface receptors leading to a reduction in tumoral blood vessel growth and blood supply. Glioma cells overexpressing GLUT3 exhibit features of bevacizumab resistance, such as increased glycolysis, impaired oxidative phosphorylation, and rapid proliferation even in low glucose environments [29]. Importantly, subsets of proneural and classical types of GBM rely on GLUT3 expression, which is aberrantly signaled by integrin αvβ3 via the activation of PAK4-YAP/TAZ [30]. Hence, αvβ3 antagonists may represent another promising therapeutic option to selectively target these chemotherapy and radioresistant populations (Figure 2).
An increase of proteins related to glucose metabolism, such as hexokinase 2 (HK2) and pyruvate dehydrogenase kinase (PDK), has also been observed in HGGs. HK2 catalyzes the phosphorylation of glucose to glucose-6-phosphate and is highly expressed in GBM. Knockdown of HK2 strongly inhibits GBM growth, indicating that HK2 is essential for GBM growth [31]. Conversely, T cells can still function when HK2 is knocked down, highlighting the possibility of selectively targeting this biochemical weakness in tumor cells [32]. Inhibition of HK2 (e.g., via 3-brompyruvate) also enhances the efficacy of temozolomide (TMZ), a cytotoxic alkylating agent that is part of the current standard of care for HGGs (Figure 2) [4, 5, 33]. HK2 inhibition likely has a dual function of sensitizing glioma cells to TMZ and downregulating p-Akt, a protein that contributes to overexpression of GLUT3 and Mcl-1, an anti-apoptotic Bcl-2 family protein that further contributes to TMZ resistance [34, 35].
In hypoxic conditions, GBM highly expresses PDK, an inhibitor of pyruvate dehydrogenase, which regulates the entry of pyruvate into the TCA cycle. In this way, energy production is shifted away from oxidative phosphorylation in favor of glycolysis [36]. Intracranial delivery of the PDK inhibitor dichloroacetate (DCA) effectively reversed the Warburg effect in TMZ resistant cells and significantly improved survival in animals with HGG (Figure 1) [33]. Furthermore, oral administration of DCA in GBM patients results in serum concentrations sufficient to inhibit PDK, thus making this approach highly translatable. However, no firm conclusions regarding DCA as a treatment for GBM in humans can be made, as only a small number of patients have been treated (NCT00540176) [37]. Agents that inhibit glucose metabolism, including the PDK inhibitor DCA, are also being explored as a potential avenue to combat glycolysis-mediated radiation resistance in HGGs. DCA works by inducing cell-cycle arrest, reducing mitochondrial reserve capacity, and increasing oxidative stress and DNA damage. The combination of DCA and radiotherapy in an orthotopic GBM mouse model significantly prolonged median survival [38], underscoring the potential utility of glucose metabolism inhibitors in overcoming TMZ/radiotherapy resistance (Figure 2).
Finally, an Achilles’ heel inherent in the continuous aerobic glycolytic cycling is the requisite downstream regeneration of NAD+, which is primarily accomplished by the conversion of pyruvate to lactate via the enzyme lactate dehydrogenase (LDH). LDH expression is high in HGGs where aerobic glycolysis produces substantial amounts of lactate, which is then secreted by cancer cells (as well as myeloid-derived suppressor cells, MDSCs) into the TME. Once secreted, lactate acts extracellularly as a potent immunosuppressive agent through acidification of the TME and intracellularly as an activator of anti-inflammatory signaling cascades in immune cells [39]. Lactate has a pan-immune suppressive effect, facilitating an anti-inflammatory phenotype in macrophages [40], NK cells [41, 42], and CD8+ T cells [42, 43], while promoting the function of pro-tumorigenic T-regulatory cells (Tregs) [43, 44]. Lactate, as well as other small molecules released by glioma cells (e.g., succinate) [45, 46], act as key oncometabolites capable of inducing an anti-inflammatory phenotype in surrounding immune cells (Figure 1). Inhibition of LDH activity may promote immune effector cell function and synergize with immunotherapies [44, 47]. It has been shown that LDH inhibition combined with IL-21 augmented the antitumor response of CD8+ stem cell memory T cells in murine models of melanoma [47]. When combined with anti-CTLA-4 therapy, LDH inhibition also forced Tregs to adopt a pro-inflammatory antitumor phenotype in murine models of mammary carcinoma [44]. Moreover, pharmacologic inhibition of LDH has been shown to prevent cancer stem cell formation and to induce differentiation and apoptosis of cancer stem cells in vitro [48]. The antitumor effects of LDH inhibition can also be augmented by inhibition of the mitochondrial Complex I, which oxidizes NADH, in the mitochondrial electron transport chain, effectively disrupting the NAD+/NADH redox couple in cancer cells [49]. Recent development of highly potent inhibitors of LDH provides an opportunity to assess LDH’s effect on tumor proliferation and immune cell recruitment and activation in animal models [50]. Further work using hyperpolarized MRI demonstrates a rapid metabolic rewiring and a prolonged median overall survival in patient-derived murine xenografts of pancreatic cancer after administration of one of these LDH inhibitors (NCI-006) [49]. Altogether, each of these selective vulnerabilities in tumor glycolytic metabolism has the potential to enhance the response to immunotherapy.
