

Abstract
Epigenetic modifications occur on genomic DNA and histones to influence gene expression. More recently, the discovery that mRNA undergoes similar chemical modifications that powerfully impact transcript turnover and translation adds another layer of dynamic gene regulation. Central to precise and synchronized regulation of gene expression is intricate crosstalk between multiple checkpoints involved in transcript biosynthesis and processing. There are more than 100 internal modifications of RNA in mammalian cells. The most common is N6-methyladenosine (m6A) methylation. Although m6A is established to influence RNA stability dynamics and translation efficiency, rapidly accumulating evidence shows significant crosstalk between RNA methylation and histone/DNA epigenetic mechanisms. These interactions specify transcriptional outputs, translation, recruitment of chromatin modifiers, as well as the deployment of the m6A methyltransferase complex (MTC) at target sites. In this review, we dissect m6A-orchestrated feedback circuits that regulate histone modifications and the activity of regulatory RNAs, such as long noncoding (lnc)RNA and chromosome-associated regulatory RNA. Collectively, this body of evidence suggests that m6A acts as a versatile checkpoint that can couple different layers of gene regulation with one another.
Historical perspective and introduction of epitranscriptomics
Methylation of specific nucleotides on RNA was thought to be a phenomenon exclusive to ribosomal and transfer RNA species [1]. Multiple lines of evidence from over half a century showed that it is unlikely that mRNA undergoes substantial methylation. Bacterial mRNA contained less than one methyl group per 3500 nucleotides [2], and a series of studies showed that incubation of eukaryotic cells with methyl-labeled methionine resulted in minimal incorporation of the label in mRNA precursors [3–5]. Ironically, it was the identification of another post-transcriptional modification that had a critical role in the discovery of mRNA methylation. The pioneering identification of widespread polyadenylation of transcriptional outputs led to the development of novel methods for large-scale isolation of purified mRNA [6–8]. Soon afterwards, Perry and Kelley demonstrated the existence of base-methylated mRNA in mouse L cells [3], followed by a study by Desrosiers et al. showing that, in contrast to the more promiscuous distribution of methylation on rRNA, methylated mRNA predominantly comprised m6A [9].
Although it was known for half a century that mRNA is chemically modified, it was only during the past decade that the field of ‘epitranscriptomics’ formally developed, mostly owing to technical and conceptual advances that allowed transcriptome-wide interrogation of m6A dynamics and, ultimately, galvanized interest in m6A function [10,11]. In particular, the identification of FTO as the first m6A demethylase in 2011 [12] revived this field, because it suggested that RNA modification can also be reversible and dynamic, analogous to DNA and histone modifications [13]. In 2012, the introduction of m6A-seq [14] and methylated RNA immunoprecipitation sequencing (MeRIP-Seq) [15] demonstrated the pervasive nature of chemical modification on mRNA and enabled functional interrogation, thus advancing our understanding of the significance of m6A modification in mammalian cell biology. Similar to epigenetics, the study of chemical modifications on DNA and histones, epitranscriptomics affects gene expression without altering the mRNA sequence (Figure 1). Although m6A is predominantly known to influence gene regulation through effects on mRNA stability and other post-transcriptional mechanisms, rapidly accumulating studies implicate m6A in epigenic control. Here, we briefly introduce post-transcriptional effects of m6A on gene regulation and then discuss rapidly evolving evidence that highlights the significance of crosstalk between epitranscriptomic and histone/DNA epigenetics mechanisms.
Figure 1. Schematic of epigenetic and epitranscriptomic mechanisms.
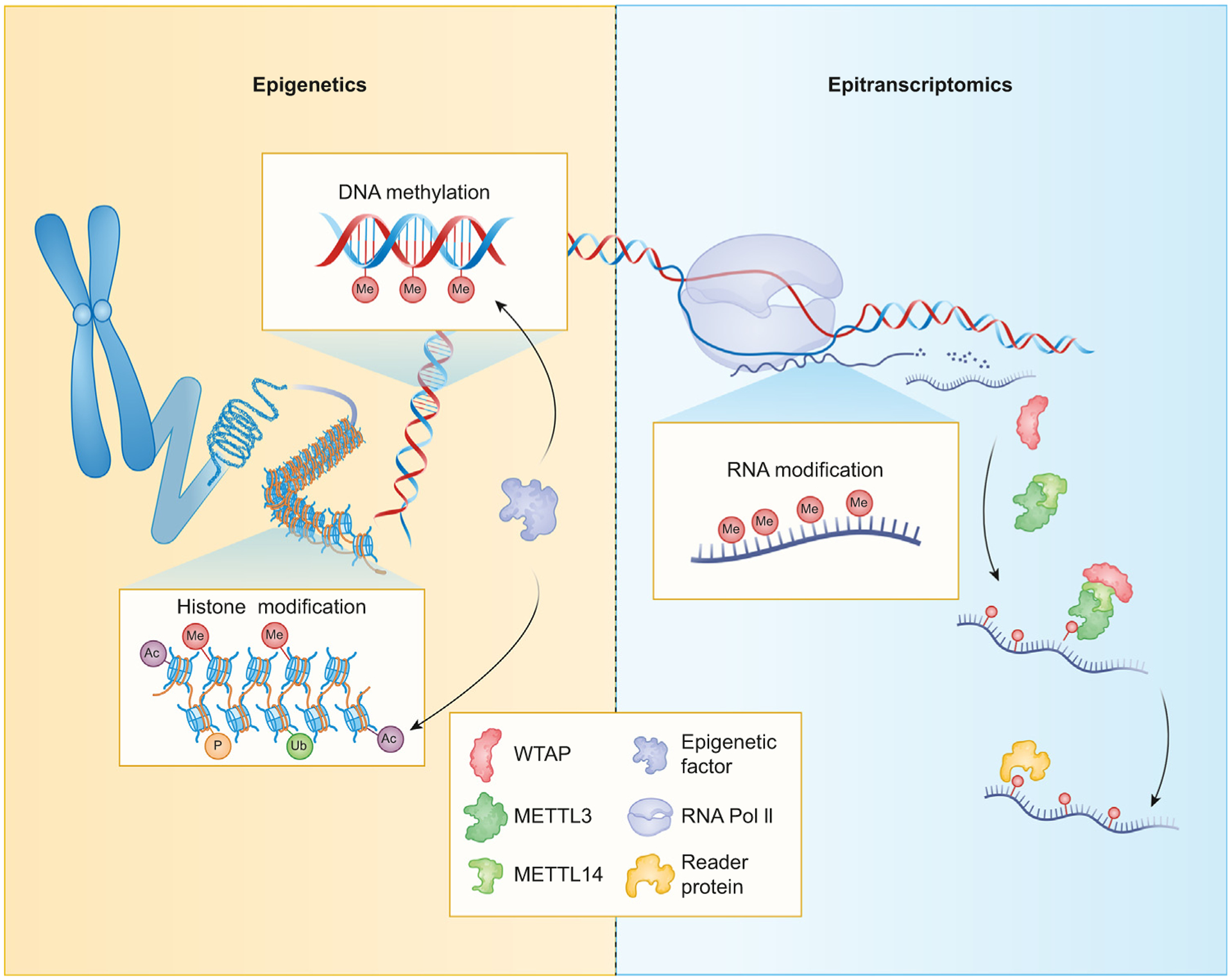
Modifications of DNA or histone proteins alter RNA polymerase II (RNA Pol II) transcriptional output. Chemical modifications on RNA, including N6-methyladenosine (m6A), are deposited cotranscriptionally by writer proteins. WTAP guides METTL3/METTL14 to m6A-binding motifs. The m6A sites are recognized by nuclear or cytoplasmic reader proteins, which alter mRNA localization, degradation, or translation.
