

Abstract

A rapid synthesis of aminoboranes from amine-boranes utilizing an iodination/dehydroiodination sequence is described. Monomeric aminoboranes are generated exclusively from several substrate adducts, following an E2-type elimination, with the added base playing a critical role in monomer vs dimer formation. Diisopropylaminoborane formed using this methodology has been applied to a one-pot palladium-catalyzed conversion of iodo- and bromoarenes to the corresponding boronates. Additionally, modification of the workup allows for isolation of the boronic acid and recovery of the utilized amine.
Introduction
Aminoboranes of the type R2N-BH2,1 commonly obtained from amine-boranes (R2NH-BH3), have received considerable attention as valuable precursors for organic and material chemistry applications.2−13 Borylations employing a variety of metal catalysts2 and leaving groups2a,2b,3 have been developed, including for C–H borylation.2d Suitable aryl,2a−2d,3,4 alkenyl,4a,5 and alkynyl2e,2f substrates can be converted to boronate esters,2b−2d,3b,6 their complexes,7 and boronic and borinic acids.3a,7,8 They have been used for the preparation of polyaminoboranes9 and boron nitride ceramics,10 with recent uses in the production of molecular sensors,11 mechanochromic materials,12 and metal thin films.13
Despite all of these developments, there is still a need for a convenient synthesis of aminoboranes. One of the earliest routes involved the reduction of aminodihaloboranes with lithium aluminum hydride [Scheme 1. (i)].14 Large-scale synthesis of aminoboranes using this protocol is limited by the highly reactive nature of the hydride and boron halide reagents. This method has since been supplanted by the thermal dehydrogenation of amine-boranes [Scheme 1. (ii)]2a and reaction of lithium aminoborohydrides with suitable organohalides [Scheme 1. (iii)].15 These routes, however, require elevated temperatures (160–220 °C) or the use of highly sensitive reagents (n-BuLi). Recently, Pucheault and co-workers reported the dehydrohalogenation of monochloroborane–amine complexes (which have been reported elsewhere16) for the preparation of aminoboranes [Scheme 1. (iv)].6b The dry, ethereal HCl utilized in this preparation is cumbersome to prepare,17 and although it is commercially available, it is expensive relative to other halogen sources. Ethereal HCl is typically prepared at concentrations of 1–2 M, as more concentrated solutions tend to expel the highly corrosive HCl gas. Release of the solute gas and the low boiling point of the solvent necessitate frequent titration, as precise stoichiometry is critical for the preparation of monochloroborane-amines. Treatment of amine-boranes with N-halosuccinimides18 or molecular halogens16a,19 and the disproportionation reaction of BH3- and BX3-amines (X = halogen)16a are some of the other procedures that exist for the conversion of amine-boranes to haloborane–amine complexes.
Scheme 1. Preparation of Aminoboranes and One-Pot Borylation.

As part of our ongoing projects on amine-boranes,20 we were interested in developing a simple protocol for the preparation of aminoboranes. It occurred to us that the halogenation of amine-boranes with molecular halogens might provide a more convenient protocol. Although known for several decades,16a the potential utility of this protocol has not been exploited fully in organic synthesis. Described herein are the details of a simple route to aminoboranes via dehydrohalogenation of appropriate monoiodoborane–amine complexes [Scheme 1. (v)], avoiding the limitations of the previous protocols. This methodology has been extended to a modified Pd-catalyzed one-pot borylation of aryl halides [Scheme 1. (vi)].
Results and Discussion
Halogenation
Over 60 years ago, Nöth described the monoiodination of trimethylamine-borane with molecular iodine in benzene.16a A similar procedure to prepare alkylhaloboranes from alkylboranes has also been reported.21 Our initial attempts sought to optimize the conditions for the halogenation of a dialkylamine-borane by reacting dimethylamine-borane (1c) with 0.5 and 1.0 equiv of either bromine or iodine in CH2Cl2 or toluene and following the reaction by 11B NMR spectroscopy.
With bromine, the nearly instantaneous halogenation readily went past the monohaloborane (Table 1 (2)) stage, providing appreciable quantities of the di- (Table 1 (3)) and trihalogenated boranes (Table 1 (4)), as detected in the 11B NMR spectrum. Using 0.5 equiv of Br2 also led to the formation of a mixture of di- and monobromoborane-amine in a 3:1 ratio. On the other hand, when 0.5 equiv of iodine was utilized, the amine-monohaloborane could be reliably produced in either solvent. Even a stoichiometric equivalent of iodine provided the monoiodoborane predominantly (80%). Halogenation of diisopropylamine-borane (1f) using N-chlorosuccinimide (NCS) and N-bromosuccinimide (NBS) was also examined as a potential route to the amine monohaloborane complexes. A full equivalent of NCS provided the desired monochloroborane, whereas NBS gave a mixture of mono- and dihalogenated products (Table 1).
Table 1. Optimization of Halogen and Solvent Studya.

| entry | halogen (equiv) | amine-borane | solvent | 11B NMR peak ratio (1:2:3:4)b |
|---|---|---|---|---|
| 1 | I2 (0.5) | 1c | DCM | 0:1:0:0 |
| 2 | I2 (1.0) | 1c | DCM | 0:4:1:0 |
| 3 | Br2 (0.5) | 1c | DCM | 1.25:1:3:0 |
| 4 | Br2 (1.0) | 1c | DCM | 0:1:1:0.75 |
| 5 | I2 (0.5) | 1c | CHCl3 | indeterminate |
| 6 | I2 (0.5) | 1c | Et2O | indeterminate |
| 7 | I2 (0.5) | 1c | PhMe | 0:1:0:0 |
| 8 | I2 (0.5) | 1c | Pentane | 1c insoluble |
| 9 | NCS (1.0) | 1f | DCM | 0:1:0:0 |
| 10 | NBS (1.0) | 1f | DCM | 1:1.6:2.1:0 |
Reactions were performed at a 2 mmol scale with respect to the amine-borane.
Ratio determined by 11B NMR (96 MHz) spectroscopy.
The scope of the iodination was demonstrated with a series of amine-boranes (1a–1q), prepared from sodium borohydride via a bicarbonate-promoted reaction of the desired amine (1b–1q)20c or by salt metathesis of the corresponding ammonium hydrochloride (1a).22 As detailed in Table 2, all of the primary and secondary amine-borane complexes underwent quantitative iodination without any difficulty.23
Table 2. Amine-Boranes Subjected to the Iodination/Dehydroiodination Sequence and Resulting Aminoboranesa,b,c.


Reactions were performed at a 2 mmol scale with respect to the amine-borane.
Conversions were >99% by 11B NMR (96 MHz) spectroscopy.
Composition determined by 11B NMR (96 MHz) spectroscopy.
Dehydrohalogenation
The dehydrohalogenation of these monoiodoborane–amine complexes was readily accomplished by the addition of a sufficiently bulky amine [i-Pr2NEt (Hünig’s base) or i-Pr2NH] to the reaction mixture.6b,16b Quantitative conversion to the elimination products was observed in the 11B NMR spectra. The monomeric or dimeric aminoborane species were typically produced, along with an equivalent of the corresponding ammonium iodide during the E2-type reaction (Table 2). Several groups2a,14,15,24 previously used NMR spectroscopy, mass spectrometry, and X-ray crystallography to fully characterize both the aminoborane monomer14,24a and dimer24,25 products. The assignment of 11B NMR peaks to either monomer or dimer products in the present reaction was based on the similarity of 11B NMR values observed from the fully characterized prior products. Similar complete characterizations have previously been made of the other proposed species detected in the present reaction including diaminoborane,26 aminodiborane,27 polyaminoborane and amine-exchange products.9,28 The proposed identities of these species are based on agreement between the 11B NMR chemical shifts observed and those previously reported for similar fully characterized products. The iodination of pyrrolidine-borane and subsequent dehydroiodination using N,N-diisopropylethylamine (Scheme 2) followed by analysis using 11B NMR spectroscopy revealed the formation of each of these products.
Scheme 2. Products of the Dehydrohalogenation Step.

The spectrum obtained from the above reaction (Scheme 2) and the proposed identities of the chemical species represented by the peaks present in the spectrum are shown in Figure 1.
Figure 1.

11B NMR (96 MHz) spectrum showing peaks that correspond to each of the dehydrohalogenation products.
It was noted that the reaction of Hünig’s base with each of the amine-iodoborane complexes, other than isopropylamine-monoiodoborane, provided at least some amount of the aminoborane monomer. However, the primary iodoborane-amines (1a, 1b) yielded very little of either the monomeric or dimeric aminoborane, but primarily a mixture of other boron species arising from the exchange of the amines present. The iodoborane complexes of secondary amines provided primarily aminoborane products, in either the monomeric or dimeric form, with the ratio dependent on the sterics of the amine in the borane complex. Compact or rigidly constrained amines, such as dimethylamine or piperidine, resulted in a higher proportion of dimeric aminoboranes (58 and 70%, respectively). Many of the iodoborane complexes of cyclic and acyclic secondary amines gave primarily the aminoborane monomer (3d–3q), with minimal formation of dimers or other products. Diisopropylaminoborane (3f), dicyclohexylaminoborane (3j), 2,6-dimethylpiperidinoborane (3l), and dibenzylaminoborane (3q) (entries 6, 10, 12, and 17 in Table 2) were each detected exclusively as the monomer (by 11B NMR). The monochloroborane example produced from 1f and NCS provided minimal monomer formation (∼1%) with 1 or 2 equiv of i-Pr2NEt, likely due to interference of the still present succinimide.
