

Abstract
Delta VOC is highly diverse with more than 120 sublineages already described as of November 30, 2021. In this study, through active monitoring of circulating severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) variants in the state of São Paulo, southeast Brazil, we identified two emerging sublineages from the ancestral AY.43 strain which were classified as AY.43.1 and AY.43.2. These sublineages were defined by the following characteristic nonsynonymous mutations ORF1ab:A4133V and ORF3a:T14I for the AY.43.1 and ORF1ab:G1155C for the AY.43.2 and our analysis reveals that they might have a likely‐Brazilian origin. Much is still unknown regarding their dissemination in the state of São Paulo and Brazil as well as their potential impact on the ongoing vaccination process. However, the results obtained in this study reinforce the importance of genomic surveillance activity for timely identification of emerging SARS‐CoV‐2 variants which can impact the ongoing SARS‐CoV‐2 vaccination and public health policies.
Keywords: AY.43, Delta VOC, emerging sublineages, genomic surveillance, SARS‐CoV‐2
1. INTRODUCTION
Currently in Brazil have been applied more than 381 million doses of anti‐severe acute respiratory syndrome coronavirus‐2 (anti‐SARS‐CoV‐2) vaccines and the individuals who have been fully vaccinated are bordering 140 million, which makes Brazil one of the most vaccinated nations in the world (https://www.gov.br/saude/pt-br/vacinacao). In Brazil, on the background of this solid process of vaccination, the first cases of Delta variant of concern (VOC) introductions were reported. 1 Despite the full substitution of the pre‐existing Gamma VOC in Brazil with the Delta VOC, no exponential growth of the new cases has been observed, most probably due to the ongoing vaccination. Delta VOC is highly diversified, and more than 120 sublineages have been classified within the Pango lineages (https://cov-lineages.org/) with the continuous description of novel lineages and sublineages.
The molecular surveillance of the SARS‐CoV‐2 variants is of crucial importance for tracking the genomic profile of this virus and the accumulation of mutations which on one hand can alter the viral functions in terms of infectivity and transmissibility and on the other might be important for the emergence of novel variants, lineages, and sublineages which can exert significant pressure on the healthcare system of a given country. 2
In this study, we identified two emerging Brazilian sublineages belonging to the ancestral AY.43 lineage, which were named AY.43.1 and AY.43.2. Most of these sequences originated from the city of São Paulo and the genome monitoring was performed by the Butantan Network for Pandemic Alert of SARS‐CoV‐2 Variants (Butantan Network) of the São Paulo State.
2. MATERIALS AND METHODS
From October 8 to October 30, 2021, nasopharyngeal swab samples were collected by Butantan Network from inhabitants suspected to be infected with SARS‐CoV‐2 from all 17 Health Divisions of the São Paulo State. Viral ribonucleic acid (RNA) was isolated from 100 µl of nasopharyngeal swab suspension using the Extracta Kit RNA viral (Loccus) in 96‐well plate automated extractor (Extracta, Loccus) following the manufacturer's instructions. All samples were tested for SARS‐CoV‐2 by targeting viral RdRp, E, and N genes using the real‐time polymerase chain reaction (PCR) assay Gene FinderTM COVID19 Plus RealAmp Kit (Osang Healthcare Co. Ltd.), and positive samples with C t below 30 were selected for whole genome sequencing. The RNA from positive samples was re‐extracted from the original nasopharyngeal swab suspension for sequencing. The SARS‐CoV‐2 sequencing libraries were prepared using the COVIDSeq Kit (Illumina Inc.), which amplifies the whole SARS‐CoV‐2 genome using the ARTIC v3 tilling PCR primer panel. 3 Paired‐end libraries were sequenced on Illumina's MiSeq (V2 kit, 2150 cycles) or NextSeq. 2000 (P2 kit, 2100 cycles) platform.