Cellular regulation of m6A methylation
It is now established that methylation of the adenosine base at the nitrogen-6 position is the most abundant internal RNA modification [16]. Thousands of mRNAs undergo internal m6A modification of the consensus motif RRACH (R being either A or G, and H can be A, C or U) [17]. The frequency of this consensus motif significantly exceeds the m6A content on RNA in mammalian cells, hinting that additional ‘codes’ beyond sequence conservation impart m6A deposition [18]. In addition, m6A modifications are not randomly enriched on mRNA. Several studies have shown that most m6A sites cluster near the 3′-terminal exon, including the 3′-untranslated region (UTR), as well as long internal exons [19].
The installation of m6A occurs via a complex of enzymes known as ‘writers’ (see Glossary), comprising METTL14, METTL3, and WTAP [20]. Loss of function in any one of these components reduces m6A, which is critical for cell function [21,22]. The catalytic activity is contained in METTL3, with METTL14 acting as an allosteric activator. WTAP is an ubiquitously expressed nuclear protein that contains no catalytic activity but acts as a guide to recruit the methyltransferase complex to target transcripts [23]. Chemical modifications on RNA are thought to be dynamic, with removal of m6A accomplished by ‘eraser’ proteins, which include FTO and ALKBH5. Recent evidence suggests that FTO also demethylates the related modification m6Am [24]. Although physiological contributions of m6A suggest a predominant role in cell differentiation and cancer regulation, m6A has been implicated in diverse biological systems, including neuroregulation, immune activation, and metabolic control [11,25–28].
m6A in RNA turnover dynamics
How does imparting m6A modifications alter the fate of mRNA? Shortly after its discovery, labeling studies in HeLa cells showed an association between m6A and mRNA stability [29]. The installation of m6A can powerfully alter the decay, nuclear export, and/or translation of a mRNA, a process that is executed by ‘reader’ proteins. Readers include HuR and YTH-containing domain proteins. For example, YTHDF2 selectively binds m6A, resulting in localization of bound mRNA from the translatable pool to mRNA decay sites [30]. Binding of YTHDF1 to m6A sites enhances the translation of mRNA [31]. Despite slightly different modes of function being ascribed to individual YTH domain-containing proteins, a more recent study suggests that the YTH group of proteins acts exclusively at the level of mRNA stability and have redundant rather than distinct roles in gene regulation in Hela cells [32]. More systematical studies are warranted to test whether this is also true in different cellular contexts and experimental conditions.
Precisely how RNA methylation can alter transcript fate is poorly understood, but multiple mechanisms have been proposed. It is thought that RNA methylation can: (i) perturb the secondary structure of a mRNA to expose or mask RNA-binding motifs; (ii) influence the affinity of m6A-binding proteins for methylated mRNAs; (iii) reduce the solvation penalty of hydrophobic side chains of RNA-binding proteins that allows for increased affinity; and (iv) enhance phase separation of mRNAs [16,33]. Regardless of the mechanism, it is clear that alterations of RNA turnover dynamics are a principal mechanism by which m6A impacts transcript levels. The presence of m6A modifications increase RNA turnover and reduce protein levels, but there are exceptions in which the opposite may also be true depending on the context [30,34]. For instance, IGF2BP proteins (IGF2BP1/2/3) have been reported to enhance the stability and translation of m6A-modified mRNA targets by recruiting the mRNA stabilizers HuR, MATR3, and PABPC1, as well as localizing to stress granules during stress [35]. Hence, the consequence of the presence of m6A on any individual mRNA will potentially be driven by the location of this modification on the mRNA, interplay between writer and eraser complexes, and specific reader proteins involved [36].
Although m6A methylation has been strongly associated with RNA stability, it has also been linked with almost every step in RNA homeostasis, from pre-mRNA processing to translational efficiency. As an example, m6A is deposited co-transcriptionally and several studies have shown that m6A methylation regulates pre-mRNA splicing [37,38]. This evidence is consistent with colocalization of m6A writers and erasers at nuclear speckles, with their depletion impacting alternative splicing [39,40]. However, other investigations found that m6A deposited on nascent transcripts dictates transcript decay but is not required for splicing [41]. Similarly, other studies found conflicting evidence for the roles of m6A methylation in regulating translational efficiency [32,42,43]. The distinct conclusions are likely due to the different cell contexts and experimental models used for the studies. Taken together, there is strong evidence suggesting that m6A impacts transcripts stability, but its precise mode of regulation in impacting other steps in RNA processing is still emerging.
m6A and epigenetic control
A hint that m6A methylation may influence physiological regulation beyond post-transcriptional mechanisms comes from knockout studies of m6A-modulating machinery. Knockout of cytoplasmic YTHDF proteins, critical regulators of m6A-imparted transcript decay, does not recapitulate the loss of nuclear m6A-installing enzymes or other nuclear readers, which are developmentally lethal at an early stage [20,44]. This effect may be due, in part, to compensation from other readers [32], but also hints that m6A modifiers in the nucleus may have other roles independent of the executive actions of cytoplasmic readers. Indeed, rapidly accumulating evidence suggests that m6A methylation is integral to the proper maintenance of RNA biogenesis through feedback on epigenetic circuits. We discuss herein the diverse mechanisms by which m6A may crosstalk with chromatin modification. A summary of the effect of m6A on specific modifications is provided in Table 1.