Targeting altered glutamine metabolism in IDH-mutant gliomas
Mutations in isocitrate dehydrogenase 1 (IDH1), a driver of early oncogenesis in many hematologic and solid malignancies, are defining genetic alteration present in ~12% of adult GBMs and nearly all secondary GBMs that progress from lower grade astrocytomas [51]. IDH normally catalyzes the conversion of isocitrate into α-ketoglutarate and NADPH. However, IDH1 mutations in cancer cells further lead to the conversion of α-ketoglutarate into 2-HG. 2-HG is an oncometabolite that dysregulates oxygen-sensing machinery through accumulation of HIF-1α and induces a hypermethylation phenotype in proneural GBM cells that promotes tumorigenesis [52, 53]. Ivosidenib (AG-120), a small molecular inhibitor of IDH1 approved by the FDA for treatment of IDH1-mutated acute myeloid leukemia, induces 2-HG suppression and is currently in clinical trials in patients with advanced solid tumors (NCT02073994) (Figure 2). [54]. In a recent case report, one patient with IDH1-mutant GBM enrolled in the study exhibited radiographically stable disease for 4 years on ivosidenib [55]. Ivosidenib is also being studied in combination with PD-1 inhibitor nivolumab (NCT04044209). Lastly, vorasidenib (AG-881), a dual IDH1/IDH2 inhibitor designed for enhanced BBB penetration is currently being trialed in patients with IDH1-mutant gliomas (NCT03343197) (Figure 2).
Of note, elevated levels of 2-HG in IDH-mutant gliomas also inhibit branched chain amino acid production, and force tumors to become increasingly dependent on glutamine as a catabolic substrate [56]. Metabolic reprogramming and upregulation of oncogenic pathways substantially increase glutamine metabolism within cancer cells. This is necessary, at least in part, for downstream production of the glutamate needed for the antioxidant glutathione, which enables cancer cells to cope with oxidative stress. Additionally, extracellular glutamine is a requirement for cellular survival and T cell signaling and function in vitro, making the inhibition of glutaminolysis in cancer cells an attractive therapeutic target [57]. Clinical attempts to target glutamine metabolism have largely focused on inhibiting glutaminase (Figure 1). CB-839, a small molecule glutaminase inhibitor, robustly depletes intracellular glutamate and glutathione in IDH-mutant glioma cell lines [56]. CB-839 is currently being tested in combination with TMZ and radiation in IDH1-mutated gliomas (NCT03528642) and CB-839 in combination with nivolumab (NCT02771626) (Figure 2).
Interestingly, recent research indicated that IDH1-mutant gliomas have reduced expression of both cytotoxic T lymphocyte associated genes and chemokines such as CXCL10 that recruit immune effector cells [58, 59]. Investigation of the molecular underpinnings of this observation revealed that 2-HG suppresses production of STAT1, a regulator of CXCL10 [58]. Further work indicated that 2-HG also interferes with nuclear factor of activated T cells (NFAT) transcriptional activity, T cell receptor signaling, and polyamine biosynthesis, providing additional evidence that 2-HG plays a role in antitumor immunity [60]. These studies suggest that IDH inhibitors may be used to synergize with other immunotherapy agents (Figure 1).
Targeting arginase in immune cells
Arginase catalyzes the hydrolysis of arginine to urea and ornithine. Two isoforms, arginase 1 (ARG1) and arginase 2 (ARG2), have been characterized and are encoded by separate but functionally equivalent genes [40, 61]. Arginase expression is regulated in immune cells, where it may act intracellularly (cytoplasmic ARG1 and mitochondrial ARG2) and extracellularly (secreted ARG1) leading to the local depletion of L-arginine in the TME [62].