Influence of the Amines on Dehydrohalogenation
To assess the influence of the amine added as a base for the elimination, another series of dehydrohalogenation reactions was performed (Table 3). Iodoborane-amines with varying steric environments were reacted with amine bases with a range of substitutions (0° (NH3), 1, 2, 3°) and sterics. The iodoboranes were prepared from diisopropylamine- (1f), dimethylamine- (1c), and piperidine-borane (1k), and their reactions with the added amines allowed for the identification of several trends in reactivity. The highly bulky iodoborane complex with diisopropylamine (2f) reacted with bulky amines, including diisopropyl-, dicyclohexyl-, diisobutyl-, and N,N-diisopropylethylamines to produce the diisopropylaminoborane monomer 3f with 98–99% conversion. Slightly less hindered triethylamine and dibenzylamine provided 80–90% conversion to 3f. The remainder of the 1° and less hindered 2° amines tested with 2f gave what are presumed to be polyaminoboranes or amine coordination products based on prior reports of these compounds made from diisopropylaminoborane.9,28 A similar species was observed when unhindered 2c was reacted with piperidine.
Table 3. Aminoboranes Formed Using Various Aminesa,b.

| entry | amine-borane | added amine | monomer (%) | dimer (%) | other (%) |
|---|---|---|---|---|---|
| 1 | 1f | i-Pr2NH | ≥99 | 0 | 0 |
| 2 | 1f | Et2NH | 0 | 0 | ≥99 |
| 3 | 1f | Propylamine | 0 | 0 | ≥99 |
| 4 | 1f | t-BuNH2 | 0 | 0 | ≥99 |
| 5 | 1f | BnNH2 | 0 | 0 | ≥99 |
| 6 | 1f | Bn2NH | 90 | 0 | 10 |
| 7 | 1f | Chx2NH | ≥99 | 0 | 0 |
| 8 | 1f | i-Bu2NH | 98 | 0 | 2 |
| 9 | 1f | Azepane | 0 | 0 | ≥99 |
| 10 | 1f | Piperidine | 0 | 0 | ≥99 |
| 11 | 1f | Morpholine | 0 | 0 | ≥99 |
| 12 | 1f | Ammonia | 0 | 0 | ≥99 |
| 13 | 1f | Et3N | 80 | 0 | 20 |
| 14 | 1f | i-Pr2EtN | ≥99 | 0 | 0 |
| 15 | 1f | Pyridine | 0 | 0 | ≥99 |
| 16 | 1c | Piperidine | 0 | 0 | ≥99 |
| 17 | 1c | Et3N | 1 | 40 | 59 |
| 18 | 1c | i-Pr2EtN | 7 | 77 | 16 |
| 19 | 1k | Piperidine | 0 | 0 | ≥99 |
| 20 | 1k | i-Pr2EtN | 2 | 89 | 9 |
Reactions were performed at the 2 mmol scale with respect to the amine-borane.
Ratio determined by 11B NMR (96 MHz) spectroscopy.
In the reactions of 2c, increasing the bulk of the added amine (triethylamine and N,N-diisopropylethylamine) led to the partial formation of the dimeric dimethylaminoborane, 40% and 77%, respectively. A small amount of monomeric aminoborane was additionally detected in each case, 1% and 7%, respectively. The reaction of 2k with piperidine gave the polyaminoborane exclusively, while N,N-diisopropylethylamine gave mainly (89%) dimeric aminoborane, with traces (2%) of the monomer present.
One-Pot Borylation
Arylboronates and boronic acids have traditionally been prepared by the reaction of trialkoxyboranes with aryl lithium29 or Grignard reagents.30 However, these organometallic reagents are highly reactive, making them incompatible with many functional groups. More recently, Miyaura and co-workers have reported the palladium-catalyzed cross-coupling reaction between aryl halides and bis(pinacolato)diboron (B2pin2).31 This was later extended by Masuda to utilize pinacolborane (HBpin).32 Although both B2pin2 and HBpin are commercially available, these reagents are costly and half of the B2pin2 reagent goes unused in the cross-coupling reaction. The preparation of B2pin233 is lengthy and uses highly reactive reagents (BBr3, Na) and HBpin34 uses unpleasant borane-dimethylsulfide. In 2003, Alcaraz and Vaultier utilized diisopropylaminoborane as an efficient source of boron for palladium catalyst borylation.2a Although the aminoborane precursor amine-boranes were expensive in 2003, there are now several simple procedures for their preparation from the corresponding amines using sodium borohydride and benign activators such as NaHCO320c and CO2.35
As a further confirmation of the formation of aminoboranes from the iodination/dehydroiodination sequence, the presumed aminoboranes were subjected to palladium-catalyzed borylation of aryl halides, as described by Pucheault and Vaultier.6b To simplify the protocol, and further persuade organic chemists to embrace this process for Suzuki coupling,36 we made two modifications to the above borylation protocol. (i) Separation of the ammonium salt formed during the dehydrohalogenation reaction was excluded since the salt from the borylation catalytic cycle does not interfere in the reaction.6a,37,21 Reactions were performed with and without filtration of the salt, and identical overall yields (95%) were observed for the borylation of 4-iodoanisole with 3f. This change makes the process one-pot. Also, (ii) “quenching” of the arylaminoborane intermediate with methanol, followed by transesterification with pinacol, was replaced with a direct pinacol “quench” without any loss in yield of the pinacol boronate (Scheme 3).
Scheme 3. Proposed Pathway for the One-Pot Synthesis of Pinacol Arylboronates from Amine-Boranes.

The progress of the borylation of 4-iodoanisole was monitored, and the proposed intermediates, shown in Scheme 3, were confirmed by 11B NMR spectroscopy. Starting from diisopropylamine-borane (δ −21.80 (q): Figure 2a), iodine and amine addition leads to the peaks at δ −17.34 (t) and δ 34.40 (t) (Figure 2b,c, respectively). After reflux, the amino(aryl)borane intermediate was detected at δ 38.20 (Figure 2d). Addition of pinacol results in a slight upfield shift to δ 30.21 (s) (Figure 2e), representative of the pinacol boronate.
Figure 2.
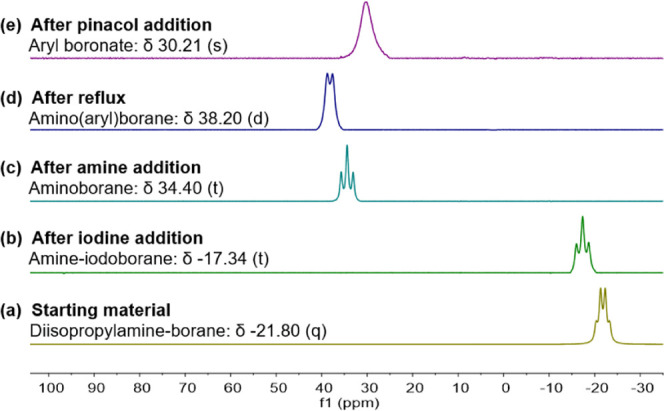
11B NMR (96 MHz) spectra depicting the progress of the one-pot conversion of amine-borane to arylboronates.
The optimized conditions shown in Scheme 3 were tested with several amine-boranes, such as monomer-forming dicyclohexylamine- (1j) and dibenzylamine-borane (1q), as well as the primarily dimer-forming dimethylamine-borane (1c) for the reaction. These complexes gave 90%, 91%, and 58% yields, respectively, as compared to the 95% yield with 1f (Table 4).
Table 4. Study of Amine-Boranes as Boron Sources for One-Pot Borylation of Aryl Halidesa.
| entry | amine-borane | monomer (%) | product obtained | yieldb (%) |
|---|---|---|---|---|
| 1 | 1c | 7–22 | 4a | 58 |
| 2 | 1f | ≥99 | 4a | 95 |
| 3 | 1j | ≥99 | 4a | 90 |
| 4 | 1q | ≥99 | 4a | 91 |
Reactions were performed using 4-iodoanisole at the 1 mmol scale with respect to the aryl halide.
Isolated yields after flash chromatography are shown.
With the 58% product recovery when using dimeric aminoborane 3c, we have demonstrated that dimers also participate in the borylation, albeit at a slower rate. Attempts are under way to improve the yields. Examination of bromine- and chlorine-containing arenes was also carried out with 4-bromo- and 4-chloroanisole. While the former provided 95% of the borylated product, the latter was unreactive, indicating that chlorine is not a suitable leaving group under the current reaction conditions (Table 5).37,38
Table 5. Leaving Groups Studied for One-Pot Borylationa.
| entry | amine-borane | starting material | product obtained | yieldb (%) |
|---|---|---|---|---|
| 1 | 1f | MeOC6H4-Cl | none | |
| 2 | 1f | MeOC6H4-Br | 9a | 95 |
| 3 | 1f | MeOC6H4-I | 9a | 95 |
Reactions were performed at the 1 mmol scale with respect to the aryl halide.
Isolated yields after flash chromatography are shown.
Following confirmation of the reaction pathway, a series of aryl iodides or bromides were subjected to the above-described reaction conditions where 4-iodoanisole provided the boronate 4a in 95% yield. Other ether-containing 4-ethoxy- (4b), 4-methoxy-2-methyl- (4c), 6-methoxynaphthyl- (4d), and 2,3-dihydrobenzofuryl- (4e) aryl halides gave equally high yields (96%, 94%, 97%, and 98%, respectively). Unadorned bromobenzene (4f), as well as its counterparts with systems of extended conjugation, 2-naphthyl- (4g), 1-naphthyl- (4h), and 9-phenanthyl- (4i) halides, and hydrocarbon substituents, 4-methyl- (4j), 4-phenyl- (4k), and 3,5-di-t-butyl- (4l) aryl halides, all gave the corresponding boronates in excellent yields (97–99%). However, the 2,4,6-substituted bromomesitylene proved to be too sterically encumbered to undergo the reaction, and no product was isolated.
Boronates of functionalized aryl halides with methylthio- (4m), nitrile- (4n), and dimethylamino- (4o) groups were obtained in 96, 65, and 70%, respectively. However, substrates with reducible functionalities (keto-, formyl-, amido-, and nitro) provided mixtures of other products along with small quantities of the expected borylated products. Borylation of aryl halides with other ring halogens (chlorine or fluorine) was also shown to be feasible. 4-Chloro- (4p), 3,5-dichloro- (4q), 4-fluoro- (4r), and 4-trifluoromethyl- (4s) iodobenzenes gave the corresponding boronates in 89–99% yields. Attempted borylation of pentafluoroiodobenzene, however, gave none of the boronate ester. The results from the study of the substrate scope are summarized in Figure 3.