Table 1.
Histone modifications impacted by m6A
| Modification | Model system(s) | Genetic manipulation | Effect on modification | Refs |
|---|---|---|---|---|
| H3K4me3 | Mouse ES cell | Mettl3 KO | Increased | [21,48] |
| Ythdc1 KO | [21,49] | |||
| Mouse neural stem cell | Mettl14 KO | [54] | ||
| H3K27ac | Mouse ES cell | Mettl3 KO | Increased | [21] |
| Ythdc1 KO | [21] | |||
| HEK293 | Increasing m6A | [56] | ||
| Mouse neural stem cell | Mettl14 KO | [54] | ||
| H3K27me3 | Mouse neural stem cell | Mettl14 KO | Increased | [54] |
| HEK293 | Increasing m6A | [56] | ||
| THP-1 | YTHDF2 KO | Decreased | [55] | |
| H3K9me2 | mESC and HEK293 | METTL3 KO | Increased | [56] |
| Mettl14 KO | [56] | |||
| YTHDC1 KD | [56] | |||
| FTO KO | Decreased | [56] | ||
| ALKHB5 KO | [56] | |||
| H3K9me3 | mESC | Mettl3KO | Decreased | [47,48] |
| Alkbh5KO | Increased | [47] | ||
| Mettl3 KO | Decreased | [47] |
Direct impact of m6A on heterochromatin regulation
Heterochromatin is an essential component of the genome because it maintains genome integrity and stable gene expression [45]. A critical function of heterochromatin is to restrain the activity of embedded satellite repeats and transposable elements [46]. In humans and mice, endogenous retroviruses (ERVs) are a prominent class of retrotransposons that require constitutive silencing by regulation machineries. Recently, three groups reported a role for m6A in the regulation of ERVs, specifically a type of repetitive element known as intracisternal A particle (IAP), in mouse embryonic stem cells (mESCs) (Figure 2) [47–49]. Knockout of Mettl3 and rescue by catalytically inactive Mettl3APPA failed to restore H3K9me3 levels, a defining molecular feature of heterochromatin, at IAP elements. Conversely, knockout of Alkbh5, a m6A eraser, significantly increased H3K9me3 levels at these sites. The m6A deposited by Mettl3 is read by Ythdc1 and contributes to heterochromatin formation. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) of Ythdc1 confirmed the enrichment at H3K9me3-rich transposable elements, and its role in mediating retrotransposon silencing and maintaining mESC identity [49]. Both IAP mRNA and protein levels are inversely correlated with that of m6A. The deposition of m6A onto the 5′UTR of the IAP mRNA recruits the YTHDF protein family for mRNA degradation. Mechanistically, Ythdc1 appears to act as an important guide for Mettl3, facilitating its interaction with chromatin, Setdb1, and Trim28, which in turn regulate H3K9me3 deposition at IAPs. Liu et al. demonstrated that, in addition to the involvement of Setdb1, m6A deposition represses Dux, the master regulator of the two-cell stage in ESCs [49]. Chelmicki et al. also observed an increase in IAP mRNA when both Mettl3 and Mettl14 were depleted; however, unlike other studies [49], H3K9me3 levels remained similar, with only a moderate increase [48]. A key difference is that Chelmicki et al. used an auxin-inducible degron (AID) system that can rapidly deplete Mettl3 and/or Mettl14 at will. Thus, it is conceivable that tampering de novo m6A deposition has a different effect on H3K9me3 levels at IAP elements than observed by Xu et al., who knocked out Mettl3 entirely. Despite some differences in the proposed mechanisms, these three studies [47–49] provide convincing evidence that links m6A and heterochromatin formation.
Figure 2. Role of METTL3 at heterochromatin.
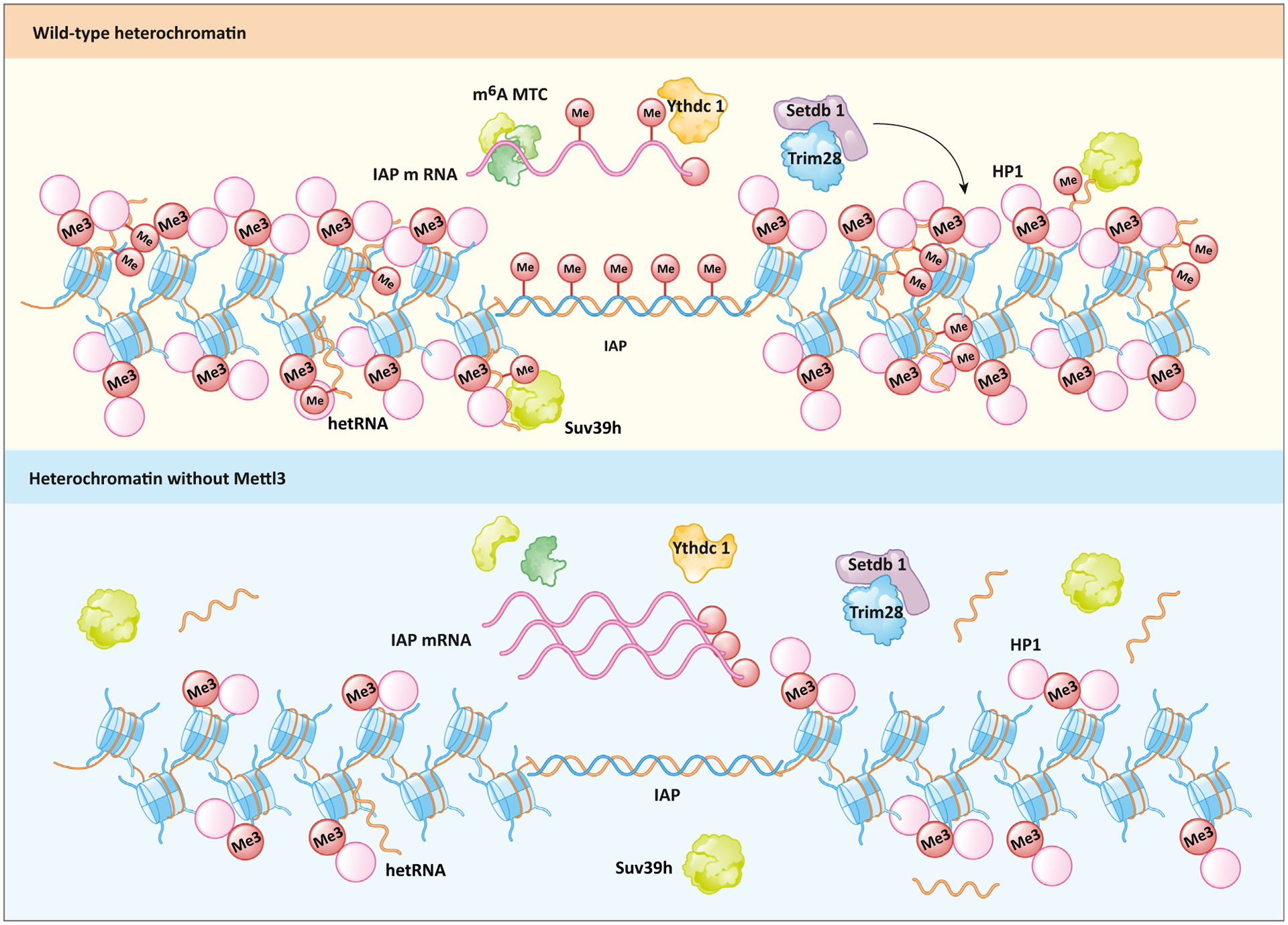
The N6-methyladenosine (m6A) methyltransferase complex (MTC) that includes METTL3 methylates intracisternal A particle (IAP) mRNA and heterochromatic RNA (hetRNA). Binding of m6A by Ythdc1 is essential for the downstream deposition of H3K9me3 by Setdb1/Trim28. Methylated hetRNA associates with chromatin and facilitates the retention of HP1 and Suv39h proteins. H3K9me3, HP1, hetRNA, and Suv39h are lost upon Mettl3 depletion.