The antitumor function of T cells is closely linked to arginine metabolism [63]. Elevated extracellular arginine levels lead to a global metabolic reprogramming in T cells that initiates an orchestrated switch from glycolysis to oxidative phosphorylation and promotes a central memory-like phenotype with enhanced T cell survival [64]. On the other hand, low arginine levels due to increased arginase activity (such as that seen in MDSCs) have been associated with inhibition of anti-tumor T cell immune responses. Indeed, higher arginase activity in MDSCs has been observed in several cancer patient populations, including GBM [63, 65], and the chemical inhibition of arginase in MDSCs has been shown to improve T cell function [65]. Thus, the use of arginase inhibitors may help overcome the immunosuppressive tumor microenvironment and achieve more robust antitumor control, particularly when used in combination with other immunotherapeutic agents and/or radiotherapy.
CB-1158, a potent oral ARG1 inhibitor, blocks myeloid cell-mediated suppression of T cell proliferation in vitro and reduced tumor growth in murine models of breast, lung, colon and skin cancers (Figure 1). CB-1158 further augments antitumor efficacy when administered in combination with other immunotherapeutic and chemotherapeutic agents [66]. CB-1158 is currently being studied in Phase I/II clinical trials alone and in combination with chemotherapeutic agents or nivolumab in patients with solid metastatic tumors (NCT0203914, NCT03314935) (Figure 2) [67].
Targeting IDO in GBM
Indoleamine 2,3-dioxygenase (IDO) is a heme-containing enzyme that converts tryptophan to kynurenine derivatives. The immunomodulatory effects of IDO are related to the increase in kynurenine and the depletion of extracellular tryptophan. Kynurenine is an endogenous agonist of the aryl hydrocarbon receptor (AhR) in both T cells and dendritic cells (DCs), where it has been shown to hinder antitumor immunity. Activation of AhR leads to conversion of T effector cells into Tregs and amplifies immunosuppressive activity of Tregs by upregulating IDO expression in DCs [68]. IDO expression also facilitates an immunosuppressive tumor microenvironment by promoting MDSCs, suppressing the function of antigen presenting cells, and decreasing cytotoxicity of CD8+ T cells [69, 70]. Further confounding the issue, IDO expression has more recently been found to increase in normal brain tissue as a consequence of aging. While still incompletely understood, data suggest that increased IDO expression in advanced age (such as in the case of typical GBM with mean age at diagnosis of 65 years old) has a negative impact on immunotherapeutic efficacy[71]. Overexpression of IDO by tumor cells leads to reduced levels of tryptophan in the microenvironment, leading to activation of the kinase general control nonderepressible 2 (GCN2) and inhibition of mTOR. Low extracellular tryptophan, coupled with the toxicity of tryptophan catabolites, inhibits tumor infiltrating lymphocytes (TILs) proliferation [72, 73].
Recent studies have investigated the clinical relevance of IDO and attempted to inhibit its expression in GBM (Figure 2). GBM have higher IDO expression compared to LGG, and there is a clear association between IDO expression in GBM and decreased overall survival [74]. Oral administration of indoximod, or 1-methyl-L-tryptophan (1-MT), an IDO inhibitor, significantly suppressed subcutaneous tumor growth in murine GL261 glioma models and synergistically enhanced antitumor efficacy when administered concomitantly with TMZ (Figure 1). However, no significant benefit was observed in intracranial GL261 glioma models treated with 1-MT and TMZ when compared to mice treated with TMZ alone. Intracranial delivery of shRNA-mediated IDO knockdown in GL261 cells significantly prolonged survival, though no significant difference in survival was observed in human U87 orthotopic xenograft models [75]. Indoximod was tested in a phase I trial in children with recurrent malignant brain tumors (NCT02502708) and a phase I/II clinical trial in adults with recurrent TMZ-resistant HGG (NCT02052648), and results are forthcoming. Currently, a phase II trial of indoximod with chemotherapy and/or radiation in progressive HGG, medulloblastoma, ependymoma, or newly-diagnosed diffuse intrinsic pontine glioma (NCT04049669) is ongoing.
Epacadostat (INCB024360) is another selective small-molecule inhibitor of IDO that is being evaluated in phase I/II clinical trials with recurrent GBM (NCT03532295) and other solid malignancies (NCT02327078 and NCT02178722) (Figure 1). Of note, in patients with unresectable or metastatic melanoma (NCT02752074), epacadostat failed to improve progression-free survival or overall survival when administered with pembrolizumab or as a monotherapy despite potent biological activity and selectivity [76]. Yet, results of a Phase I/II trial of epacadostat and nivolumab in glioma patients demonstrated that the combination is generally well-tolerated and resulted in a disease control rate of 70% (NCT02327078) [77]. Additional trials are testing epacadostat in combination with radiation and bevacizumab (NCT03532295). These results highlight the need for additional pre-clinical investigations and clinical trials to define the optimal use of IDO inhibitors.