Figure 3.

Scope of the one-pot borylation from amine-boranes.a,b aReactions were performed at the 1 mmol scale with respect to the aryl halide. bIsolated yields after flash chromatography are shown.
Amine Recycling
The reaction sequences in Scheme 3 suggested that the ammonium salt byproduct from both the borylation cycle and aminoborane synthesis could be recovered and recycled to regenerate the amine-borane. Applying an alteration in the workup procedure (Scheme 4) for the borylation of 4-iodoanisole provided 83% of 4-methoxyphenylboronic acid (5a) along with 86% of diisopropylammonium chloride. The recovered ammonium salt can be converted to the starting amine-borane via the salt metathesis protocol.19 Though diisopropylamine is relatively inexpensive, the demonstrated recovery of the amine can be useful when a more valuable amine is utilized.
Scheme 4. Recovery of Diisopropylammonium Salt/Boronic Acid, and Regeneration of Amine-Borane,

Reaction was performed at the 1 mmol scale with respect to the aryl halide.
Isolated yields after aqueous workup are shown.
Conclusions
In summary, we have described the preparation of aminoboranes, within minutes, at room temperature, in reagent-grade solvents from amine-boranes via an iodination-dehydroiodination sequence. Monomeric or dimeric aminoboranes can be produced by alteration of the coordinated amine, and the amine used for dehydrohalogenation, with the monomers being formed exclusively in several cases. Application of these monomeric aminoboranes has been demonstrated for a one-pot palladium-catalyzed conversion of aryl iodides and bromides containing substituents with varying steric and electronic environments to the corresponding boronate esters and boronic acids.
Experimental Section
General Information
Unless otherwise noted, all manipulations were carried out under open air conditions. 11B,19F,13C, and 1H NMR spectra were recorded at room temperature, on a Varian INOVA 300 MHz NMR spectrophotometer. Chemical shifts (δ values) are reported in parts per million relative to BF3·Et2O for 11B NMR. Data are reported as: δ value, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; h, hextet; hept, heptet; m, multiplet; br, broad) and integration. All solvents for routine isolation of products were reagent-grade. Sodium borohydride (powder, purity >99% by hydride estimation 1) was purchased from Oakwood Chemical. Tetrahydrofuran (THF, ACS reagent >99.0% containing 0.004% water and 0.025% BHT), toluene (anhydrous, ≥99.8%), iodine (ACS reagent, ≥99.8%), bromine (reagent grade), N-chlorosuccinimide (ReagentPlus, 99%), and N-bromosuccinimide (ReagentPlus, 99%) were purchased from Sigma-Aldrich. All amines, aryl halides, and pinacol were purchased from commercial sources and used without further purification. Flash chromatography was performed using silica gel 40–63 um, 60 Å with diethyl ether as the eluent.
Preparation of Amine Boranes via Sodium Bicarbonate (AB Procedure 1)
Sodium borohydride (1.51 g, 2 equiv, 40 mmol) and powdered sodium bicarbonate (6.72 g, 4 equiv, 80 mmol) were transferred to a 100 mL dry round-bottom flask, charged with a magnetic stir-bar. The corresponding amine (1 equiv, 20 mmol) was charged into the reaction flask followed by addition of reagent-grade tetrahydrofuran (20 mL) at rt. Under vigorous stirring, water (0.36 mL, 4 equiv, 80 mmol) was added dropwise to prevent excessive frothing. Reaction progress was monitored by 11B NMR spectroscopy. (Note: A drop of anhydrous DMSO is added to the reaction aliquot before running the 11B NMR experiment to solubilize NaBH4.) Upon completion of the reaction (4–48 h, as determined by 11B NMR), the reaction contents were filtered through sodium sulfate and celite and the solid residue was washed with THF. Removal of the solvent in vacuo from the filtrate yielded the corresponding amine-borane (1b, 1d–1r). The residual solvent was removed by placing under a high vacuum for ∼12 h.
Isopropylamine-borane (1b)
1b was synthesized using AB Procedure 1, obtained as a white solid (91%, 1.327 g). 1H NMR (300 MHz, chloroform-d) δ 3.75 (s, 2H), 2.98 (dp, J = 12.9, 6.4 Hz, 1H), 1.22 (d, J = 6.5 Hz, 6H); 13C NMR (75 MHz, chloroform-d) δ 50.2, 21.8; 11B NMR (96 MHz, chloroform-d) δ −20.99 (q, J = 95.3 Hz).
Diethylamine-borane (1d)
1d was synthesized using AB Procedure 1, obtained as a colorless liquid (95%, 1.652 g). 1H NMR (300 MHz, chloroform-d) δ 3.58 (s, 1H), 2.72 (qd, J = 7.3, 5.6 Hz, 4H), 1.16 (t, J = 7.3 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 48.8, 11.6.11B NMR (96 MHz, chloroform-d) δ −17.44 (q, J = 95.9, 95.4 Hz).
Dipropylamine-borane (1e)
1e was synthesized using AB Procedure 1, obtained as a colorless oil (92%, 2.116 g). 1H NMR (300 MHz, chloroform-d) δ 3.56 (s, 1H), 2.67–2.52 (m, 4H), 1.69–1.54 (m, 4H), 0.82 (t, J = 7.4 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 56.8, 19.5, 11.3. 11B NMR (96 MHz, chloroform-d) δ −16.67 (q, J = 96.4, 95.4 Hz).
Diisopropylamine-borane (1f)
1f was synthesized using AB Procedure 1, obtained as a colorless oil (89%, 2.315 g). 1H NMR (300 MHz, chloroform-d) δ 3.10 (hd, J = 6.6, 3.4 Hz, 3H), 1.15 (t, J = 6.3 Hz, 12H). 13C NMR (75 MHz, chloroform-d) δ 52.0, 20.9, 18.9. 11B NMR (96 MHz, chloroform-d) δ -21.80 (q, J = 96.4 Hz).
Dibutylamine-borane (1g)
1g was synthesized using AB Procedure 1, obtained as a colorless oil (90%, 2.575 g). 1H NMR (300 MHz, chloroform-d) δ 3.54 (s, 1H), 2.73–2.58 (m, 4H), 1.65–1.53 (m, 4H), 1.32–1.19 (m, 4H), 0.86 (t, J = 7.4 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 54.9, 28.3, 20.2, 13.8. 11B NMR (96 MHz, chloroform-d) δ −16.58 (q, J = 99.9, 98.6 Hz).
Diisobutylamine-borane (1h)
1h was synthesized using AB Procedure 1, obtained as a colorless oil (87%, 2.489 g). 1H NMR (300 MHz, chloroform-d) δ 3.11 (s, 1H), 2.60 (dt, J = 12.3, 6.9 Hz, 2H), 2.40 (ddd, J = 12.6, 8.0, 5.2 Hz, 2H), 2.19 (dh, J = 7.8, 6.6 Hz, 2H), 0.90 (dd, J = 6.6, 2.1 Hz, 12H). 13C NMR (75 MHz, chloroform-d) δ 63.6, 24.6, 20.2, 19.8. 11B NMR (96 MHz, chloroform-d) δ -16.20 (q, J = 96.5, 95.9 Hz).
Dipentylamine-borane (1i)
1i was synthesized using AB Procedure 1, obtained as a white solid (90%, 3.080 g). 1H NMR (300 MHz, chloroform-d) δ 3.25 (s, 1H), 2.72 (dddd, J = 15.7, 13.9, 12.4, 6.9 Hz, 4H), 1.66 (dddd, J = 16.2, 9.3, 6.3, 2.6 Hz, 4H), 1.38–1.19 (m, 8H), 0.89 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 55.3, 29.1, 26.1, 22.4, 14.0. 11B NMR (96 MHz, chloroform-d) δ −16.44 (q, J = 98.9 Hz).
Dicyclohexylamine-borane (1j)
1j was synthesized using AB Procedure 1, obtained as a white solid (88%, 3.434 g). 1H NMR (300 MHz, chloroform-d) δ 3.08–2.75 (m, 3H), 1.94–1.76 (m, 8H), 1.64 (tdd, J = 12.3, 8.9, 3.7 Hz, 6H), 1.22 (ddddd, J = 28.0, 21.8, 15.7, 8.7, 3.8 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 60.7, 31.0, 29.7, 25.8, 25.5, 25.3. 11B NMR (96 MHz, chloroform-d) δ −20.69 (q, J = 97.8, 97.3 Hz).
Piperidine-borane (1k)
1k was synthesized using AB Procedure 1, obtained as a white solid (99%, 1.959 g). 1H NMR (300 MHz, chloroform-d) δ 3.75 (s, 1H), 3.39–3.10 (m, 2H), 2.47 (tdd, J = 13.5, 11.3, 2.7 Hz, 2H), 1.81–1.73 (m, 3H), 1.51 (tdd, J = 13.3, 10.8, 3.7 Hz, 2H), 1.31 (dddd, J = 16.1, 12.4, 8.6, 4.2 Hz, 1H). 13C NMR (75 MHz, chloroform-d) δ 53.4, 25.4, 22.6. 11B NMR (96 MHz, chloroform-d) δ -15.55 (q, J = 95.9, 95.2 Hz).
2,6-Dimethylpiperidine-borane (1l)
1l was synthesized using AB Procedure 1, obtained as a white solid (87%, 2.210 g). 1H NMR (300 MHz, chloroform-d) δ 3.79 (s, 1H), 3.07–2.85 (m, 2H), 2.71 (dddd, J = 21.0, 12.3, 6.2, 2.8 Hz, 2H), 2.49 (s, 1H), 2.35 (s, 1H), 1.97 (tdd, J = 14.3, 10.7, 3.1 Hz, 2H), 1.84 (s, 0H), 1.86–1.67 (m, 5H), 1.68–1.56 (m, 1H), 1.57–1.41 (m, 3H), 1.39 (d, J = 6.3 Hz, 6H), 1.34 (td, J = 6.1, 4.0 Hz, 3H), 1.29 (d, J = 6.6 Hz, 6H), 1.27–1.16 (m, 2H), 1.16–0.65 (m, 2H). 13C NMR (75 MHz, chloroform-d) δ 59.4, 59.4, 34.7, 25.8, 24.1, 23.1, 22.3, 20.7. 11B NMR (96 MHz, chloroform-d) δ −17.62 (q, J = 96.5, 95.8 Hz), −25.85 (q, J = 96.3 Hz).