Another role of m6A in stabilizing heterochromatin lies in major satellite repeats (MSR). In mouse chromatin, the centromeres are occupied by heterochromatin, which contains MSR and is characterized by H3K9me3, HP1 proteins, and DNA methylation [45]. Some MSR can be transcribed by RNA polymerase II (Pol II) and the resulting RNA is categorized as heterochromatic RNA (hetRNA) [50]. These hetRNA form RNA:DNA duplexes that help retain HP1 and Suv39h, which are histone lysine methyltransferases. MSR RNA, especially the sense strand, which contains six RRACH motifs compared with one in the antisense strand, is a preferred substrate for the METTL3/METTL14 complex in vitro. In Mettl3/Mettl14-knockout mESCs, MSR transcripts have reduced m6A levels, impaired chromatin association, as assayed by RT-qPCR of different cellular fractions, as well as decreased RNA:DNA hybrid formation [51]. Although the biological effect of this phenomenon is unclear, this study shows another mode by which m6A can regulate chromatin structure.
m6A and chromosome-associated regulatory RNA
Various RNA species can physically associate with chromatin, including promoter-associated RNAs (paRNA), enhancer RNAs (eRNA), and repeat RNAs transcribed from transposons. These chromosome-associated regulatory RNAs (carRNAs) have been shown to contribute to genome organization, transcript regulation, and nuclear subdomains [52]. A study by Liu et al. expanded our understanding of the destabilizing nature of m6A, showing that RNA modifications on carRNAs regulate local chromatin accessibility and downstream transcription (Figure 3, Key figure) [21]. Deletion of Mettl3 in mESCs enhanced the stability of carRNAs, leading to more-open chromatin and active transcription [21]. Congruent with these observations, the authors found elevations in histone marks associated with active transcription, such as H3K4me3 and H3K27ac. A similar phenotype was observed with nucleus-localizing Ythdc1 conditional knockout, but not cytoplasmic Ythdf2. YTHDC1 associated with RBM7 and ZCCHC8, key components of the Nuclear Exosome Targeting (NEXT) complex [53]. Hence, the study suggests that METTL3 tags carRNAs with m6A for degradation by YTHDC1 through the NEXT complex.
An intriguing observation is that an increase in chromatin accessibility following Mettl3 knockout was more prevalent at genes that normally had upstream m6A-tagged carRNAs. The biggest transcription rate differences were observed at genes that were involved in stem cell maintenance (Esrrb and Ranbp17) and chromatin remodeling (Prdm9 and Kmt2d), forming a positive-feedback loop in opening up chromatin [21]. Hypomethylation of carRNAs enhances the recruitment of EP300, a histone acetyltransferase, and YY1, a multifunctional transcription factor. In addition, RNA methylation loss decreased the binding of JARID2, a component of the Polycomb Repressive Complex 2 (PRC2), to carRNAs. Together, these findings suggest that methylation of carRNAs stabilizes closed chromatin by recruiting deactivating factors and parrying activating ones, thus adding another twist to the role of m6A in gene regulation.
Among the repeat RNA families that showed m6A-enrichment changes, are Long Interspersed Element −1 (LINE1) elements. These abundant retrotransposons showed more significant fold changes in methylation and altered decay upon Mettl3 depletion. Consistent with other carRNAs, LINE1 was highly enriched upon Mettl3 knockout and contributed to the global increase in chromatin accessibility. The authors provide direct evidence of functional effects of LINE1 methylation through use of CRISPR/dCas13b-fused FTO. Analogous to modified cas9 guides for DNA, the dCas13b-FTO fusion protein can achieve site-specific RNA demethylation when deployed with a guide RNA [21]. Using this approach, Liu et al. demonstrated a specific decrease in m6A levels at LINE1 RNAs as well as enhanced chromatin accessibility, carRNA abundance, and downstream transcription. Finally, the authors provide evidence of the functional relevance of RNA modifications on LINE RNAs by showing that this targeted strategy can decrease differentiation potential and increase self-renewal in mESC. Collectively, this work strongly supports the idea that m6A modifications gate chromatin accessibility and downstream transcription through modulation of carRNAs [21].
Key figure
Mechanisms of crosstalk between N6-methyladenosine (m6A) and epigenetic checkpoints
Figure 3.
