

Abstract
Background
Non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers. Berberine (BBR), an isoquinoline alkaloid, is commonly used in traditional Chinese medicine. Previous studies have shown that BBR has a potential anti-tumor effect. However, the mechanisms of BBR on mitochondrial function in anti-lung cancer remain unknown. The aim of this study was to explore mitochondrial function in anti-tumor mechanisms of BBR in NSCLC.
Methods
The NSCLCs were cultured and treated with various doses (40, 80, 120 µg/mL) of BBR for 24 and 48 h. Cell viability was evaluated using Cell Counting Kit-8 (CCK-8). Cell apoptosis, reactive oxygen species (ROS) and mitochondrial membrane potential (MMP) were detected by flow cytometry. Relative protein expression was examined by western blot and immunohistochemical (IHC) analysis.
Results
BBR potently suppressed NSCLC cells growth by inducing apoptosis in a dose-and time-dependent manner. BBR induced apoptosis in NSCLC cells as evidenced by caspase-3 cleavage, cytochrome c release, and mitochondrial membrane depolarization. BBR-induced, dose-dependent induction of apoptosis was accompanied by sustained phosphorylation of c-jun-NH2-kinase (JNK) and the JNK inhibitor (SP600125) significantly suppressed BBR-induced apoptosis, N-acetyl cysteine (NAC), a ROS scavenger, was sufficient to both suppress apoptosis signal-regulating kinase 1 (ASK1) and JNK activation and disrupt apoptotic induction.
Conclusions
The results suggest that BBR induces apoptosis of NSCLC cells via ROS-mediated ASK1/JNK activation and the mitochondrial pathway.
Keywords: Berberine (BBR), apoptosis, mitochondria, reactive oxygen species (ROS), non-small cell lung cancer (NSCLC)
Introduction
With an estimated 228,820 new cases and nearly 136,000 deaths associated with this condition in the USA alone in 2020, lung cancer is among the most prevalent and deadly cancers (1). Lung cancer rates have risen substantially in China in recent years, and thus lung cancer represents a major threat to public health (1). This type of cancer is typically treated by a combination of surgical tumor resection, adjuvant chemotherapy, and adjuvant immunotherapy. However, non-small cell lung cancer (NSCLC) is often chemo-resistant, and individuals with advanced NSCLC tend to have poor outcomes due to an absence of reliable curative treatments (2). Thus, further research needs to be conducted to identify effective approaches to treating NSCLC.
Berberine (BBR) is an isoquinoline alkaloid found in Coptidis Rhizoma, which has been shown to induce cell death in a range of cancer cell models by regulating the cell cycle, autophagy, apoptosis, and the surrounding tumor microenvironment (3). Although many studies had shown that BBR against NSCLC mainly focused on different cell signaling pathways, including Sin3A/TOP2B (4), Bcl-2/Bax (5), mTOR (6) and NF-κB/COX-2, Akt/ERK (7), etc. However, the specific mechanisms by which BBR drives the apoptosis of NSCLC cells is still worth exploring further. Moreover, BBR have been reported to stimulate reactive oxygen species (ROS) generation and drives the apoptosis in some cancer cell lines, including breast cancer (8), prostate cancer (9) and colonic carcinoma (10). However, little study has focused on NSCLC, and thus we sought to explore whether BBR drives the apoptosis of NSCLC cells through ROS and what are the possible mechanisms involved.
Apoptosis is a key regulator of diverse physiological and pathological processes, including cancer development and treatment (11). Chemotherapeutic drugs and many different stimuli can induce apoptotic cell death, often by inducing ROS production and thereby driving death receptor signaling and mitochondrial pathway activation (12). Research has shown that ROS triggers the activation of the apoptosis signal-regulating kinase 1 (ASK1)/mitogen-activated protein kinase (MAPK) signaling pathway (13). ASK1, a serine/threonine protein kinase, participates in cell differentiation and apoptosis (14). Once activated, ASK1 dissociates from thioredoxin-1 and induces cell death by activating the c-jun-NH2-kinase (JNK) and p38 MAPK pathways (15). Targeting ROS generation is thus a promising strategy for the development of novel anti-cancer drugs.