Targeting adenosine metabolism in immune cells and non-immune cells
The TME contains elevated levels of extracellular adenosine, which upregulates anti-inflammatory molecules and immunoregulatory cells, collectively establishing a durable immunosuppressive environment [78, 79]. Mechanistically, tumor hypoxia, arising from disorganized tumor vasculature and the high oxygen demand required to sustain rapid cellular proliferation, increases extracellular adenosine production via induction of the CD39-CD73 axis [80]. CD39 is expressed by various immune cells and non-immune cells (such as endothelial cells and cancer cells). CD73 is an extracellular adenosine-generating enzyme that is involved in cell growth, maturation and differentiation and can be co-opted by tumor cells to facilitate cell adhesion, proliferation, invasion, and angiogenesis [81, 82]. Both CD39 and CD73 are also overexpressed on Tregs, and the adenosine formed by their catalysis engages A2A receptors on T cells. Experimentally, activation of A2A suppresses T cell proliferation [83]. Within the TME, accumulated extracellular ATP can either stimulate cell inflammation activity via type 2 purinergic receptors (P2XRs and P2YRs) or be degraded to immunosuppressive adenosine by the sequential action of the ectonucleotidases CD39 and CD73 [79].
Adenosine transmits an immunosuppressive signal through four G protein-coupled adenosine receptors (A1, A2A, A2B, and A3) that work in concert to exert tumorigenic effects [78, 84]. The adenosine A2A receptor (A2AR) is upregulated in TILs and CD11b+ tumor-infiltrating monocytes/macrophages in patient-derived GBM models [85]. Purinergic signaling and the CD73/A2BR axis play multifaceted roles in gliomagenesis and tumor progression and is therefore under investigation as a novel target for GBM therapy [86, 87]. Interestingly, Yan and colleagues established a GBM mouse model in which CD73 was spatially expressed only on endothelial cells in CD73 knockout mice (CD73-FLK). While CD73−/− mice had decreased tumor size, tumor vessel density, and tumor invasiveness as compared to wild-type mice, CD73-FLK tumors were more invasive, resulting in complete distortion of brain morphology, and showed a 20-fold upregulation of A2BAR relative to controls [82]. Treatment of glioma cells with a CD73 inhibitor, APCP (α,β-methylene ADP), reduced glioma cell proliferation by 30% [81]. Though A2AR inhibitors have not yet been studied in glioma patients, there are a number of ongoing clinical trials investigating its use in combination with anti-PD-1/PD-L1 therapy or in the setting of anti-PD-1/PD-L1 treatment resistance (Figure 2).
Targeting Toll-like Receptors in innate immune cells
Toll-like receptors (TLRs) are type I transmembrane proteins that belong to the pattern recognition receptor family and mediate pro-inflammatory responses in innate immune cells. TLRs also play a complex role in tumorigenesis and cancer progression via the modulation of inflammation in the TME [88]. TLR-mediated reprogramming of macrophages and DCs orchestrates a metabolic switch from oxidative phosphorylation to glycolysis, allowing for cancer cell biosynthesis and shunting of glucose away from TILs to maintain a suppressive pro-tumor microenvironment [89]. Critically, TLRs can also promote an anti-tumor response by priming the immune system against malignant cells.
Therefore, TLR-based cancer immunotherapy is under active development. Treatment of patient-derived GBM cell lines with TLR3 agonist polyinosine-polycytidylic acid (poly [I:C]) stimulates expression of PD-L1 and PD-L2 and a pro-inflammatory secretome comprising TNF-α and various chemokines [90]. Poly (I:C) treatment strengthened the response of immune checkpoint inhibitors and increased attraction of CD8+ T cells, and to a lesser extent CD4+ T cells, via a CXCR3- and CCR5-mediated mechanism [91]. Poly-ICLC (poly [I:C]) stabilized by lysine and carboxymethylcellulose), a synthetic double-stranded RNA viral mimic that binds TLR3, MDA5 and other pathogen receptors, has been tested in a phase II clinical trial (NCT00262730) (Figure 2) [92]. In combination with radiation and TMZ in adults with newly diagnosed GBM, poly-ICLC did not produce any significant toxicities. The combination also resulted in a median overall survival of 18.3 months, which compares favorably to the 14.6 months reported by the phase 3 EORTC trial with radiotherapy and TMZ [4], suggesting that poly-ICLC may improve the efficacy of chemo-radiation [93].