2,2,6,6-Tetramethylpiperidine-borane (1m)
1m was synthesized using AB Procedure 1, obtained as a white solid (85%, 2.636 g). 1H NMR (300 MHz, chloroform-d) δ 2.82 (s, 1H), 1.86–1.63 (m, 4H), 1.58–1.43 (m, 2H), 1.38 (s, 6H), 1.33 (s, 6H). 13C NMR (75 MHz, chloroform-d) δ 58.6, 41.0, 34.0, 20.7, 16.7. 11B NMR (96 MHz, chloroform-d) δ −22.00 (q, J = 97.8 Hz).
Morpholine-borane (1n)
1n was synthesized using AB Procedure 1, obtained as a white solid (97%, 1.958 g). 1H NMR (300 MHz, chloroform-d) δ 4.40 (s, 1H), 3.91 (dd, J = 12.7, 3.6 Hz, 2H), 3.55 (td, J = 12.3, 2.3 Hz, 2H), 3.13–2.97 (m, 2H), 2.75 (dtd, J = 13.9, 11.5, 3.6 Hz, 2H), 2.31–0.65 (m, 3H). 13C NMR (75 MHz, chloroform-d) δ 65.8, 52.0. 11B NMR (96 MHz, chloroform-d) δ −15.47 (q, J = 95.9 Hz).
Azepane-borane (1o)
1o was synthesized using AB Procedure 1, obtained as a white solid (93%, 2.102 g). 1H NMR (300 MHz, chloroform-d) δ 4.06 (s, 1H), 3.21 (ddt, J = 13.8, 6.8, 3.1 Hz, 2H), 2.74 (tdd, J = 12.1, 10.1, 5.3 Hz, 2H), 1.81 (ddq, J = 12.8, 6.0, 3.0 Hz, 2H), 1.64 (dddd, J = 17.1, 11.3, 9.4, 5.6 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 55.1, 27.2, 26.7. 11B NMR (96 MHz, chloroform-d) δ −14.56 (q, J = 95.9 Hz).
Pyrrolidine-borane (1p)
1p was synthesized using AB Procedure 1, obtained as a white solid (88%, 1.495 g). 1H NMR (300 MHz, chloroform-d) δ 4.64 (s, 1H), 3.32–3.02 (m, 2H), 2.60 (ttd, J = 10.7, 7.4, 6.4, 3.0 Hz, 2H), 1.96–1.82 (m, 2H), 1.76 (dqd, J = 8.0, 4.6, 2.3 Hz, 2H). 13C NMR (75 MHz, chloroform-d) δ 54.2, 24.7. 11B NMR (96 MHz, chloroform-d) δ −17.25 (q, J = 94.7 Hz).
Dibenzylamine-borane (1q)
1q was synthesized using AB Procedure 1, obtained as a white solid (92%, 3.884 g). 1H NMR (300 MHz, chloroform-d) δ 7.41–7.29 (m, 6H), 7.27–7.16 (m, 4H), 4.19 (s, 1H), 4.00 (dd, J = 13.1, 5.2 Hz, 2H), 3.87–3.69 (m, 2H), 2.31–1.24 (m, 3H). 13C NMR (75 MHz, chloroform-d) δ 134.2, 129.7, 128.8, 128.6, 58.4. 11B NMR (96 MHz, chloroform-d) δ −15.15.
Triethylamine-borane (1r)
1r was synthesized using AB Procedure 1, obtained as a colorless liquid (97%, 2.232 g). 1H NMR (300 MHz, chloroform-d) δ 2.61 (q, J = 7.3 Hz, 6H), 1.02 (t, J = 7.3 Hz, 9H). 13C NMR (75 MHz, chloroform-d) δ 52.3, 8.6. 11B NMR (96 MHz, chloroform-d) δ −13.81 (q, J = 97.2, 96.8 Hz).
Preparation of Amine Boranes via Salt Metathesis (AB Procedure 2)
Sodium borohydride (0.76 g, 20 mmol) and the appropriate ammonium salt (20 mmol) were transferred to a 100 mL dry round-bottom flask, charged with a magnetic stir-bar. This was followed by addition of reagent-grade tetrahydrofuran (20.0 mL) at rt. Reaction progress was monitored by 11B NMR spectroscopy. (Note: A drop of anhydrous DMSO is added to the reaction aliquot before running the 11B NMR experiment to solubilize NaBH4.) Upon completion of the reaction (1–24 h, as determined by 11B NMR), the reaction contents were filtered through sodium sulfate and celite and the solid residue was washed with THF. Removal of the solvent in vacuo from the filtrate yielded the corresponding amine-borane. No further purification was necessary in the examples (1a, 1c) presented here.
Ethylamine-borane (1a)
1a was synthesized using AB Procedure 2, obtained as a white solid (79%, 0.930 g). 1H NMR (300 MHz, chloroform-d) δ 3.83 (s, 2H), 3.00–2.69 (m, 2H), 1.23 (t, J = 7.3 Hz, 3H), 1.45 (q, J = 86.1 Hz, 3H); 13C NMR (75 MHz, chloroform-d) δ 43.6, 14.5; 11B NMR (96 MHz, chloroform-d) δ −20.09 (q, J = 95.2 Hz).
Dimethylamine-borane (1c)
1c was synthesized using AB Procedure 2, obtained as a white solid (93%, 1.096 g). 1H NMR (300 MHz, chloroform-d) δ 4.30 (s, 1H), 2.46 (d, J = 5.8 Hz, 6H), 1.42 (dd, J = 188.2, 91.9 Hz, 3H). 13C NMR (75 MHz, chloroform-d) δ 44.4. 11B NMR (96 MHz, chloroform-d) δ −14.76 (q, J = 95.5 Hz).
General Amine-Iodoborane Synthesis Procedure
In a 25 mL round-bottom flask, containing a stir-bar, the amine-borane (2 mmol, 1 equiv) was weighed. This was followed by addition of dichloromethane (4 mL). After dissolution of the amine-borane, iodine (1 mmol, 0.5 equiv) was added portionwise at rt. After stirring for 5 min at rt, the reaction mixture was analyzed using 11B NMR spectroscopy. (All iodoboranes are unisolated intermediates identified by 11B NMR spectroscopy.)
Ethylamine-iodoborane (2a)
11B NMR (96 MHz, chloroform-d) δ −18.10 (t, J = 129.8 Hz).
Isopropylamine-iodoborane (2b)
11B NMR (96 MHz, chloroform-d) δ −18.41 (t, J = 131.2 Hz).
Dimethylamine-iodoborane (2c)
11B NMR (96 MHz, chloroform-d) δ −13.53 (t, J = 130.0 Hz).
Diethylamine-iodoborane (2d)
11B NMR (96 MHz, chloroform-d) δ −15.14 (t, J = 129.8 Hz).
Dipropylamine-iodoborane (2e)
11B NMR (96 MHz, chloroform-d) δ −14.63 (t, J = 130.5 Hz).
Diisopropylamine-iodoborane (2f)
11B NMR (96 MHz, chloroform-d) δ −17.34 (t, J = 131.7 Hz).
Diisopropylamine-chloroborane (2f-Cl)
11B NMR (96 MHz, chloroform-d) δ −7.64 (t, J = 123.5 Hz).
Dibutylamine-iodoborane (2g)
11B NMR (96 MHz, chloroform-d) δ −14.59 (t, J = 134.5 Hz).
Diisobutylamine-iodoborane (2h)
11B NMR (96 MHz, chloroform-d) δ −14.38 (t, J = 131.4 Hz).
Dipentylamine-iodoborane (2i)
11B NMR (96 MHz, chloroform-d) δ −13.92 (d, J = 136.2 Hz).
Dicyclohexylamine-iodoborane (2j)
11B NMR (96 MHz, chloroform-d) δ −17.36 (d, J = 140.9 Hz).
Piperidine-iodoborane (2k)
11B NMR (96 MHz, chloroform-d) δ −14.16 (t, J = 131.0 Hz).
2,6-Dimethylpiperidine-iodoborane (2l)
11B NMR (96 MHz, chloroform-d) δ −15.89 (t, J = 132.6 Hz), −20.18 (t, J = 131.9 Hz).
2,2,6,6-Tetramethylpiperidine-iodoborane (2m)
11B NMR (96 MHz, chloroform-d) δ −17.68 (t, J = 132.6 Hz).
Morpholine-iodoborane (2n)
11B NMR (96 MHz, chloroform-d) δ −14.57 (t, J = 130.5 Hz).
Azepane-iodoborane (2o)
11B NMR (96 MHz, chloroform-d) δ −13.59 (t, J = 127.8 Hz).
Pyrrolidine-iodoborane (2p)
11B NMR (96 MHz, chloroform-d) δ −15.07 (t, J = 129.5 Hz).
Dibenzylamine-iodoborane (2q)
11B NMR (96 MHz, chloroform-d) δ −14.51.
Triethylamine-iodoborane (2r)
11B NMR (96 MHz,) δ −14.31 (t, J = 100.5 Hz).
General Aminoborane Synthesis Procedure
In a 25 mL round-bottom flask containing a stir-bar, the amine-borane (2 mmol, 1 equiv) was weighed. This was followed by addition of dichloromethane (4 mL). After dissolution of the amine-borane, iodine (1 mmol, 0.5 equiv) was added portionwise at rt. After complete formation of the amine-iodoborane complex, as evidenced by a return to colorlessness of the reaction mixture, diisopropylethylamine (2 mmol, 1 equiv) was added dropwise to the stirred reaction mixture at rt. After stirring for 5 min at rt, the reactions were complete. (All aminoboranes are unisolated intermediates identified by 11B NMR spectroscopy.)