In the present study, we demonstrated BBR induced apoptosis in NSCLC via ROS generation and the subsequent activation of ASK1/JNK signaling and the mitochondrial apoptosis pathway. Altogether, we provide a new mechanistic evidence that BBR-induced apoptosis in NSCLC is mediated by ROS/ASK1/JNK pathway. We present the following article in accordance with the ARRIVE and MDAR reporting checklists (available at https://atm.amegroups.com/article/view/10.21037/atm-22-1298/rc).
Methods
Cells and treatment
The A549 and PC9 cells were provided by the Cell Bank of Shanghai Institute of Cell Biology, Shanghai, China. BBR was obtained from Sigma and was dissolved at a concentration of 40 mM in dimethyl sulfoxide (DMSO; Sigma-Aldrich) as a stock solution (stored at −20 ℃). It was then diluted to working concentrations with cell culture medium. The antibodies specific for coxIV, p-JNK, JNK, p-ASK1, ASK1, Bax, Bcl-2, caspase-3, cytochrome c, and β-actin were provided by Abcam (Boston, MA, USA). The horseradish peroxidase (HRP)-labeled anti-mouse and anti-rabbit IgG were provided by Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The radioimmunoprecipitation assay (RIPA) buffer, Hoechst33342, and DCFH-DA were provided by Sigma-Aldrich Inc. (Saint Louis, MO, USA). The JC-1 was provided by Life Technologies Inc. (Carlsbad, California, USA). A Caspase-3 Colorimetric Assay Kit was provided by Nanjing Keygen Biotech Co., Ltd. (Nanjing, China). A High Pure Mitochondria Isolation Kit was provided by Shanghai Genmed Biotech Co., Ltd. (Shanghai, China). All the other materials were provided by Bio-Rad (Hercules, CA, USA).
Cell Counting Kit-8 (CCK-8) assays
A CCK-8 (Dojindo, Kumamoto, Japan) approach was used to monitor cell proliferation. Briefly, 2×103 cells were added to 96-well plates with a range of BBR concentrations (0, 20, 40, 80, and 160 µM). After 24, 48, or 72 h, the cells were incubated for 2 h with CCK-8 reagent at 37 ℃, and absorbance was then measured by spectrophotometry (BioTek, Winooski, Vermont, USA).
Flow cytometry
The cells were treated for 48 h with BBR (0, 40, and 80 µmol/L), rinsed with pre-cooled phosphate buffered solution (PBS) and stained for 20 min with Annexin V-FITC/PI (BD Biosciences, San Jose, CA, USA) in the dark, after which the analysis was conducted by flow cytometry (BD Biosciences).
Intracellular ROS measurement
The membrane-permeable DCFH-DA probe was used to quantify the ROS levels of the cells. Exposure to intracellular esterase results in the inability of the hydrolyzed DCFH to exit the cell, while exposure to peroxides results in oxidation to yield DCF, which is fluorescent. Following treatment with BBR (0, 40, and 80 µmol/L), the cells were treated for 30 min with DCFH-DA (50 µM). After 2 washes with PBS, the cells were lysed and analyzed via flow cytometry (BD Biosciences).
Mitochondrial membrane potential (MMP) analysis
MMP was assessed with the JC-1 probe. Briefly, the cells were initially treated for 48 h with a range of BBR concentrations (i.e., 0, 40, and 80 µM), followed and then incubated for 20 min with JC-1 (10 µM) at 37 ℃ in the dark. The cells were then washed before the analysis via flow cytometry (BD Biosciences) with JC-1 aggregates (red) that were detected at 590 nm, and JC-1 monomers (green) that were detected at 529 nm. The resultant ratio of red to green fluorescence was then reported.