In addition, MEDI9197, a dual TLR7/8 agonist, is in a phase I trial as part of a combination therapy regimen for other metastatic or locally advanced solid malignancies (Figure 2) (NCT02556463). Preliminary results have shown that the agent is well-tolerated, though survival data has not yet been published [94]. Quantitative image analysis and RNAseq analysis of paired tumor biopsies showed an enrichment in CD8+ T cells, CD40+ myeloid and B cells (CD40), and CD56+ NK cells, as well as an increase in tumoral PD-1 and PD-L1 gene expression following treatment [95].
Combination therapy and future directions
Clinical benefit from monotherapy with immune and antimetabolic agents, while promising, is still limited and combination approaches will likely be required to provide greater efficacy. For example, immune checkpoint inhibitors such as pembrolizumab (monoclonal antibody against the PD-1 receptor) have been studied in GBM. It has failed to show efficacy in randomized phase II trials for unselected cases of recurrent GBM either alone or with concomitant bevacizumab therapy [96]. Similarly, results from the CheckMate randomized phase III trial demonstrate that treatment with nivolumab (monoclonal antibody against the PD-1 receptor) does not lead to improved survival compared with bevacizumab therapy in recurrent GBM [97]. Vaccine therapies have had similarly disappointing results. For example, the ACT IV multicenter randomized phase III trial evaluated rindopepimut (EGFRvIII peptide vaccine) with standard dose TMZ versus control failed to show benefit to overall survival at interim analysis [98].
Combinatorial approaches that are being explored include combination with other immunotherapies including oncolytic virotherapy, vaccines, and chimeric antigen receptor (CAR) T cell therapy. Oncolytic virotherapy has been shown to upregulate immunosuppressive metabolites and ligands in the tumor stroma [99, 100], stimulating a durable innate and adaptive immune response [101]. Adjuvant therapies targeting oncometabolism and the immunosuppressive microenvironment have potential to enhance both the direct killing effect of oncolytic viruses and the immune response following virotherapy-induced oncolysis [100]. A variety of cancer vaccine technologies are also being developed and tested in adults and children including dendritic cells, peptides, nucleic acids, and viral vectors [102]. One approach being studied to enhance peptide vaccines is combining the vaccine with a potent TLR immunostimulant such as poly-ICLC or using a peptide-TLR-7/8 agonist conjugate vaccine. Poly-ICLC has also been used as an adjuvant agent for HLA-A2+ children with recurrent low-grade glioma or high-grade glioma receiving vaccination with peptide epitopes derived from glioma-associated antigens, EphA2, IL-13 receptor alpha2, and survivin [103, 104]. Immunoreactivity to at least one of the antigens was seen in most patients. Finally, recent evidence suggests signaling of specific co-receptor domains on CAR T cells can alter T cell metabolism and promote CD8+ central memory T cells with enhanced respiratory capacity and increased fatty acid oxidation [105]. Together, these data suggest that immunotherapies may be enhanced through combinatorial approaches designed to target metabolism of tumor cells, immune cells, or both.
Conclusions
Major advances in the understanding of tumor biology, molecular biochemistry, and genetics have led to therapeutic breakthroughs in cancer treatment. The advent of immunotherapy is poised to further transform patient outcomes in the coming decades. The successful application of immunotherapeutic and immuno-metabolic strategies in adult and pediatric malignant brain tumors like HGG, however, faces significant challenges given the immunologically ‘cold’ and frequently immunosuppressive microenvironment of tumors and the immune privileged location. Opportunities are being explored both preclinically and clinically to specifically target metabolic processes in tumor cells and immune cells in the TME to improve the efficacy of immunotherapy. Combinational treatment approaches that exploit the interaction between heterogeneous tumors and their microenvironments have shown great clinical promise. Barriers to successful implementation of such combinatorial approaches rest on the genetic heterogeneity of all tumors, methods of drug delivery to achieve penetration into the tumor, optimal timing of adjuvant therapy with respect to delivery of immunotherapy that will promote the subsequent immune response, and the challenge of precisely targeting metabolism of tumor cells in the TME. As such, future research should focus on overcoming these barriers as well as on identifying synergies between metabolism and immunotherapy to maximize the antitumor potential of personalized treatment strategies.