To obtain the final SARS‐CoV‐2 genome sequences, the following bioinformatics approaches were performed. The raw sequencing data obtained were submitted to quality control analysis using the FastaQC 4 software version 0.11.8. To select the sequences with the best quality score (>30), quality filtering was performed using Trimmomatic 5 version 0.3.9. We mapped the quality‐filtered sequences against the SARS‐CoV‐2 reference (Genbank RefSeq NC_045512.2) using BWA 6 and used SAMtools 7 for indexing the mapping results. The mapped files were submitted for improvement using Pilon 8 to correct possible deletions and insertions caused by the mapping process. The quality‐filtered sequences were subjected to a remapping against the genome improved by Pilon. Finally, we use bcftools 9 for variant calling and seqtk 10 for the assembly of the consensus SARS‐CoV‐2 genomes. Positions covered by fewer than 10 reads (DP < 10) and bases with a quality score lower than 30 were considered as an assembly gap and thus converted into Ns. Coverage values for each genome were calculated using SAMtools v1.12. We assessed the consensus genome sequence quality using Nextclade v0.8.1. 11
Phylogenetic analysis was performed using the Nextstrain v3.0.3 SARS‐CoV‐2 workflow. 11 In brief, this workflow aligns the input sequences using nextalign and then reconstructs the phylogenetic trees using the IQTree v.2. 12 Subsequently, TreeTime 13 is used to reroot the resulting tree, resolve polytomies, prune sequences, infer internal node dates and label them. To reconstruct the phylogeny of the AY.43 hypothetical sublineages we used 1616 SARS‐CoV‐2 sequences generated from the Butantan Network distributed between the epidemiological Weeks 41−43 (GISAID accession ID in File S1) as input for the Nextstrain workflow. To use as a background for our analysis, a representative global data set was retrieved from GISAID (3711 sequences, downloaded from nextregions global on November 10, 2021—GISAID accession ID in File S2) (https://www.gisaid.org/).
3. RESULTS
In the performed phylogenetic analysis we observed a large cluster of the AY.43 lineage containing 492 sequences, the majority of which (n = 352, 71.5%) were from the Butantan Network, present inside the Brazilian clade (Figure 1). The AY.43 sublineage can be distinguished from other AY sublineages of Delta VOC by the presence of mutations N:Q9L and ORF9b:S6C (https://www.pango.network/summary-of-designated-ay-lineages/). We observed two subdivisions of the AY.43 cluster based on the presence of the nonsynonymous mutations, labeling them as AY.43.1 (ORF1ab: A4133V and ORF3a:T14I) and AY.43.2 (ORF1ab:G1155C). The subcluster of AY.43.1 was composed of 100 strains (Figure 1—purple box), the majority of which were obtained from the city of São Paulo (46.0%). The AY.43.2 subcluster was composed of 99 strains (Figure 1—blue box), of which 97 of them were sequenced by Butantan Network with most sequences from the city of São Paulo (53%).
Figure 1.
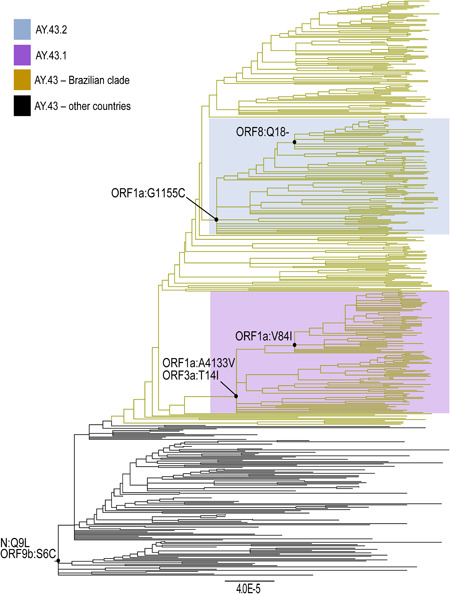
Phylogenetic analysis of the AY.43 lineage clade containing 492 genomes. The newly identified sublineages are highlighted with boxes: AY.43.1 (purple box) and AY.43.2 (blue box), and their characterizing mutations are represented on the newly formed branches. The original tree from which the AY.43 cluster was initially can be observed in Figure S1
The rest of the AY.43 sequences from Butantan Network (n = 155) were mainly obtained from the city of São Paulo (60.0%) and represented the highest number of sequences composing the AY.43 sublineage cluster. The AY.43 sequences, including the newly identified sublineages, were distributed in several cities of the São Paulo State with higher concentrations in the city of São Paulo (Figure 2).
Figure 2.