Mitochondrial isolation
The cellular mitochondria were collected with a High Pure Mitochondria Isolation Kit following a 48-h BBR treatment based on the instructions provided. Briefly, 107 cells were rinsed using chilled reagent A, after which they were lysed on ice with the prepared lysis reagent. The samples were then spun at 800 g at 4 ℃ for 10 min, after which the supernatants were spun again at 13,000 g at 4 ℃ for 10 min to collect the mitochondria.
Western blotting
RIPA buffer containing protease inhibitors (NCM Biotech, Suzhou, China) was used to lyse cells, after which a BCA Protein Assay Kit (Beyotime, Shanghai, China) was used to quantify protein levels. Equal protein amounts from each sample were then separated via 10% SDS-PAGE (Haochen Biotechnology Co., Ltd., Shanghai, China), and transferred to nitrocellulose membranes. Blots were blocked for 1 h with 3% BSA (Shanghai Yihui Biotechnology Co., Ltd., Shanghai, China), after which they were probed overnight with anti-β-actin (1:2,000), anti-Bax, anti-Bcl-2, anti-caspase-3, anti-p-JNK, anti-JNK, anti-cytochrome c, and anti-coxIV (1:1,000) at 4 ℃. After 3 washes in phosphate buffered solution with tween (PBST), the blots were probed with secondary antibodies for 1 h at room temperature, and visualized with an Odyssey scanner (LI-COR Biosciences, Lincoln, Nebraska, USA).
Immunohistochemical (IHC) analysis
IHC testing technology was applied to detect the expression level of the candidate target genes. The samples were de-waxed, paraffin-embedded, and incubated in 3% hydrogen peroxide for 30 min to suppress endogenous peroxidase activities. Citrate buffer was used to infiltrate these sections, followed by a 10-min heating process in a microwave oven to retrieve the antigen. Afterwards, these sections underwent an overnight-incubation in primary antibodies (1:2,000) at 4 ℃, rinsed with PBS, treated with peroxidase-labeled goat anti-mouse secondary antibodies at room temperature for 1 h, stained with hematoxylin and 3'-diaminobenzidine tetrahydrochloride, and finally visualized.
Nude mice xenograft tumor assays
Sixteen BALB/c nude mice were bought from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). All the mice were 4-week-old and about 20 g weight. A total of 1×107 A549 cells in 100 µL of serum-free media were subcutaneously injected into nude mice. Subsequently, these mice were divided into two groups, control group (saline) and BBR (500 mg/kg) group. Mice were given saline or BBR by gavage every other day. All the mice were sacrificed after 30 days. Then, tumors were dissected and analyzed. Animal experiments were performed under a project license (No. SHDSYY-2015-3298) granted by the Ethics Committee of Shanghai Tenth People’s Hospital (Tenth People’s Hospital Affiliated to Tongji University) and in compliance with the institutional guidelines for the care and use of animals. A protocol was prepared before the study without registration.
Statistical analysis
All the results represented at least three independent experiments, and are presented as the mean ± standard deviation, and were compared via one-way analyses of variance. The significance threshold for this study was P≤0.05.
Results
BBR inhibited proliferation and induced apoptosis in NSCLC cells
The A549 and PC9 cells were treated with 0–160 µM of BBR, and the cell viability was assayed after 24, 48, and 72 h using CCK8 assays. BBR markedly induced cell death in the A549 and PC9 cells in a concentration- and time-dependent manner (see Figure 1A). IC50 was determined to be 80–100 µM at 48 h of exposure to BBR. These cells also exhibited marked morphological changes following a 48-h treatment with BBR (see Figure 1B). Based on these experiments, we selected a BBR dose of 80 µM and a treatment time of 48 h for subsequent experiments to examine the mechanistic basis for the changes in NSCLC cell viability.
Figure 1.