Acknowledgements:
GKF is supported by grants from the U.S. Food and Drug Administration (R01FD005379 and R01FD006368), the Rally Foundation for Childhood Cancer Research, Hyundai Hope on Wheels, Andrew McDonough B+ Foundation, and Cannonball Kids’ cancer Foundation. LP-J has been supported by a senior research fellowship FISM - Fondazione Italiana Sclerosi Multipla - cod. 2017/B/5 and financed or co financed with the ‘5 per mille’ public funding, a Wellcome Trust CRCD Fellowship (RG G105713), and the Addenbrooke’s Charitable Trust (RG 97519). EMT is supported by the Department of Defense (CA171067), The Musella Foundation for Brain Tumor Research and Information, and the Pediatric Brain Tumor Foundation. We thank Adam A. Dmytriw for reviewing the manuscript.
Footnotes
Conflicts of interest: JDB has an equity position in Treovir LLC, an oHSV clinical stage company and is a member of the POCKiT Diagnostics Board of Scientific Advisors. The remaining authors declared that no conflict of interest exists.
References:
- 1.Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol 2019; 21: v1–v100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rogers TW, Toor G, Drummond K, Love C, Field K, Asher R et al. The 2016 revision of the WHO Classification of Central Nervous System Tumours: retrospective application to a cohort of diffuse gliomas. J Neurooncol 2018; 137: 181–189. [DOI] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 2021; 23: 1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med 2005; 352: 987–996. [DOI] [PubMed] [Google Scholar]
- 5.Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol (Clinical Trial, Phase II) 2011; 13: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mooney J, Bernstock JD, Ilyas A, Ibrahim A, Yamashita D, Markert JM et al. Current Approaches and Challenges in the Molecular Therapeutic Targeting of Glioblastoma. World Neurosurg 2019; 129: 90–100. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Kroemer G. Preface: oncometabolism: a new field of research with profound therapeutic implications. Methods Enzymol 2014; 542: xix–xxiii. [DOI] [PubMed] [Google Scholar]
- 8.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010; 465: 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sciacovelli M, Frezza C. Fumarate drives EMT in renal cancer. Cell Death Differ 2017; 24: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov 2013; 12: 829–846. [DOI] [PubMed] [Google Scholar]
- 11.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab 2016; 23: 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kareva I, Hahnfeldt P. The emerging “hallmarks” of metabolic reprogramming and immune evasion: distinct or linked? Cancer Res 2013; 73: 2737–2742. [DOI] [PubMed] [Google Scholar]
- 13.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol 1927; 8: 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill BG, Shiva S, Ballinger S, Zhang J, Darley-Usmar VM. Bioenergetics and translational metabolism: implications for genetics, physiology and precision medicine. Biol Chem 2019; 401: 3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 2013; 19: 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kathagen-Buhmann A, Schulte A, Weller J, Holz M, Herold-Mende C, Glass R et al. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol 2016; 18: 1219–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR et al. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol 2017; 19: 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Libby CJ, Tran AN, Scott SE, Griguer C, Hjelmeland AB. The Pro-tumorigenic Effects of Metabolic Alterations in Glioblastoma Including Brain Tumor Initiating Cells. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2018. [DOI] [PMC free article] [PubMed]
- 19.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010; 28: 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 2013; 14: 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002; 16: 769–777. [DOI] [PubMed] [Google Scholar]
- 22.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab 2018; 27: 977–987 e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015; 162: 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021; 593: 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou W, Wahl DR. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers (Basel) 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 2013; 16: 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 2014; 20: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Libby CJ, Zhang S, Benavides GA, Scott SE, Li Y, Redmann M et al. Identification of Compounds That Decrease Glioblastoma Growth and Glucose Uptake in Vitro. ACS Chem Biol 2018; 13: 2048–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuang R, Jahangiri A, Mascharak S, Nguyen A, Chandra A, Flanigan PM et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2017; 2: e88815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cosset E, Ilmjarv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF et al. Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell 2017; 32: 856–868 e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanzey M, Abdul Rahim SA, Oudin A, Dirkse A, Kaoma T, Vallar L et al. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS One 2015; 10: e0123544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta MM, Weinberg SE, Steinert EM, Chhiba K, Martinez CA, Gao P et al. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab 2018; 6: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wicks RT, Azadi J, Mangraviti A, Zhang I, Hwang L, Joshi A et al. Local delivery of cancer-cell glycolytic inhibitors in high-grade glioma. Neuro Oncol 2015; 17: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Calve B, Rynkowski M, Le Mercier M, Bruyere C, Lonez C, Gras T et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010; 12: 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velpula KK, Guda MR, Sahu K, Tuszynski J, Asuthkar S, Bach SE et al. Metabolic targeting of EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma. Oncotarget 2017; 8: 35639–35655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuen CA, Asuthkar S, Guda MR, Tsung AJ, Velpula KK. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: a new target gleaned from an old concept. CNS Oncol 2016; 5: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 2: 31ra34. [DOI] [PubMed] [Google Scholar]
- 38.Shen H, Hau E, Joshi S, Dilda PJ, McDonald KL. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol Cancer Ther 2015; 14: 1794–1804. [DOI] [PubMed] [Google Scholar]
- 39.Certo M, Tsai CH, Pucino V, Ho PC, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol 2021; 21: 151–161. [DOI] [PubMed] [Google Scholar]
- 40.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014; 513: 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A 2014; 111: 12823–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 2016; 24: 657–671. [DOI] [PubMed] [Google Scholar]
- 43.Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 2017; 25: 1282–1293 e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 2021; 591: 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Molecular cell 2020; 77: 213–227 e215. [DOI] [PubMed] [Google Scholar]
- 46.Krzak G, Willis CM, Smith JA, Pluchino S, Peruzzotti-Jametti L. Succinate Receptor 1: An Emerging Regulator of Myeloid Cell Function in Inflammation. Trends in immunology 2021; 42: 45–58. [DOI] [PubMed] [Google Scholar]
- 47.Hermans D, Gautam S, Garcia-Canaveras JC, Gromer D, Mitra S, Spolski R et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8(+) T cell stemness and antitumor immunity. Proc Natl Acad Sci U S A 2020; 117: 6047–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daniele S, Giacomelli C, Zappelli E, Granchi C, Trincavelli ML, Minutolo F et al. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci Rep 2015; 5: 15556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oshima N, Ishida R, Kishimoto S, Beebe K, Brender JR, Yamamoto K et al. Dynamic Imaging of LDH Inhibition in Tumors Reveals Rapid In Vivo Metabolic Rewiring and Vulnerability to Combination Therapy. Cell Rep 2020; 30: 1798–1810 e1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rai G, Urban DJ, Mott BT, Hu X, Yang SM, Benavides GA et al. Pyrazole-Based Lactate Dehydrogenase Inhibitors with Optimized Cell Activity and Pharmacokinetic Properties. J Med Chem 2020; 63: 10984–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009; 324: 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012; 483: 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mellinghoff I, Maher E, Wen P, Cloughesy T, Peters K, Choi C et al. Rbtt-03. a Phase 1, Multicenter, Randomized, Open-Label, Perioperative Study of Ag-120 (Ivosidenib) and Ag-881 in Patients With Recurrent, Nonenhancing, Idh1-Mutant, Low-Grade Glioma. Neuro Oncol 2018; 20: vi234–vi234. [Google Scholar]
- 55.Tejera D, Kushnirsky M, Gultekin SH, Lu M, Steelman L, de la Fuente MI. Ivosidenib, an IDH1 inhibitor, in a patient with recurrent, IDH1-mutant glioblastoma: a case report from a Phase I study. CNS Oncol 2020; 9: CNS62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018; 175: 101–116 e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang J, Srivastava S, Zhang J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers (Basel) 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest 2017; 127: 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu F, Huang J, Liu X, Cheng Q, Luo C, Liu Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int 2020; 20: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 2018; 24: 1192–1203. [DOI] [PubMed] [Google Scholar]
- 61.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010; 32: 593–604. [DOI] [PubMed] [Google Scholar]
- 62.Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D et al. Myeloid Cell-Derived Arginase in Cancer Immune Response. Frontiers in immunology 2020; 11: 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol 2016; 18: 1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016; 167: 829–842 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol 2011; 13: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer 2017; 5: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Papadopoulos KP, Tsai FY-C, Bauer TM, Muigai L, Liang Y, Bennett MK et al. CX-1158–101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. Journal of Clinical Oncology 2017; 35: 3005–3005. [Google Scholar]
- 68.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol 2016; 37: 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon K-S, Auffinger B et al. IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts Survival. Clinical Cancer Research 2012; 18: 6110–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer 2015; 3: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhai L, Bell A, Ladomersky E, Lauing KL, Bollu L, Sosman JA et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front Immunol 2020; 11: 1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012; 1: 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003; 9: 1269–1274. [DOI] [PubMed] [Google Scholar]