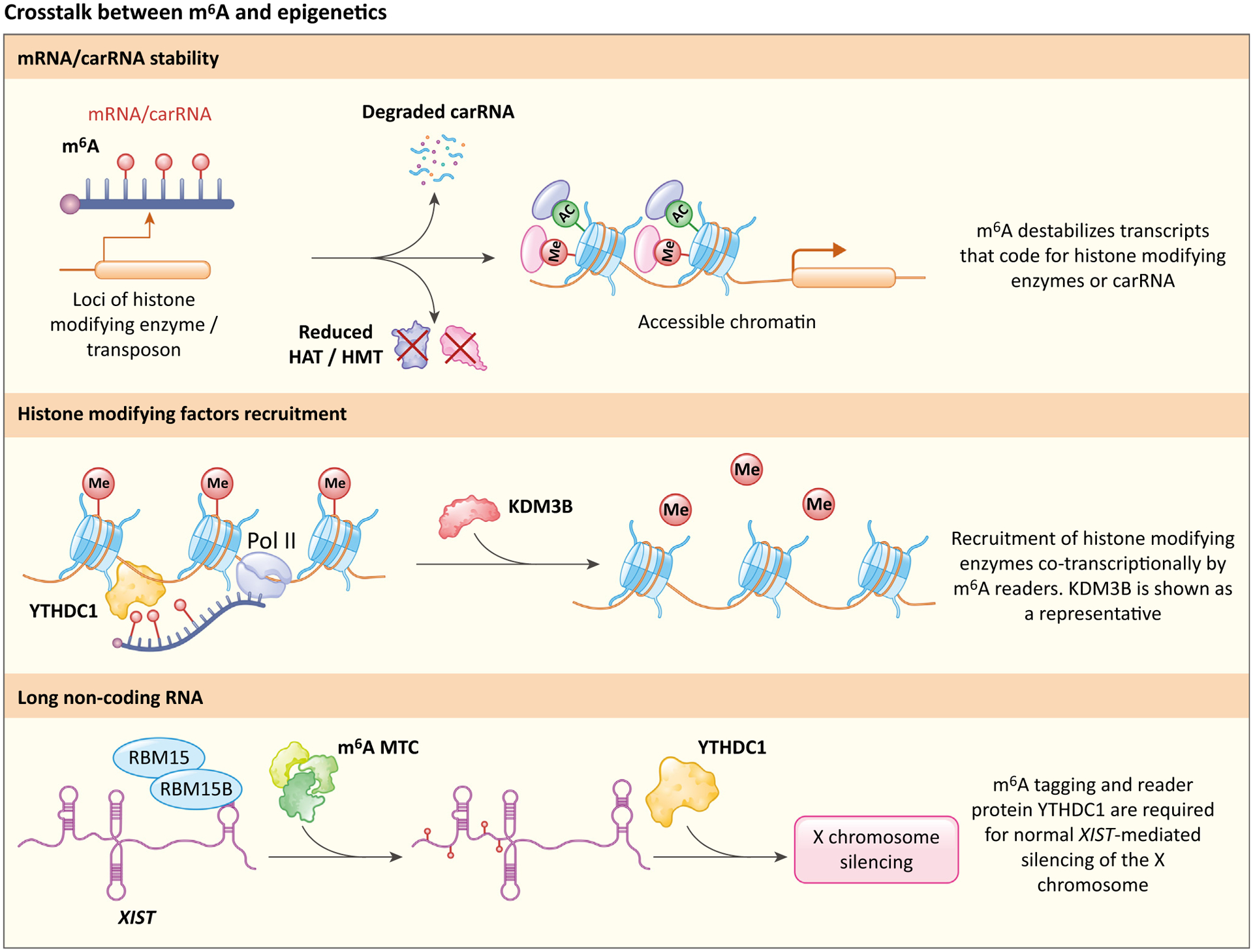
Chromosome-associated regulatory RNAs (carRNAs) or mRNA that encode histone-modifying enzymes are methylated and tagged for degradation, causing a decrease in carRNA and histone acetyltransferases (HATs) or histone methyltransferases (HMTs), ultimately leading to accessible chromatin. KDM3B is shown as a representative enzyme that is recruited by m6A. Other histone-modifying factors, such as Setdb1 and Trim28, are recruited in the same fashion. RBM15/RBM15B-mediated m6A deposition and subsequent YTHDC1 recruitment are essential for X-chromosome silencing.
m6A and H3K27ac
One mechanism by which RNA modifications can feed back on critical epigenetic checkpoints is through deposition of m6A on transcripts that encode histone-modifying proteins (Figure 3). Wang et al. discovered that m6A regulates gene expression in neural stem cells (NSCs) by destabilizing transcripts that encode the critical histone acetyltransferases P300/CBP [54]. The loss of Mettl14 was associated with developmental abnormalities, including reduced proliferation and premature differentiation. In homozygous mettl14f/fnes-cre mice, levels of H3K27ac and H3K27me3 were significantly upregulated. Compared with controls, mettl14f/fnes-cre mice showed increased expression of CBP and P300, with m6A-RNA immunoprecipitation showing minimal m6A enrichment of CBP and P300 transcripts. These results hint that the change in the activity of P300/CBP may be due to the destabilizing effects of m6A installation and that changes in these histone-modifying regulators underlie the alteration of chromatin architecture. Two key histone marks, H3K27ac and H3K4me3, were associated with gene activation, whereas H3K27me3 was associated with gene repression. In line with these results, unbiased transcriptome analysis did not favor global gene activation or repression in Mettl14-knockout NSCs, but Gene Ontology (GO) analysis indicated that the upregulated genes have a role in NSC differentiation, while the downregulated genes relate to proliferation. Treatment with the small-molecule Ezh2 inhibitor GSK343 partially rescued the proliferation defect in the loss-of-function cells. Although P300/CBP mediate acetylation of H3K27 at gene promoters and enhancers, it is unclear how a change in these factors specifically influences the methylation state of histone. Notably, the authors did not observe changes in the methylation state of PRC2 components, hinting that there must be distinct mechanisms by which m6A regulates histone modifications. Nonetheless, the aforementioned evidence suggests that a key mechanism by which m6A regulates the pluripotency of NSC is by altering the transcript stability of proteins that deposit histone chemical modifications [54].