Ethylaminoborane (3a)
11B NMR (96 MHz, chloroform-d) δ 34.96–28.30 (m), 20.83, 4.78 to −1.67 (m), −8.19 (dd, J = 244.6, 98.1 Hz), −19.94 (dd, J = 181.8, 88.0 Hz), −21.84 to −27.21 (m).
Isopropylaminoborane (3b)
11B NMR (96 MHz, chloroform-d) δ 34.08–23.60 (m), 20.60, −10.73, −18.75 to −31.65 (m).
Dimethylaminoborane (3c)
11B NMR (96 MHz, chloroform-d) δ 37.07 (t, J = 128.4 Hz, aminoborane monomer), 4.71 (t, J = 112.1 Hz, aminoborane dimer), −0.53 to −4.43 (m).
Diethylaminoborane (3d)
11B NMR (96 MHz, chloroform-d) δ 36.16 (t, J = 128.0 Hz, aminoborane monomer), 1.63 (t, J = 112.8 Hz, aminoborane dimer), −4.27 (t, J = 107.2 Hz).
Dipropylaminoborane (3e)
11B NMR (96 MHz, chloroform-d) δ 36.59 (t, J = 127.9 Hz, aminoborane monomer), −3.45.
Diisopropylaminoborane (3f)
11B NMR (96 MHz, chloroform-d) δ 34.40 (t, J = 126.8 Hz, aminoborane monomer).
Dibutylaminoborane (3g)
11B NMR (96 MHz, chloroform-d) δ 36.56 (t, J = 125.4 Hz, aminoborane monomer), −3.42.
Diisobutylaminoborane (3h)
11B NMR (96 MHz, chloroform-d) δ 36.99 (t, J = 128.9 Hz, aminoborane monomer), −2.99.
Dipentylaminoborane (3i)
11B NMR (96 MHz, chloroform-d) δ 36.53 (t, J = 129.2 Hz, aminoborane monomer), −3.56.
Dicyclohexylaminoborane (3j)
11B NMR (96 MHz, chloroform-d) δ 34.60 (t, J = 126.2 Hz, aminoborane monomer).
Piperidinoborane (3k)
11B NMR (96 MHz, chloroform-d) δ 35.26 (t, J = 126.4 Hz, aminoborane monomer), 1.52 (t, J = 111.0 Hz, aminoborane dimer), −1.87.
2,6-Dimethylpiperidinoborane (3l)
11B NMR (96 MHz, chloroform-d) δ 31.21 (t, J = 126.9 Hz, aminoborane monomer).
2,2,6,6-Tetramethylpiperidinoborane (3m)
11B NMR (96 MHz, chloroform-d) δ 36.05 (t, J = 129.1 Hz, aminoborane monomer), −17.70 (t, J = 132.4 Hz), −20.62 to −24.28 (m).
Morpholinoborane (3n)
11B NMR (96 MHz, chloroform-d) δ 35.94 (t, J = 132.3 Hz, aminoborane monomer), 1.53 (t, J = 112.7 Hz, aminoborane dimer), −2.08, −15.23 (d, J = 96.8 Hz).
Azepanoborane (3o)
11B NMR (96 MHz, chloroform-d) δ 37.34 (t, J = 127.3 Hz, aminoborane monomer), 2.80 (t, J = 113.4 Hz, aminoborane dimer), −1.92, −14.19 (d, J = 98.3 Hz), −18.39.
Pyrrolidinoborane (3p)
11B NMR (96 MHz, chloroform-d) δ 34.55 (t, J = 128.5 Hz, aminoborane monomer), 25.91 (d, J = 126.1 Hz, diaminoborane), 2.52 (t, J = 109.6 Hz, aminoborane dimer), −4.55 (t, J = 110.4 Hz, exchange/coordination compound), −18.77 (td, J = 128.6, 32.3 Hz, aminodiborane).
Dibenzylaminoborane (3q)
11B NMR (96 MHz, chloroform-d) δ 39.22 (t, J = 149.2 Hz, aminoborane monomer).
General Procedure for Boronate Ester Synthesis (General Procedure 1)
In a 25 mL round-bottom flask containing a stir-bar, the amine-borane (2 mmol, 2 equiv) was weighed. This was followed by addition of dichloromethane (5 mL). After dissolution of the amine-borane, iodine (1 mmol, 1 equiv) was added portionwise at rt. After complete formation of the iodoborane–amine complex, as evidenced by a return to colorlessness of the reaction mixture, diisopropylamine (5 mmol, 5 equiv) was added to the stirred reaction mixture at rt. After stirring for 5 min at rt, the reaction was complete. Then, by stirring toluene (5 mL), the aryl halide substrate (1 mmol, 1 equiv) and PdCl2(dppp) (0.05 mmol, 0.05 equiv) were added to the reaction mixture at rt. A reflux condenser was affixed to the flask, and the mix was brought to reflux. After completion (∼12–16 h), the reaction mixture was cooled to rt and then brought to 0 °C using an ice-water bath. At 0 °C, diethyl ether (3 mL) was added to the mixture, followed by pinacol (1.1 mmol, 1.1 equiv). The mixture was stirred for 4 h while being allowed to warm to rt. After completion, the reaction mixture was diluted with diethyl ether (10 mL) and the crude mixture was passed through a pad of silica gel contained in a fritted glass Büchner funnel and eluted with diethyl ether as necessary. The resulting filtrate was condensed by rotary evaporation followed by drying in vacuo for 12 h.
2-(4-Methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4a)
4a was synthesized using General Procedure 1, obtained as a yellow oil (95%, 222 mg). 1H NMR (300 MHz, chloroform-d) δ 7.78 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 3.82 (s, 3H), 1.35 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 162.00, 136.43, 113.26, 83.55, 55.12, 24.98. 11B NMR (96 MHz, chloroform-d) δ 30.46. Compound characterization is in accordance with previous reports.6b
2-(4-Ethoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4b)
4b was synthesized using General Procedure 1, obtained as a yellow oil (96%, 238 mg). 1H NMR (300 MHz, chloroform-d) δ 7.76 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 4.06 (q, J = 7.0 Hz, 2H), 1.42 (t, J = 7.0 Hz, 3H), 1.34 (s, 11H). 13C NMR (75 MHz, chloroform-d) δ 161.38, 136.41, 113.76, 83.54, 63.24, 24.97, 14.91. 11B NMR (96 MHz, chloroform-d) δ 30.40. Compound characterization is in accordance with previous reports.39
2-(4-Methoxy-2-methylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4c)
4c was synthesized using General Procedure 1, obtained as a yellow oil (94%, 233 mg). 1H NMR (300 MHz, chloroform-d) δ 7.78 (d, J = 8.9 Hz, 1H), 6.76 (s, 2H), 3.83 (s, 3H), 2.58 (s, 3H), 1.37 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 161.59, 147.14, 137.81, 115.47, 110.09, 83.13, 55.03, 25.03, 22.61. 11B NMR (96 MHz, chloroform-d) δ 30.71. Compound characterization is in accordance with previous reports.40
2-(6-Methoxynaphthalen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4d)
4d was synthesized using General Procedure 1, obtained as a yellow oil (97%, 275 mg). 1H NMR (300 MHz, chloroform-d) δ 8.32 (s, 1H), 7.85–7.71 (m, 3H), 7.14 (d, J = 8.3 Hz, 2H), 3.93 (s, 3H), 1.40 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 158.39, 136.34, 135.91, 131.03, 130.17, 128.29, 125.86, 118.65, 105.56, 83.83, 55.34, 25.04.11B NMR (96 MHz, chloroform-d) δ 30.96. Compound characterization is in accordance with previous reports.41
2-(2,3-Dihydrobenzofuran-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4e)
4e was synthesized using General Procedure 1, obtained as a yellow oil (98%, 241 mg). 1H NMR (300 MHz, chloroform-d) δ 7.67 (s, 1H), 7.62 (d, J = 8.1 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 4.58 (t, J = 8.7 Hz, 2H), 3.19 (t, J = 8.7 Hz, 2H), 1.34 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 162.70, 135.46, 126.44, 108.94, 83.50, 71.37, 29.30, 24.97. 11B NMR (96 MHz, chloroform-d) δ 30.39. Compound characterization is in accordance with previous reports.42
4,4,5,5-Tetramethyl-2-phenyl-1,3,2-dioxaborolane (4f)
4f was synthesized using General Procedure 1, obtained as a yellow oil (98%, 200 mg). 1H NMR (300 MHz, chloroform-d) δ 7.89–7.80 (m, 2H), 7.52–7.44 (m, 1H), 7.43–7.35 (m, 2H), 1.37 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 134.66, 131.19, 127.64, 83.78, 25.00. 11B NMR (96 MHz, chloroform-d) δ 30.67. Compound characterization is in accordance with previous reports.39
4,4,5,5-Tetramethyl-2-(naphthalen-2-yl)-1,3,2-dioxaborolane (4g)
4g was synthesized using General Procedure 1, obtained as a yellow oil (99%, 251 mg). 1H NMR (300 MHz, chloroform-d) δ 8.44 (d, J = 1.1 Hz, 1H), 7.97–7.81 (m, 4H), 7.52 (dqd, J = 8.4, 6.8, 1.6 Hz, 2H), 1.43 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 136.21, 134.97, 132.75, 130.35, 128.61, 127.67, 126.95, 125.76, 83.96, 25.08. 11B NMR (96 MHz, chloroform-d) δ 30.75. Compound characterization is in accordance with previous reports.41
4,4,5,5-Tetramethyl-2-(naphthalen-1-yl)-1,3,2-dioxaborolane (4h)
4h was synthesized using General Procedure 1, obtained as a yellow oil (99%, 250 mg). 1H NMR (300 MHz, chloroform-d) δ 8.85 (dd, J = 8.4, 1.3 Hz, 1H), 8.16 (dd, J = 6.8, 1.4 Hz, 1H), 8.03–7.94 (m, 1H), 7.90–7.86 (m, 1H), 7.60 (ddd, J = 8.4, 6.8, 1.6 Hz, 1H), 7.55–7.50 (m, 2H), 1.48 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 136.88, 135.63, 133.17, 131.60, 128.38, 128.32, 126.32, 125.81, 125.47, 124.95, 83.79, 25.13. 11B NMR (96 MHz, chloroform-d) δ 31.23. Compound characterization is in accordance with previous reports.41
4,4,5,5-Tetramethyl-2-(phenanthren-9-yl)-1,3,2-dioxaborolane (4i)