BBR inhibited proliferation and induced apoptosis in NSCLC cells. (A) The A549 and PC9 cells were treated with various concentrations of BBR for 24, 48, and 72 h. Cell viability was analyzed by CCK-8 assays. (B) Morphological changes were assessed via microscopy after a 48-h treatment (20×). (C) The A549 and PC9 cells were treated with a range of BBR concentrations (0, 40, and 80 μM) for 48 h, after which Annexin V/PI staining was used to evaluate apoptotic death by flow cytometry. (D) The apoptotic rates were quantified. (E) After the same treatment described in (C), CL-caspase-3, Bax, and Bcl-2 in the NSCLC cells were measured by western blotting. (F) The protein levels from (E) were quantified. The data are presented as the mean ± standard deviation for the three different experiments with triplicate sets in each assay. *P<0.05, ***P<0.001, and nsP>0.05. BBR, berberine; NSCLC, non-small cell lung cancer; CCK-8, Cell Counting Kit-8.
To explore whether BBR-induced cell growth inhibition was related to apoptosis, we performed Annexin V-FITC/PI double staining, and found that the A549 and PC9 cells underwent significant apoptosis after treatment with increasing concentrations of BBR over a 48-h period. Roughly 7% of the A549 cells and 21% of the PC9 cells treated with the 80-µM dose exhibited signs of apoptosis as opposed to only 1.3% and 1.5% of the cells in the control groups, respectively (see Figure 1C,1D). Further, to determine the mechanism underlying BBR-induced apoptosis, we examined the expression of apoptosis-related proteins. Western blotting revealed dose-dependent increases in caspase-3 and Bax levels, and dose-dependent reductions in Bcl-2 levels, which led to an increase in the proapoptotic/anti-apoptotic (Bax/Bcl-2) ratio in the A549 and PC9 cells (see Figure 1E,1F). Taken together, these findings indicated that BBR induced cell growth inhibition by apoptosis.
BBR activated ASK1/JNK and the mitochondrial apoptotic pathway in NSCLC cells
Next, we explored how BBR induces the apoptosis of NSCLC cells. As is well established, the MAPK signaling pathway is one of the most important apoptotic pathways, and ASK1 is a member of the MAPK family. Thus, we tested the ability of BBR-induced ASK1 and JNK activation in the A549 and PC9 cells, and found a marked increase in the phosphorylation of the Thr 845 residue of ASK and JNK following a 48-h treatment with 80 µM of BBR in both cell types (see Figure 2A,2B). We also examined the A549 and PC9 cells to establish whether the apoptosis was associated with changes in mitochondrial phenotypes. We found that BBR induced a loss of MMP in a dose-dependent fashion in both the tested cell lines (see Figure 2C,2D). We also found that a high-dose (40 and 80 µM) BBR treatment for 48 h resulted in a reduction in mitochondrial cytochrome c levels, and a concomitant increase in cytosolic cytochrome c levels (see Figure 2E,2F). This suggested that mitochondrial dysfunction played a role in the apoptotic death of BBR-treated NSCLC cells.
Figure 2.

BBR activated the ASK1/JNK and mitochondrial apoptotic pathway in NSCLC cells. (A) The A549 and PC9 cells were treated with BBR (0, 40, and 80 μM) for 48 h, after which the ASK1, p-ASK1, JNK, and p-JNK levels were assessed by western blotting, (B) The protein levels from (A) were quantified. (C) After the same treatment described in (A), changes in MMP were measured in the NSCLC cells. (D) Cells with low MMP are shown in Q2, and were quantified. (E) After the same treatment described in (A), the A549 and PC9 cells were separated into mitochondrial and cytosolic fractions, and the cytochrome c levels in these fractions were measured by Western blotting. (F) The protein levels from (E) were quantified. The data are presented as the mean ± standard deviation for the three different experiments with triplicate sets in each assay. *P<0.05, **P<0.01, and ***P<0.001. BBR, berberine; ASK1, apoptosis signal-regulating kinase 1; JNK, c-jun-NH2-kinase; NSCLC, non-small cell lung cancer; MMP, mitochondrial membrane potential.