m6A and H3K27me3
A recent study interrogated bacteria-induced inflammatory responses, finding that tight m6A control guards against unrestrained inflammation [55]. The authors found that loss of the m6A reader protein YTHDF2 led to enhanced stability of the histone demethylase KDM6B transcript, thereby increasing KDM6B translation and decreasing H3K27me3 levels at loci that encode proinflammatory cytokines, such as IL6, IL12B, and ICAM1. Enhanced hypomethylation of H3K27me3 in turn increases the transcription of inflammatory cytokines. In addition, the authors surmised that bidirectional crosstalk between m6A and H3K27me3 acts as a critical regulatory checkpoint that controls inflammatory gene expression. In heat-killed Salmonella typhimurium (HKST)-treated THP-1 cells, m6A peaks were mostly enriched in regions in which H3K27me3 was sparse. Chemically inhibiting KDM6B by GSK-J4 also reduced global m6A levels. Furthermore, HA-KDM6B co-immunoprecipitated with FLAG-METTL3 and FLAG-METTL14, hinting that KDM6B acts as guide for m6A installing machinery. Indeed, GSK-J4-treated cells showed impaired binding of METTL14 at the KDM6B locus, forming a negative-feedback loop to regulate H3K27me3 levels by modulating the stability of KDM6B [55]. Thus, it appears that H3K27me3 deposition, which is tightly regulated by the demethylase KDM6B, protects against m6A installation. In summary, this study suggests that the demethylation of H3K27me3 is crucial for the co-transcriptional recruitment of the methyltransferase complex METTL3/METTL14.
m6A and H3K9me2
Similar to the mode of regulation imparted by H3K27me3 and KDM6B crosstalk, a recent study by Li et al. suggests that METTL3/METTL14 regulates the repressive histone mark H3K9me2 [56]. The authors utilized an elegant screening system using a tetracycline-inducible reporter in human Flp-In HEK293 cells to directly investigate the effects of m6A on histone modifications. The reporter system comprised two plasmids, one with the m6A consensus motif GGAC repeats and the other with GGTC repeats as a control. An array of histone modifications was measured and analyzed after tetracycline treatment, and H3K9me2 showed the most drastic decrease (approximately fourfold) compared with the control reporter. The authors also observed minor upregulation of H3K27ac and H3K27me3, which would conflict with results reported by Wang et al. [54], who observed upregulation of H3K27me3 in Mettl14-knockout NSCs. However, the experimental conditions and contexts used by the two studies were drastically different. Li et al. validated their main findings showing that a loss of METTL3 or METTL14 increased global levels of H3K9me2, and such upregulation was reversed by induced expression of wild-type METTL3, but not the catalytically inactive METTL3D395A. m6A was shown to recruit KDM3B to demethylate H3K9me2, typically a repressive mark, through the reader protein YTHDC1. YTHDC1 coimmunoprecipitated with KDM3B, and a large fraction of endogenous YTHDC1 colocalized with KDM3B within the nucleus. Ythdc1 knockdown also showed a similar decrease in KDM3B binding to the chromatin as METTL3D395A. In addition, specific targeting of dPspCas13b-YTHDC1 increased local KDM3B abundance and a collateral decrease in H3K9me2 levels. KDM3B-occupied genes and m6A-modified genes showed large overlap, and distance analysis of KDM3B-ChIP showed enrichment at m6A RIP and METTL3 peak centers. Furthermore, such recruitment occurred cotranscriptionally, as supported by the enrichment of KDM3B at Pol II peaks, and addition of the Pol II inhibitor 5,6-dichlorobenzimidazole-1- β-D-ribofuranoside (DRB) significantly decreased the abundance of chromatin-bound KDM3B. Collectively, the body of work in this study provides direct mechanistic insights that link m6A deposition with functionally relevant histone modifications.
m6A and long noncoding RNA
Long noncoding RNAs (lncRNAs) have emerged as critical regulators of epigenetic mechanisms [57]. Expression of Xist, arguably the best-characterized lncRNA, ‘coats’ the X chromosome in females to silence gene expression [58]. Thus, Xist is critical for maintaining dose compensation between genders. A recent study by Patil et al. suggested that Xist is a target of m6A regulation and that chemical modifications on Xist are required for its function [59]. Mapping m6A sites at high resolution using m6A individual nucleotide-resolution crosslinking and immunoprecipitation (miCLIP) showed 78 putative m6A sites on XIST near the A repeat region. The authors further surmised that Xist-binding partners, RBM15 and RBM15B, are required for gene silencing by guiding the m6A methylation complex to the RNA. Co-immunoprecipitation assays showed a robust interaction between RBM15/RBM15B and METTL3 in a WTAP-dependent manner. Using a doxycycline-inducible Xist expression system, knockdown of either RBM15 or RBM15B did not achieve silencing of the Xist targets Gpc4 and Atrx, whereas Rbm15/Rbm15b double knockdown resulted in a substantial increase in Gpc4 and Atrx transcription. These results suggest that RBM15 and RBM15B have redundant functions. In addition, knockdown of Rbm15/Rbm15b significantly reduced the amount of m6A on XIST, a result recapitulated with knockdown of METTL3. Finally, the authors showed that the reader protein YTHDC1 is critical for m6A recognition on Xist and downstream transcription silencing. Altogether, these data support the model that RBM15/RBM15B recruit the METTL3-WTAP complex to XIST for targeted methylation. In addition, this work positions m6A as a dynamic regulator of physiological gene silencing and that the methylation state of ncRNA may be critical to their function.
m6A and H3K36me3
Accumulating evidence suggests that m6A feeds back on chromatin modifiers and histones, but the reverse is also true. H3K36me3, a transcription elongation marker that is enriched mainly in coding sequences and near the 3′ end, has been shown to guide m6A deposition [60]. Consistent with previous results, H3K36me3, but not H3K9me3 or H3K27me3, is over-represented at m6A peaks under distance analysis. This distribution is also consistent with the observation that H3K9me3 and H3K27me3 are mainly found in heterochromatin, in which transcription activity is inherently low. In various normal and cancer cell lines, the mRNA expression of SETD2, the histone methyltransferase of H3K36, positively correlated with the expression of the m6A writers METTL3, METTL14, and WTAP. Knockdown of SETD2 or overexpression of the histone demethylase KDM4A drastically decreased global m6A levels, as well as representative genes, such as MYC and ACTB.