- 74.Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery 2013; 72: 1031–1038; discussion 1038–1039. [DOI] [PubMed] [Google Scholar]
- 75.Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A, Kinouchi H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J Neurosurg 2016; 124: 1594–1601. [DOI] [PubMed] [Google Scholar]
- 76.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol 2019; 20: 1083–1097. [DOI] [PubMed] [Google Scholar]
- 77.Perez RP, Riese MJ, Lewis KD, Saleh MN, Daud A, Berlin J et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. Journal of Clinical Oncology 2017; 35: 3003–3003. [Google Scholar]
- 78.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 2006; 103: 13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moesta AK, Li XY, Smyth MJ. Targeting CD39 in cancer. Nature reviews Immunology 2020; 20: 739–755. [DOI] [PubMed] [Google Scholar]
- 80.Bullen JW, Tchernyshyov I, Holewinski RJ, DeVine L, Wu F, Venkatraman V et al. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci Signal 2016; 9: ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bavaresco L, Bernardi A, Braganhol E, Cappellari AR, Rockenbach L, Farias PF et al. The role of ecto-5’-nucleotidase/CD73 in glioma cell line proliferation. Mol Cell Biochem 2008; 319: 61–68. [DOI] [PubMed] [Google Scholar]
- 82.Yan A, Joachims ML, Thompson LF, Miller AD, Canoll PD, Bynoe MS. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A2B Adenosine Receptor Signaling. J Neurosci 2019; 39: 4387–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204: 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol 2014; 5: 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sheth S, Brito R, Mukherjea D, Rybak LP, Ramkumar V. Adenosine Receptors: Expression, Function and Regulation. International Journal of Molecular Sciences 2014; 15: 2024–2052-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu S, Shao QQ, Sun JT, Yang N, Xie Q, Wang DH et al. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro Oncol 2013; 15: 1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ott M, Tomaszowski KH, Marisetty A, Kong LY, Wei J, Duna M et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol 2003; 21: 335–376. [DOI] [PubMed] [Google Scholar]
- 89.Huang L, Xu H, Peng G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cellular & Molecular Immunology 2018; 15: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kees T, Lohr J, Noack J, Mora R, Gdynia G, Todt G et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol 2012; 14: 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Waele J, Marcq E, Van Audenaerde JR, Van Loenhout J, Deben C, Zwaenepoel K et al. Poly(I:C) primes primary human glioblastoma cells for an immune response invigorated by PD-L1 blockade. Oncoimmunology 2018; 7: e1407899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kyi C, Roudko V, Sabado R, Saenger Y, Loging W, Mandeli J et al. Therapeutic Immune Modulation Against Solid Cancers with Intratumoral Poly-ICLC: A Pilot Trial. Clinical Cancer Research 2018; 24: clincanres.1866.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosenfeld MR, Chamberlain MC, Grossman SA, Peereboom DM, Lesser GJ, Batchelor TT et al. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol 2010; 12: 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gupta S, Grilley-Olson J, Hong D, Marabelle A, Munster P, Aggarwal R et al. Abstract CT091: Safety and pharmacodynamic activity of MEDI9197, a TLR 7/8 agonist, administered intratumorally in subjects with solid tumors2017: CT091-CT091-CT091
- 95.Siu L, Brody J, Gupta S, Marabelle A, Jimeno A, Munster P et al. Safety and clinical activity of intratumoral MEDI9197 alone and in combination with durvalumab and/or palliative radiation therapy in patients with advanced solid tumors. J Immunother Cancer 2020; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin Cancer Res 2021; 27: 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 2020; 6: 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017; 18: 1373–1385. [DOI] [PubMed] [Google Scholar]
- 99.Bernstock JD, Vicario N, Rong L, Valdes PA, Choi BD, Chen JA et al. A novel in situ multiplex immunofluorescence panel for the assessment of tumor immunopathology and response to virotherapy in pediatric glioblastoma reveals a role for checkpoint protein inhibition. Oncoimmunology 2019; 8: e1678921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Totsch SK, Schlappi C, Kang KD, Ishizuka AS, Lynn GM, Fox B et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene 2019; 38: 6159–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. The New England journal of medicine 2021; 384: 1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Olsen HE, Lynn GM, Valdes PA, Cerecedo Lopez CD, Ishizuka AS, Arnaout O et al. Therapeutic cancer vaccines for pediatric malignancies: advances, challenges, and emerging technologies. Neurooncol Adv 2021; 3: vdab027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Normolle DP et al. Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro Oncol 2016; 18: 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Muller S, Agnihotri S, Shoger KE, Myers MI, Smith N, Chaparala S et al. Peptide vaccine immunotherapy biomarkers and response patterns in pediatric gliomas. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr. et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016; 44: 380–390. [DOI] [PubMed] [Google Scholar]