4i was synthesized using General Procedure 1, obtained as a yellow oil (98%, 298 mg). 1H NMR (300 MHz, chloroform-d) δ 9.02 (d, J = 8.0 Hz, 1H), 8.80–8.71 (m, 2H), 8.58 (s, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.76 (t, J = 7.5 Hz, 3H), 7.69 (d, J = 7.8 Hz, 1H), 1.55 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 138.25, 134.54, 131.95, 131.04, 129.98, 129.37, 129.18, 127.84, 126.79, 126.59, 126.51, 126.23, 122.71, 122.55, 83.97, 25.20. 11B NMR (96 MHz, chloroform-d) δ 30.73. Compound characterization is in accordance with previous reports.43
4,4,5,5-Tetramethyl-2-(p-tolyl)-1,3,2-dioxaborolane (4j)
4j was synthesized using General Procedure 1, obtained as a yellow oil (97%, 211 mg). 1H NMR (300 MHz, chloroform-d) δ 7.75 (d, J = 7.9 Hz, 2H), 7.22 (d, J = 7.6 Hz, 2H), 2.40 (s, 3H), 1.37 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 141.30, 134.75, 128.46, 83.64, 25.00. 11B NMR (96 MHz, chloroform-d) δ 30.65. Compound characterization is in accordance with previous reports.6b
2-([1,1′-Biphenyl]-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4k)
4k was synthesized using General Procedure 1, obtained as a yellow oil (99%, 277 mg). 1H NMR (300 MHz, chloroform-d) δ 7.96 (d, J = 8.1 Hz, 2H), 7.71–7.64 (m, 4H), 7.48 (dd, J = 8.3, 6.5 Hz, 2H), 7.41 (d, J = 7.1 Hz, 1H), 1.42 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 143.83, 140.92, 135.23, 128.73, 127.53, 127.19, 127.12, 126.44, 83.88, 25.05. 11B NMR (96 MHz, chloroform-d) δ 30.47. Compound characterization is in accordance with previous reports.39
2-(3,5-Di-tert-butylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4l)
4l was synthesized using General Procedure 1, obtained as a pale-yellow solid (99%, 313 mg). 1H NMR (300 MHz, chloroform-d) δ 7.71 (d, J = 2.1 Hz, 2H), 7.65–7.54 (m, 1H), 1.39 (s, 18H), 1.38 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 149.71, 128.73, 125.51, 83.56, 34.93, 31.65, 25.01. 11B NMR (96 MHz, chloroform-d) δ 30.58. Compound characterization is in accordance with previous reports.44
4,4,5,5-Tetramethyl-2-(4-(methylthio)phenyl)-1,3,2-dioxaborolane (4m)
4m was synthesized using General Procedure 1, obtained as a yellow oil (96%, 240 mg). 1H NMR (300 MHz, chloroform-d) δ 7.79–7.67 (m, 2H), 7.27–7.18 (m, 2H), 2.48 (s, 3H), 1.34 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 142.50, 134.99, 124.88, 83.74, 24.98, 15.15. 11B NMR (96 MHz, chloroform-d) δ 30.48. Compound characterization is in accordance with previous reports.39
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (4n)
4n was synthesized using General Procedure 1, obtained as an orange solid (65%, 149 mg). 1H NMR (300 MHz, chloroform-d) δ 7.86 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 1.42–1.24 (m, 12H). 13C NMR (75 MHz, chloroform-d) δ 134.99, 130.99, 118.75, 114.43, 84.46, 24.96. 11B NMR (96 MHz, chloroform-d) δ 30.10. Compound characterization is in accordance with previous reports.39
N,N-Dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (4o)
4o was synthesized using General Procedure 1, obtained as a yellow oil (70%, 173 mg). 1H NMR (300 MHz, chloroform-d) δ 7.70 (d, J = 8.8 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 2.99 (s, 6H), 1.34 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 152.40, 136.04, 111.18, 83.16, 40.21, 24.97. 11B NMR (96 MHz, chloroform-d) δ 30.38. Compound characterization is in accordance with previous reports.43
2-(4-Chlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4p)
4p was synthesized using General Procedure 1, obtained as an orange oil (99%, 236 mg). 1H NMR (300 MHz, chloroform-d) δ 7.75 (dd, J = 8.3, 1.6 Hz, 2H), 7.35 (dd, J = 8.4, 1.6 Hz, 2H), 1.41–1.29 (m, 12H). 13C NMR (75 MHz, chloroform-d) δ 137.45, 136.05, 127.93, 84.03, 24.97. 11B NMR (96 MHz, chloroform-d) δ 30.30. Compound characterization is in accordance with previous reports.41
2-(3,5-Dichlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4q)
4q was synthesized using General Procedure 1, obtained as an orange oil (89%, 243 mg). 1H NMR (300 MHz, chloroform-d) δ 7.65 (d, J = 2.0 Hz, 2H), 7.42 (t, J = 2.0 Hz, 1H), 1.34 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 134.61, 132.61, 130.98, 84.51, 24.94. 11B NMR (96 MHz, chloroform-d) δ 29.74. Compound characterization is in accordance with previous reports.45
2-(4-Fluorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4r)
4r was synthesized using General Procedure 1, obtained as an orange oil (99%, 220 mg). 1H NMR (300 MHz, chloroform-d) δ 7.91–7.72 (m, 2H), 7.13–6.98 (m, 2H), 1.35 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 164.93 (d, J = 250.3 Hz), 136.88 (d, J = 8.1 Hz), 114.78 (d, J = 20.1 Hz), 83.91, 24.96. 11B NMR (96 MHz, chloroform-d) δ 30.19. 19F NMR (282 MHz, chloroform-d) δ −109.90 (p, J = 7.8 Hz). Compound characterization is in accordance with previous reports.39
4,4,5,5-Tetramethyl-2-(4-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane (4s)
4s was synthesized using General Procedure 1, obtained as a pale-orange solid (98%, 266 mg). 1H NMR (300 MHz, chloroform-d) δ 7.94 (d, J = 7.5 Hz, 2H), 7.62 (d, J = 7.8 Hz, 2H), 1.36 (s, 12H). 13C NMR (75 MHz, chloroform-d) δ 134.92, 132.71 (q, J = 32.0 Hz), 124.18 (q, J = 4.1 Hz), 84.22, 24.87. 11B NMR (96 MHz, chloroform-d) δ 30.27. 19F NMR (282 MHz, chloroform-d) δ −64.57. Compound characterization is in accordance with previous reports.6b
Procedure for Synthesis of Boronic Acid and Amine Recovery (General Procedure 2)
In a 25 mL round-bottom flask containing a stir-bar, the amine-borane (2 mmol, 2 equiv) was weighed. This was followed by addition of dichloromethane (5 mL). After dissolution of the amine-borane, iodine (1 mmol, 1 equiv) was added portionwise at rt. After complete formation of the iodoborane–amine complex, as evidenced by a return to colorlessness of the reaction mixture, diisopropylamine (5 mmol, 5 equiv) was added to the stirred reaction mixture at rt. After stirring for 5 min at rt, the reaction was complete. Then, by stirring toluene (5 mL), the aryl halide substrate (1 mmol, 1 equiv) and PdCl2(dppp) (0.05 mmol, 0.05 equiv) were added to the reaction mixture at rt. A reflux condenser was affixed to the flask, and the mix was brought to reflux. After completion (∼12–16 h), the reaction mixture was cooled to rt and then brought to 0 °C using an ice-water bath. At 0 °C, methanol (8 mL) was added to the mixture, and the solvent was removed by rotary evaporation. The residue was then dissolved with sodium hydroxide (3 M, 8 mL). The aqueous layer was washed with hexanes (3 × 10 mL), and the hexane layers were set aside. The aqueous layer was then acidified with 3 M HCl until reaching a pH of 1. The slurry was extracted with diethyl ether (4 × 15 mL). The combined organic portions were dried over sodium sulfate, filtered through cotton, and condensed via rotary evaporation followed by drying in vacuo for 12 h to retrieve the boronic acid. To the earlier separated hexane fractions was added 2 M ethereal HCl (5 mL) precipitating the ammonium salt. The salt was collected on a filter paper in a Hirsch funnel and washed with hexanes (2 × 10 mL). The solid was transferred to a preweighed flask and dried in vacuo for 12 h.
(4-Methoxyphenyl)boronic Acid (5a)
5a was synthesized using General Procedure 2, obtained as an off-white solid (83%, 126 mg). 1H NMR (300 MHz, chloroform-d) δ 8.15 (d, J = 8.3 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 3.88 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 162.98, 137.38, 113.42, 55.19. 11B NMR (96 MHz, chloroform-d) δ 29.25. Compound characterization is in accordance with previous reports.3a
Diisopropylammonium Chloride
Diisopropylammonium chloride was recovered using General Procedure 2, obtained as a white solid (86%, 828 mg). 1H NMR (300 MHz, chloroform-d) δ 9.17 (s, 2H), 3.38 (hept, J = 6.4 Hz, 2H), 1.48 (d, J = 6.5 Hz, 12H). 13C NMR (75 MHz, chloroform-d) δ 47.42, 19.24.
Acknowledgments
The authors thank the Hebert C. Brown Center for Borane Research for financial support. S.M. thanks WINStep Forward for the S. N. Bose Indo-US scholarship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01461.