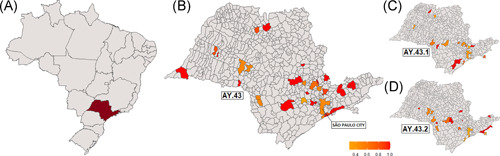
Dissemination of the AY.43 lineage and the newly identified sublineages in the state of São Paulo (A). Map of Brazil with emphasis on the state of São Paulo (B). Dissemination of the AY.43 lineage (C). Dissemination of the AY.43.1 sublineage and (D). Dissemination of the AY.43.2 sublineage. Bar: percentage of the sequenced genomes
A whole phylogenetic interactive tree, from which the AY.43 cluster was initially observed, can be accessed at the following link: https://nextstrain.org/fetch/repositorio.butantan.gov.br/bitstream/butantan/3990/1/ncov_EpiWeek_forty-one-to-forty-three.json
4. DISCUSSION
In this study, we provide a report regarding the characterization of two novels AY.43 sublineages with a likely Brazilian origin, characterized by a specific mutational profile. Considering the number of mutations (localization in ORF1a or ORF3a) we proposed the identification of two emerging sublineages designed as AY.43.1 and AY.43.2. They were additionally recognized as such by the official Pango designation committee (https://github.com/cov-lineages/pango-designation/issues/319) and were released on pangoLEARN release v1.2.96. The obtained data shows the importance of the SARS‐CoV‐2 genomic surveillance for the identification of emerging lineages. This is particularly important because SARS‐CoV‐2 emerging lineages can exert an enormous impact on the public health systems due to increased infectivity and transmission. 2
In the newly characterized sublineages, we defined the mutations A4133V and G1155C in the ORF1a and the T14I mutation in the ORF3a, which were nonsynonymous. To our knowledge, the impact of these mutations is still unknown, except for the T14I, which shows deleterious effects on the viral proteins. 14 A nonsynonymous mutation in ORF3a, which is a conserved protein involved in viral replication and release, 15 may affect viral functions in addition to the mutational constellation defined for the Delta VOC.
We additionally observed other subdivisions within the AY.43 clade: one in the AY.43.1 clade, presenting a nonsynonymous mutation at ORF1a:V84I (Figure 1—purple box); and another at the AY.43.2 clade, presenting a deletion at ORF8:Q18‐. However, at the moment those subdivisions were characterized by a limited number of sequences and did not show sufficient support to be suggestive as novel sublineages of AY.43. Based on the performed analysis the newly classified AY.43.1 and AY.43.2 sublineages probably might have a Brazilian origin. Further studies are necessary to investigate their dissemination within Brazilian regions, but preliminary results show that the majority of AY.43.1 and AY.43.2 sequences originated from the city of São Paulo.
In conclusion, we show that SARS‐CoV‐2 genomic monitoring is crucial for the prompt characterization of SARS‐CoV‐2 novel lineages and sublineages. By this approach, we can timely detect the presence of novel SARS‐CoV‐2 variants and implement strategies for preventing their dissemination which can have further implications on the ongoing SARS‐CoV‐2 vaccination and public health policies.
ETHICS STATEMENT
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Ethics Committee of the Faculty of Medicine of Ribeirão Preto, University of São Paulo (CAAE: 50367721.7.1001.5440).