The authors used a catalytically inactive CRISPR/dCas9 system fusion protein targeted to ectopic loci on the genome to test the causal relationship between H3K36me3 and m6A. Targeting of dCas9-KDM4A to the coding region instability determinant (CRD) region of MYC, in which H3K36me3 is enriched, resulted in a substantial decrease in H3K36me3 and m6A levels; conversely, targeting of dCas9-SETD2 to the gene body of GNG4, where there is no H3K36me3, resulted in an increase in H3K36me3 and m6A. In addition, the authors found that METTL14 acts as a key player to recognize and bind H3K36me3 in the MTC, linking H3K36me3 and m6A deposition. HA-METTL14 showed significant chromatin binding in HepG2 cells, as shown by ChIP-seq and, not surprisingly, co-enrichment with H3K36me3 mostly at gene bodies. Photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) followed by sequencing also confirmed that METTL14-binding sites overlapped with H3K36me3 and m6A. METTL3/METTL14/WTAP co-immunoprecipitated with H3K36me3 and the elongating form of Pol II (phosphorylated at Ser2 of the CTD). In mESCs, doxycycline-induced knockdown of Setd2 recapitulated the loss of m6A, as seen in other cell types, as well as an increase in the mRNA and protein levels of pluripotency factors Oct4, Sox2, and Nanog, consistent with the role of m6A in destabilizing transcripts. Collectively, this supports the model that H3K36me3 guides the binding of the m6A MTC to elongating transcripts and methylates nascent RNA co-transcriptionally [60].
Concluding remarks
In recent years, we have witnessed an explosion in the number of studies dissecting the mechanisms of m6A and its physiological contributions. Accumulating evidence has substantiated the significance of m6A in controlling gene expression and disease progression. While not the initial focus of m6A studies, the interaction between RNA methylation and other epigenetic regulatory players has received well-deserved attention and has inspired several intriguing questions moving forward. It is of great interest to understand whether and how the defects in such interactions or crosstalk contribute to the development of various types of disease. For example, altered ERV regulation is associated with abnormal development, cancer, and neurodegeneration, pathological states highly linked to m6A. Thus, the recent link between MTC activity and repetitive element requires further probing as to the extent to which m6A-endowed epigenetic mechanisms contribute to normal development and pathological states. While most studies have focused on roles of m6A in protein-coding transcripts, it is clear that we are just scratching the surface as far as appreciating the role of m6A in noncoding gene regulation is concerned (see Outstanding questions). The poor signal-to-noise ratio associated with commonly used m6A-mapping techniques is a major barrier to progress on that front, particularly since noncoding transcripts tend to be less abundant, short lived, and expressed in a context-specific fashion. Newer techniques are rapidly being disseminated, which should improve signal at a base pair resolution and provide detailed stochiometric analysis. Given that noncoding gene modulation is known to have a regulatory role in epigenetic control, further probing of this area will likely be fruitful. Currently available m6A-profiling methods often need a large amount of RNA material, which hampers the application of such methods to profiling m6A distributions in the transcriptome of primary patient samples, especially cancer stem cells and the primitive/progenitor cells of normal tissues. Development of novel technologies that only need a limited amount of RNA material and provide base-resolution m6A profiles with better quantitative information will substantially advance research in this field. In addition, the use of CRISPR genome editing and CRISPR-mediated RNA modification can help clarify the epistatic relationship between RNA methylation and chromatin dynamics. More recently, extensive evidence has shown that the introduction or removal of m6A at precise RNA sites is readily achievable using a guide RNA and dCas13-tethered modified methylase complex (e.g., METTL3:METTL14) with minimal off-target effects [21,61,62]. This opens exciting possibilities for RNA modification-based therapy and testing directly for crosstalk with other layers of gene regulation at precise transcripts. In addition, it is evident that more-refined definitions of epigenetic and epitranscriptomic landscapes are necessary [63]. For example, single-cell technologies are slowly being leveraged in epitranscriptomic applications, which can unmask heterogeneity and enable deeper investigation of spatial differences in RNA methylation patterns. Finally, it is unclear how m6A can have seemingly opposing effects on epigenetic control under different contexts. It could make the chromatin more or less accessible, as well as increase or decrease transcription and translation. Despite an increase in the number of studies and depth, we clearly need continued investment to investigate the molecular underpinnings of m6A effects.
Outstanding questions.
What is the role of RNA methylation of noncoding transcripts in gene regulation? Newer techniques will provide a better resolution of the m6A interactome of non-coding transcripts and enhance applications in multiple fields.
How does the deposition of m6A on RNA integrate with different layers of gene regulation at precise transcripts?
The recent evidence that m6A is required to restrain aberrant ERV activity provides evidence that effects of METTL3 on heterochromatin architecture may underlie its role in organismal development. However, what are the relative contributions of heterochromatin-dependent versus independent effects to mammalian physiology? What underlies the differences in heterochromatin formation in different cells and conditions? How does the MTC interface with transcriptional enablers at heterochromatin?
Are there other RNA modifications beyond m6A that feed back on epigenetic checkpoints?
Are the effects of m6A on epigenetic landscapes durable over a range of conditions and cell types? Can they override discordant effects on transcript stability or gene translation or are their biological effects more subtle?
Methylation residues on Xist tether other proteins that promote transcriptional repression. How does the binding of collaborative factors, such RBM15 and YTHDC1, lead to gene silencing? Are there other regulatory RNAs that exploit RNA modification sites as decoys or guides for binding of epigenetic factors?
Given the development of novel approaches that enable targeted in vivo control of RNA modification, can we exploit these technologies to alter the epigenetic landscape relevant in health and disease states?
Highlights.
Chemical modifications on histones, DNA, and RNA robustly impact gene regulation. Installation of the RNA modification m6A leads to altered mRNA stability and translation. Emerging data suggest that perturbations in m6A feedback on epigenetic checkpoints and vice versa.
RNA methylation of intracisternal A particle, a type of repetitive element, is required for proper heterochromatin formation and maintain genome integrity.
Loss of m6A in mouse embryonic stem cells enhances the stability of chromatin-associated RNAs, leading to more-open chromatin and active transcription.
m6A destabilizes transcripts that encode histone-modifying enzymes and complexes, including KDM6B, CBP, and P300.
The histone elongation mark H3K36me3 guides m6A deposition.
Xist-mediated silencing of the X chromosome requires m6A deposition and recognition by reader proteins.
m6A influence extends beyond mRNA stability, and investigation of functional effects of m6A on RNA biogenesis should be considered in physiological and mechanistic studies.
Acknowledgments
T.S. is funded by grants from NIDDK (DK118086), NHLBI (HL139549, HL149766), American Heart Association, and Burroughs Wellcome Fund. J.C. is funded by grants from NCI (CA236399, CA214965, and CA243386) and is a Leukemia & Lymphoma Society (LLS) Scholar. Figures created using BioRender (BioRender.com).