Experimental details and characterization data (PDF)
Author Present Address
† Summer undergraduate researcher from Department of Chemistry, University of Delhi, 110021 New Delhi, India
The authors declare no competing financial interest.
Supplementary Material
References
- For a recent synthesis of aminoboranes of the type R2B-NH2, see;; Ramachandran P. V.; Drolet M. P.; Kulkarni A. S. A non-dissociative open-flask hydroboration with ammonia borane: ready synthesis of ammonia-trialkylboranes and aminodialkylboranes. Chem. Commun. 2016, 52, 11897–11900. 10.1039/C6CC06151F. [DOI] [PubMed] [Google Scholar]
- a Euzenat L.; Horhant D.; Ribourdouille Y.; Duriez C.; Alcaraz G.; Vaultier M. Monomeric (dialkylamino)boranes: a new and efficient boron source in palladium catalyzed C-B bond formation with aryl halides. Chem. Commun. 2003, 133, 2280–2281. 10.1039/B306874A. [DOI] [PubMed] [Google Scholar]; b Marciasini L. D.; Richy N.; Vaultier M.; Pucheault M. Iron-Catalysed Borylation of Arenediazonium Salts to Give Access to Arylboron Derivatives via Aryl(amino)boranes at Room Temperature. Adv. Synth. Catal. 2013, 355, 1083–1088. 10.1002/adsc.201200942. [DOI] [Google Scholar]; c Marciasini L. D.; Vaultier M.; Pucheault M. Borylation using group IV metallocene under mild conditions. Tetrahedron Lett. 2014, 55, 1702–1705. 10.1016/j.tetlet.2014.01.080. [DOI] [Google Scholar]; d Tobisu M.; Igarashi T.; Chatani N. Iridium/N-heterocyclic carbene-catalyzed C-H borylation of arenes by diisopropylaminoborane. Beilstein J. Org. Chem. 2016, 12, 654–661. 10.3762/bjoc.12.65. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Birepinte M.; Liautard V.; Chabaud L.; Pucheault M. Zirconium-Catalyzed Synthesis of Alkenylaminoboranes: From a Reliable Preparation of Alkenylboronates to a Direct Stereodivergent Access to Alkenyl Bromides. Org. Lett. 2020, 22, 2838–2843. 10.1021/acs.orglett.0c00908. [DOI] [PubMed] [Google Scholar]; f Birepinte M.; Liautard V.; Chabaud L.; Pucheault M. Magnesium-Catalyzed Tandem Dehydrogenation-Dehydrocoupling: An Atom Economical Access to Alkynylboranes. Chem. - Eur. J. 2020, 26, 3236–3240. 10.1002/chem.201905772. [DOI] [PubMed] [Google Scholar]
- a Haddenham D.; Bailey C. L.; Vu C.; Nepomuceno G.; Eagon S.; Pasumansky L.; Singaram B. Lithium aminoborohydrides 17. Palladium catalyzed borylation of aryl iodides, bromides, and triflates with diisopropylaminoborane prepared from lithium diisopropylaminoborohydride. Tetrahedron 2011, 67, 576–583. 10.1016/j.tet.2010.11.065. [DOI] [Google Scholar]; b Guerrand H. D. S.; Marciasini L. D.; Jousseaume M.; Vaultier M.; Pucheault M. Borylation of Unactivated Aryl Chlorides under Mild Conditions by Using Diisopropylaminoborane as a Borylating Reagent. Chem. - Eur. J. 2014, 20, 5573–5579. 10.1002/chem.201304861. [DOI] [PubMed] [Google Scholar]
- a Bailey C. L.; Murphy C. L.; Clary J. W.; Eagon S.; Gould N.; Singaram B. Reaction of Grignard reagents with diisopropyl-aminoborane. Synthesis of alkyl, aryl, heteroaryl and allyl boronic acids from organo(diisopropyl)-aminoborane by a simple hydrolysis. Heterocycles 2012, 86, 331–341. 10.3987/com-12-s(n)14. [DOI] [Google Scholar]; b Marciasini L.; Richy N.; Vaultier M.; Pucheault M. Aminoborylation/Suzuki-Miyaura tandem cross coupling of aryl iodides as efficient and selective synthesis of unsymmetrical biaryls. Chem. Commun. 2012, 48, 1553–1555. 10.1039/C1CC14605J. [DOI] [PubMed] [Google Scholar]
- Euzénat L.; Horhant D.; Brielles C.; Alcaraz G.; Vaultier M. Stereospecific palladium-catalyzed borylation reaction of 1-alkenyl halides with diispropylaminoborane. J. Organomet. Chem. 2005, 690, 2721–2724. 10.1016/j.jorganchem.2005.01.054. [DOI] [Google Scholar]
- a Guerrand H. D. S.; Marciasini L. D.; Gendrineau T.; Pascu O.; Marre S.; Pinet S.; Vaultier M.; Aymonier C.; Pucheault M. Sequential dehydrogenation-arylation of diisopropylamine-borane complex catalyzed by palladium nanoparticles. Tetrahedron 2014, 70, 6156–6161. 10.1016/j.tet.2014.04.036. [DOI] [Google Scholar]; b Guerrand H. D. S.; Vaultier M.; Pinet S.; Pucheault M. Amine-Borane Complexes: Air-and Moisture-Stable Partners for Palladium-Catalyzed Borylation of Aryl Bromides and Chlorides. Adv. Synth. Catal. 2015, 357, 1167–1174. 10.1002/adsc.201401153. [DOI] [Google Scholar]
- Marciasini L.; Cacciuttolo B.; Vaultier M.; Pucheault M. Synthesis of Borinic Acids and Borinate Adducts Using Diisopropylaminoborane. Org. Lett. 2015, 17, 3532–3535. 10.1021/acs.orglett.5b01620. [DOI] [PubMed] [Google Scholar]
- Igarashi T.; Tobisu M.; Chatani N. Catalytic Double Carbon-Boron Bond Formation for the Synthesis of Cyclic Diarylborinic Acids as Versatile Building Blocks for pi-Extended Heteroarenes. Angew. Chem., Int. Ed. 2017, 56, 2069–2073. 10.1002/anie.201612535. [DOI] [PubMed] [Google Scholar]
- De Albuquerque Pinheiro C. A.; Roiland C.; Jehan P.; Alcaraz G. Solventless and Metal-Free Synthesis of High-Molecular-Mass Polyaminoboranes from Diisopropylaminoborane and Primary Amines. Angew. Chem., Int. Ed. 2018, 57, 1519–1522. 10.1002/anie.201710293. [DOI] [PubMed] [Google Scholar]
- Thévenot F.; Doche C.; Mongeot H.; Guilhon F.; Miele P.; Cornu D.; Bonnetot B. Boron nitride obtained from molecular precursors: Aminoboranes used as a BN source for coatings, matrix, and Si3N4BN composite ceramic preparation. J. Solid State Chem. 1997, 133, 164–168. 10.1006/jssc.1997.7422. [DOI] [Google Scholar]
- Qi Y. Y.; Xu W. J.; Kang R.; Ding N. N.; Wang Y. L.; He G.; Fang Y. Discrimination of saturated alkanes and relevant volatile compounds via the utilization of a conceptual fluorescent sensor array based on organoboron-containing polymers. Chem. Sci. 2018, 9, 1892–1901. 10.1039/C7SC05243J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y. Y.; Ding N. N.; Wang Z. L.; Xu L.; Fang Y. Mechanochromic Wide-Spectrum Luminescence Based on a Monoboron Complex. ACS Appl. Mater. Interfaces 2019, 11, 8676–8684. 10.1021/acsami.8b21617. [DOI] [PubMed] [Google Scholar]
- Barrière C.; Alcaraz G.; Margeat O.; Fau P.; Quoirin J. B.; Anceau C.; Chaudret B. Copper nanoparticles and organometallic chemical liquid deposition (OMCLD) for substrate metallization. J. Mater. Chem. 2008, 18, 3084–3086. 10.1039/b804460k. [DOI] [Google Scholar]
- Maringgele W.; Noltemeyer M.; Teichgraber J.; Meiler A. Reduction of piperidino- and related sec.amino(dihalogeno)boranes with LiAlH4 in toluene and related reactions. Main Group Met. Chem. 2000, 23, 735–760. 10.1515/MGMC.2000.23.12.735. [DOI] [Google Scholar]
- Pasumansky L.; Haddenham D.; Clary J. W.; Fisher G. B.; Goralski C. T.; Singaram B. Lithium aminoborohydrides. 16. Synthesis and reactions of monomeric and dimeric aminoboranes. J. Org. Chem. 2008, 73, 1898–1905. 10.1021/jo702271c. [DOI] [PubMed] [Google Scholar]
- a Nöth H.; Beyer H. Beitrage zur chemie des bors. 4. Darstellung von N-alkyl-B-monohalogen-borazanen. Chem. Ber. Recl. 1960, 93, 2251–2263. 10.1002/cber.19600931011. [DOI] [Google Scholar]; b Metters O. J.; Chapman A. M.; Robertson A. P. M.; Woodall C. H.; Gates P. J.; Wass D. F.; Manners I. Generation of aminoborane monomers RR′N=BH2 from amine-boronium cations RR′NH-BH2L(+): metal catalyst-free formation of polyaminoboranes at ambient temperature. Chem. Commun. 2014, 50, 12146–12149. 10.1039/C4CC05145A. [DOI] [PubMed] [Google Scholar]
- Brown H. C.; Chandrasekharan J.; Ramachandran P. V. Chiral synthesis via organoboranes. 14. Selective reductions. 41. Diisopinocampheylchloroborane, an exceptionally efficient chiral reducing agent. J. Am. Chem. Soc. 1988, 110, 1539–1546. 10.1021/ja00213a030. [DOI] [Google Scholar]
- Douglass J. E. Amine haloboranes. Reaction of N-halosuccinimide with amine boranes. J. Org. Chem. 1966, 31, 962–963. 10.1021/jo01341a510. [DOI] [Google Scholar]
- a Das M. K.; Saha U.; Banerjee R. Kinetics of iodination of some amine-boranes with molecular iodine in toluene. Indian J. Chem. A 1996, 35, 513–516. [Google Scholar]; b Ryschkewitsch G. E. W.; J W. Trimethylaminehaloboranes. Inorg. Synth. 1970, 12, 116–126. [Google Scholar]
- a Ramachandran P. V.; Hamann H. J. Ammonia-borane as a Catalyst for the Direct Amidation of Carboxylic Acids. Org. Lett. 2021, 23, 2938–2942. 10.1021/acs.orglett.1c00591. [DOI] [PubMed] [Google Scholar]; b Ramachandran P. V.; Hamann H. J.; Choudhary S. Amine-boranes as Dual-Purpose Reagents for Direct Amidation of Carboxylic Acids. Org. Lett. 2020, 22, 8593–8597. 10.1021/acs.orglett.0c03184. [DOI] [PubMed] [Google Scholar]; c Ramachandran P. V.; Kulkarni A. S.; Zhao Y.; Mei J. G. Amine-boranes bearing borane-incompatible functionalities: application to selective amine protection and surface functionalization. Chem. Commun. 2016, 52, 11885–11888. 10.1039/C6CC06031E. [DOI] [PubMed] [Google Scholar]; d Ramachandran P. V.; Kulkarni A. S.; Pfeil M. A.; Dennis J. D.; Willits J. D.; Heister S. D.; Son S. F.; Pourpoint T. L. Amine-Boranes: Green Hypergolic Fuels with Consistently Low Ignition Delays. Chem. - Eur. J. 2014, 20, 16869–16872. 10.1002/chem.201405224. [DOI] [PubMed] [Google Scholar]
- Brown H. C.; Ramachandran P. V.; Chandrasekharan J. Selective reductions. 56. Exploration of the B-haloisopinocampheylboranes for asymmetric reduction of ketones. Heteroat. Chem. 1995, 6, 117–131. 10.1002/hc.520060206. [DOI] [Google Scholar]
- a Schaeffer G. W.; Anderson E. R. The preparation of trimethylamine-borine, N-trimethylborazole and N-dimethylaminoborine. J. Am. Chem. Soc. 1949, 71, 2143–2145. 10.1021/ja01174a065. [DOI] [Google Scholar]; b Taylor M. D.; Grant L. R.; Sands C. A. A convenient preparation of pyridine-borane. J. Am. Chem. Soc. 1955, 77, 1506–1507. 10.1021/ja01611a031. [DOI] [Google Scholar]
- A tertiary amine-borane complex (Et3N-BH3, 1r) was also included to show the generality of the iodination procedure.