AUTHOR CONTRIBUTIONS
Alex Ranieri Jerônimo Lima, Gabriela Ribeiro, Vincent Louis Viala, Loyze Paola Oliveira de Lima, Antonio Jorge Martins, Claudia Renata dos Santos Barros, Elaine Cristina Marqueze, Jardelina de Souza Todao Bernardino, Debora Botequio Moretti, Evandra Strazza Rodrigues, Elaine Vieira Santos, Ricardo Augusto Brassaloti, Raquel de Lello Rocha Campos Cassano, Pilar Drummond Sampaio Corrêa Mariani, Luan Gaspar Clemente, Patricia Akemi Assato, Felipe Allan da Silva da Costa, Mirele Daiana Poleti, Jessika Cristina Chagas Lesbon, Elaine Cristina Marqueze, Cecilia Artico Banho, Lívia Sacchetto, Marília Mazzi Moraes, Melissa Palmieri, Maiara Martininghi, Luiz Artur Vieira Caldeira, Fabiana Erica Vilanova da Silva, Rejane Maria Tommasini Grotto, and Jayme A. Souza‐Neto designed and performed the experiments. Alex Ranieri Jerônimo Lima, Gabriela Ribeiro, Svetoslav Nanev Slavov, Maria Carolina Elias, Marta Giovanetti, Luiz Carlos Junior Alcantara, Sandra Coccuzzo Sampaio, and Simone Kashima analyzed the data and wrote the article. Maiara Martininghi, Luiz Artur Vieira Caldeira, and Fabiana Erica Vilanova da Silva evaluated the clinical/epidemiological data and reviewed the article. Alex Ranieri Jerônimo Lima, Gabriela Ribeiro, Svetoslav Nanev Slavov, Maria Carolina Elias, and Marta Giovanetti edited and reviewed the article. Maurício Lacerda Nogueira, Heidge Fukumasu, Luiz Lehmann Coutinho, Simone Kashima, Raul Machado Neto, Dimas Tadeu Covas, Svetoslav Nanev Slavov, Sandra Coccuzzo Sampaio, and Maria Carolina Elias supervised this study. All the authors agreed on the submission of the final manuscript.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We thank all contributors from GISAID. We also thank Gabriela Mauric Frossard Ribeiro and Glaucia Borges for their help with the Instituto Butantan's local database repository. This study was supported by the Fundação Butantan, Fundação de Amparo à Pesquisa do Estado de São Paulo (Grant Numbers: 2020/10127‐1; 2020/06441‐2) and Fundação Hemocentro Ribeirão Preto.
Lima ARJ, Ribeiro G, Viala VL, et al. SARS‐COV‐2 genomic monitoring in the state of São Paulo unveils two emerging AY.43 sublineages. J Med Virol. 2022;94:3394‐3398. 10.1002/jmv.27674
Alex Ranieri Jerônimo Lima and Gabriela Ribeiro authors contributed equally to this study.
Contributor Information
Svetoslav Nanev Slavov, Email: svetoslav.slavov@hemocentro.fmrp.usp.br.
Sandra Coccuzzo Sampaio, Email: sandra.coccuzzo@butantan.gov.br.
Maria Carolina Elias, Email: carolina.eliassabbaga@butantan.gov.br.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in GISAID at https://www.gisaid.org/
REFERENCES
- 1. Patané J, Viala V, Lima L, et al. SARS‐CoV‐2 Delta variant of concern in Brazil—multiple introductions, communitary transmission, and early signs of local evolution. medRxiv. Published online October 2021. 10.1101/2021.09.15.21262846 [DOI] [Google Scholar]
- 2. Khateeb J, Li Y, Zhang H. Emerging SARS‐CoV‐2 variants of concern and potential intervention approaches. Crit Care. 2021;25:244. 10.1186/s13054-021-03662-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DNA Pipelines R&D, Defilippis EM, Sinnenberg L, et al. COVID‐19 ARTIC v3 Illumina library construction and sequencing protocol. ProtocolsIo. 2020;5:1‐16. 10.17504/protocols.io.bibtkann [DOI] [Google Scholar]
- 4. Andrews S. FastQC: a quality control tool for high throughput sequence data. Published online 2019. Acessed March 08, 2022. http://www.bioinformatics.barbaham.ac.uk/projects/fastqc/
- 5. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114‐2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987‐2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li H. seqtk Toolkit for processing sequences in FASTA/Q formats. GitHub. Published online 2018. Accessed March 08, 2022. https://github.com/lh3/seqtk [Google Scholar]
- 11. Hadfield J, Megill C, Bell SM, et al. NextStrain: real‐time tracking of pathogen evolution. Bioinformatics. 2018;34:4121‐4123. 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2015;32(1):268‐274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sagulenko P, Puller V, Neher RA. TreeTime: maximum‐likelihood phylodynamic analysis. Virus Evol. 2018;4(1):1‐9. 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Issa E, Merhi G, Panossian B, Salloum T, Tokajian S. SARS‐CoV‐2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems. 2020;5:e00266‐e00320. 10.1128/msystems.00266-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bianchi M, Borsetti A, Ciccozzi M, Pascarella S. SARS‐Cov‐2 ORF3a: mutability and function. Int J Biol Macromol. 2021;170:820‐826. 10.1016/j.ijbiomac.2020.12.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are openly available in GISAID at https://www.gisaid.org/