Glossary
- Erasers
proteins that remove m6A; include ALKBH5 and FTO, although FTO may preferentially target m6Am
- Nuclear speckles
subnuclear domains enriched in pre-mRNA splicing factors that are located in the interchromatin regions of the nucleoplasm of mammalian cells
- Readers
proteins that bind m6A and impact the stability or localization of mRNAs
- Writers
protein complexes that install m6A and that include the core protein METTL3 and the adaptor protein METTL14
Footnotes
Declaration of interests
J.C. is a scientific advisor for Race Oncology and a scientific founder of Genovel Biotech Corp., in which he also holds equities.
References
- 1.Starr JL and Sells BH (1969) Methylated ribonucleic acids. Physiol. Rev 49, 623–669 [DOI] [PubMed] [Google Scholar]
- 2.Moore PB (1966) Methylation of messenger RNA in Escherichia coli. J. Mol. Biol 18, 38–47 [DOI] [PubMed] [Google Scholar]
- 3.Perry RP and Kelley DE (1974) Existence of methylated messenger RNA in mouse L cells. Cell 1, 37–42 [Google Scholar]
- 4.Greenberg H and Penman S (1966) Methylation and processing of ribosomal RNA in HeLa cells. J. Mol. Biol 21, 527–535 [DOI] [PubMed] [Google Scholar]
- 5.Perry RP and Kelley DE (1970) Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J. Cell. Physiol 76, 127–139 [DOI] [PubMed] [Google Scholar]
- 6.Nakazato H and Edmonds M (1972) The isolation and purification of rapidly labeled polysome-bound ribonucleic acid on polythymidylate cellulose. J. Biol. Chem 247, 3365–3367 [PubMed] [Google Scholar]
- 7.Sheldon R et al. (1972) Detection of polyadenylic acid sequences in viral and eukaryotic RNA. Proc. Natl. Acad. Sci. U. S. A 69, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brawerman G et al. (1972) A procedure for the isolation of mammalian messenger ribonucleic acid. Biochemistry 11, 637–641 [DOI] [PubMed] [Google Scholar]
- 9.Desrosiers R et al. (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. U. S. A 71, 3971–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frye M et al. (2018) RNA modifications modulate gene expression during development. Science 361, 1346–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer KD and Jaffrey SR (2014) The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol 15, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia G et al. (2011) N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol 7, 885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng X et al. (2018) RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 28, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominissini D et al. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 [DOI] [PubMed] [Google Scholar]
- 15.Meyer KD et al. (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roundtree IA et al. (2017) Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y and Zhao JC (2016) Update: mechanisms underlying N(6)-methyladenosine modification of eukaryotic mRNA. Trends Genet. 32, 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linder B et al. (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ke S et al. (2015) A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev. 29, 2037–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaccara S et al. (2019) Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol 20, 608–624 [DOI] [PubMed] [Google Scholar]
- 21.Liu J et al. (2020) N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H et al. (2020) The biogenesis and precise control of RNA m(6)A methylation. Trends Genet. 36, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ping XL et al. (2014) Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mauer J et al. (2019) FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat. Chem. Biol 15, 340–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H et al. (2020) m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37, 270–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weng H et al. (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng X et al. (2018) Role of N(6)-methyladenosine modification in cancer. Curr. Opin. Genet. Dev 48, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H et al. (2018) m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sommer S et al. (1978) The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J. Mol. Biol 124, 487–499 [DOI] [PubMed] [Google Scholar]
- 30.Wang X et al. (2014) N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han D et al. (2019) Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zaccara S and Jaffrey SR (2020) A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell 181, 1582–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ries RJ et al. (2019) m(6)A enhances the phase separation potential of mRNA. Nature 571, 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nachtergaele S and He C (2018) Chemical modifications in the life of an mRNA transcript. Annu. Rev. Genet 52, 349–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H et al. (2018) Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol 20, 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patil DP et al. (2018) Reading m(6)A in the transcriptome: m(6) A-binding proteins. Trends Cell Biol. 28, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou KI et al. (2019) Regulation of co-transcriptional pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol. Cell 76, 70–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartosovic M et al. (2017) N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3′-end processing. Nucleic Acids Res. 45, 11356–11370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J et al. (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louloupi A et al. (2018) Transient N-6-methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep. 23, 3429–3437 [DOI] [PubMed] [Google Scholar]
- 41.Ke S et al. (2017) m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao Y et al. (2019) m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun 10, 5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z et al. (2020) Genetic analyses support the contribution of mRNA N(6)-methyladenosine (m(6)A) modification to human disease heritability. Nat. Genet 52, 939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geula S et al. (2015) Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 45.Janssen A et al. (2018) Heterochromatin: guardian of the genome. Annu. Rev. Cell Dev. Biol 34, 265–288 [DOI] [PubMed] [Google Scholar]
- 46.Allshire RC and Madhani HD (2018) Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol 19, 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W et al. (2021) METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591, 317–321 [DOI] [PubMed] [Google Scholar]
- 48.Chelmicki T et al. (2021) m(6)A RNA methylation regulates the fate of endogenous retroviruses. Nature 591, 312–316 [DOI] [PubMed] [Google Scholar]
- 49.Liu J et al. (2021) The RNA m(6)A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591, 322–326 [DOI] [PubMed] [Google Scholar]
- 50.Velazquez Camacho O et al. (2017) Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA–nucleosome association and RNA:DNA hybrid formation. eLife 6, e25293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duda KJ et al. (2021) m6A RNA methylation of major satellite repeat transcripts facilitates chromatin association and RNA: DNA hybrid formation in mouse heterochromatin. Nucleic Acids Res. 49, 5568–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X and Fu XD (2019) Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat. Rev. Genet 20, 503–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lubas M et al. (2011) Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell 43, 624–637 [DOI] [PubMed] [Google Scholar]
- 54.Wang Y et al. (2018) N 6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci 21, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu C et al. (2020) Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci. Adv 6, eaba0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y et al. (2020) N(6)-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat. Genet 52, 870–877 [DOI] [PubMed] [Google Scholar]
- 57.Wang KC and Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sahakyan A et al. (2018) The role of Xist in X-chromosome dosage compensation. Trends Cell Biol. 28, 999–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patil DP et al. (2016) m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang H et al. (2019) Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson C et al. (2020) Programmable m(6)A modification of cellular RNAs with a Cas13–directed methyltransferase. Nat. Biotechnol 38, 1431–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu XM et al. (2019) Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol 15, 865–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allis CD and Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat. Rev. Genet 17, 487–500 [DOI] [PubMed] [Google Scholar]