- a Maringgele W.; Noltemeyer M.; Schmidt H. G.; Meiler A. Sterically encumbered monomeric sec.amino-(halogeno)hydroboranes and the corresponding dihalogeno- and dihydroborane precursors. Main Group Met. Chem. 1999, 22, 715–732. 10.1515/MGMC.1999.22.12.715. [DOI] [Google Scholar]; b Robertson A. P. M.; Leitao E. M.; Manners I. Catalytic Redistribution and Polymerization of Diborazanes: Unexpected Observation of Metal-Free Hydrogen Transfer between Aminoboranes and Amine-Boranes. J. Am. Chem. Soc. 2011, 133, 19322–19325. 10.1021/ja208752w. [DOI] [PubMed] [Google Scholar]
- Jaska C. A.; Temple K.; Lough A. J.; Manners I. Transition metal-catalyzed formation of boron-nitrogen bonds: Catalytic dehydrocoupling of amine-borane adducts to form aminoboranes and borazines. J. Am. Chem. Soc. 2003, 125, 9424–9434. 10.1021/ja030160l. [DOI] [PubMed] [Google Scholar]
- McLellan R.; Kennedy A. R.; Orr S. A.; Robertson S. D.; Mulvey R. E. Lithium Dihydropyridine Dehydrogenation Catalysis: A Group 1 Approach to the Cyclization of Diamine Boranes. Angew. Chem., Int. Ed. 2017, 56, 1036–1041. 10.1002/anie.201610905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. N. A.; Zhao J. C.; Shore S. G. Facile Synthesis of Aminodiborane and Inorganic Butane Analogue NH3BH2NH2BH3. J. Am. Chem. Soc. 2010, 132, 10658–10659. 10.1021/ja104938v. [DOI] [PubMed] [Google Scholar]
- Devillard M.; Pinheiro C. A. D.; Caytan E.; Roiland C.; Dinoi C.; Del Rosal I.; Alcaraz G. Uncatalyzed Formation of Polyaminoboranes from Diisopropylaminoborane and Primary Amines: a Kinetically Controlled Polymerization Reaction. Adv. Synth. Catal. 2021, 363, 2417–2426. 10.1002/adsc.202001458. [DOI] [Google Scholar]
- Brown H. C.; Srebnik M.; Cole T. E. Organoboranes. 48. Improved procedures for the preparation of boronic and borinic esters. Organometallics 1986, 5, 2300–2303. 10.1021/om00142a020. [DOI] [Google Scholar]
- Snyder H. R.; Kuck J. A.; Johnson J. R. Organoboron compounds, and the study of reaction mechanisms - Primary aliphatic boronic acids. J. Am. Chem. Soc. 1938, 60, 105–111. 10.1021/ja01268a033. [DOI] [Google Scholar]
- Ishiyama T.; Murata M.; Miyaura N. Palladium(0)-Catalyzed cross-coupling reaction of alkoxydiboron with haloarenes- A direct procedure for arylboronic esters. J. Org. Chem. 1995, 60, 7508–7510. 10.1021/jo00128a024. [DOI] [Google Scholar]
- Murata M.; Oyama T.; Watanabe S.; Masuda Y. Palladium-catalyzed borylation of aryl halides or triflates with dialkoxyborane: A novel and facile synthetic route to arylboronates. J. Org. Chem. 2000, 65, 164–168. 10.1021/jo991337q. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; M M.; A T.; Ahiko N. Miyaura Bis(pinacolato)diboron. Org. Synth. 2000, 77, 176–182. 10.15227/orgsyn.077.0176. [DOI] [Google Scholar]
- Tucker C. E.; Davidson J.; Knochel P. Mild and stereoselective hydroborations of functionalized alkynes and alkenes using pinacolborane. J. Org. Chem. 1992, 57, 3482–3485. 10.1021/jo00038a044. [DOI] [Google Scholar]
- Ramachandran P. V.; Hamann H. J.; Lin R. Activation of sodium borohydride via carbonyl reduction for the synthesis of amine- and phosphine-boranes. Dalton Trans. 2021, 50, 16770–16774. 10.1039/D1DT03495B. [DOI] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc. Chem. Commun. 1979, 866–867. 10.1039/c39790000866. [DOI] [Google Scholar]
- Lam K. C.; Marder T. B.; Lin Z. Y. Mechanism of the Palladium-Catalyzed Borylation of Aryl Halides with Pinacolborane. Organometallics 2010, 29, 1849–1857. 10.1021/om9010802. [DOI] [Google Scholar]
- Previous work on borylation of aryl halides suggest that the inclusion of potassium iodide could be a possible solution to allow for the inclusion of chloro- substrates.
- Xu J. H.; Cao J. L.; Wu X. Y.; Wang H.; Yang X. N.; Tang X. X.; Toh R. W.; Zhou R.; Yeow E. K. L.; Wu J. Unveiling Extreme Photoreduction Potentials of Donor-Acceptor Cyanoarenes to Access Aryl Radicals from Aryl Chlorides. J. Am. Chem. Soc. 2021, 143, 13266–13273. 10.1021/jacs.1c05994. [DOI] [PubMed] [Google Scholar]
- Clary J. W.; Rettenmaier T. J.; Snelling R.; Bryks W.; Banwell J.; Wipke W. T.; Singaram B. Hydride as a Leaving Group in the Reaction of Pinacolborane with Halides under Ambient Grignard and Barbier Conditions. One-Pot Synthesis of Alkyl, Aryl, Heteroaryl, Vinyl, and Allyl Pinacolboronic Esters. J. Org. Chem. 2011, 76, 9602–9610. 10.1021/jo201093u. [DOI] [PubMed] [Google Scholar]
- Li H. Y.; Ma B.; Liu Q. S.; Wang M. L.; Wang Z. Y.; Xu H.; Li L. J.; Wang X.; Dai H. X. Transformations of Aryl Ketones via Ligand-Promoted C-C Bond Activation. Angew. Chem., Int. Ed. 2020, 59, 14388–14393. 10.1002/anie.202006740. [DOI] [PubMed] [Google Scholar]
- Lyu H. R.; Kevlishvili I.; Yu X.; Liu P.; Dong G. B. Boron insertion into alkyl ether bonds via zinc/nickel tandem catalysis. Science 2021, 372, 175–182. 10.1126/science.abg5526. [DOI] [PubMed] [Google Scholar]
- Geng S. S.; Zhang J.; Chen S.; Liu Z. L.; Zeng X. Q.; He Y.; Feng Z. Development and Mechanistic Studies of Iron-Catalyzed Construction of Csp(2)-B Bonds via C-O Bond Activation. Org. Lett. 2020, 22, 5582–5588. 10.1021/acs.orglett.0c01937. [DOI] [PubMed] [Google Scholar]
- Genov G. R.; Douthwaite J. L.; Lahdenpera A. S. K.; Gibson D. C.; Phipps R. J. Enantioselective remote C-H activation directed by a chiral cation. Science 2020, 367, 1246–1251. 10.1126/science.aba1120. [DOI] [PubMed] [Google Scholar]
- Slack E. D.; Colacot T. J. Understanding the Activation of Air-Stable Ir(COD)(Phen)Cl Precatalyst for C-H Borylation of Aromatics and Heteroaromatics. Org. Lett. 2021, 23, 1561–1565. 10.1021/acs.orglett.0c04210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.