BBR induced NSCLC cell apoptosis via the activation of the ASK1/JNK pathway
To examine whether the activation of the ASK1/JNK pathway played a role in BBR-induced NSCLC cells apoptosis, we next treated these cells with the JNK inhibitor SP600125. The results showed that tumor cells with SP600125 was partially reduced in BBR-induced p-JNK, CL-caspase-3 and Bax/Bcl-2 levels (see Figure 3A,3B), and apoptosis assay also confirmed that JNK inhibitors SP600125 with BBR can partially inhibit the apoptosis of NSCLC cells (see Figure 3C). These findings suggested that the activation of the ASK1/JNK pathway was associated with the BBR-induced apoptosis of NSCLC cells.
Figure 3.

BBR induced NSCLC cell apoptosis via the activation of the ASK1/JNK pathway. (A) The A549 and PC9 cells were treated with or without 40 μM of SP600125 for 1 h before treatment with 80 μM of BBR for 48 h. The protein levels of p-JNK, JNK, CL-caspase-3, Bax, and Bcl-2 in the NSCLC cells were measured by western blotting. (B) The protein levels were quantified. (C) Cell viability was measured by CCK-8 assays. The data are presented as the mean ± standard deviation for the three different experiments with triplicate sets in each assay. *P<0.05, **P<0.01, and ***P<0.001. +, added with indicated agent; −, none. BBR, berberine; NSCLC, non-small cell lung cancer; ASK1, apoptosis signal-regulating kinase 1; JNK, c-jun-NH2-kinase; CCK-8, Cell Counting Kit-8.
BBR induced NSCLC cell apoptosis via the activation of the ROS/ASK1/JNK pathway
The JNK signaling pathway is known to be sensitive to intracellular redox state and ROS production. To establish the effect of BBR treatment on ROS generation, we incubated the A549 and PC9 cells with DCFH-DA, and found that exposure to BBR (80 µM) for 48 h resulted in elevated ROS production of 78.0%±0.4% and 77.9%±4.1%, respectively (see Figure 4A,4B). The ROS induction was also dose-dependent. The above findings suggest that ROS plays a key role in BBR-induced ASK1/JNK activation, and the consequent apoptosis in NSCLC cells. To test this possibility, we treated cells with N-acetyl cysteine (NAC), which significantly reduced BBR-induced ROS production in both tested cell lines (see Figure 4C,4D).
Figure 4.

BBR induced NSCLC cell apoptosis via the activation of the ROS/ASK1/JNK pathway in vitro. (A) The A549 and PC9 cells were then treated with BBR (0, 40, and 80 μM) for 48 h, and the ROS levels in the A549 and PC9 cells were measured by flow cytometry. (B) The data from (A) were quantified. (C) Following a 1-h pretreatment with NAC (500 μM), the cells were treated for 48 h with 80 μM of BBR, and the ROS levels were then assessed by flow cytometry. (D) The data from (C) were quantified. (E) The cells were treated as described in (C), and the protein levels were then assessed by western blotting. (F) The data are presented as the mean ± standard deviation for the three different experiments with triplicate sets in each assay. *P<0.05, and ***P<0.001. +, added with indicated agent; −, none. BBR, berberine; ROS, reactive oxygen species; NAC, N-acetyl cysteine; ASK1, apoptosis signal-regulating kinase 1; JNK, c-jun-NH2-kinase; NSCLC, non-small cell lung cancer.
We also found that NAC treatment reduced BBR-induced ASK1 and JNK phosphorylation in these NSCLC cells, indicating that ASK1/JNK activation is ROS-dependent in both tested cell lines, and simultaneously suppresses BBR-mediated caspase-3 and Bax/Bcl-2 upregulation. These findings suggested that ROS induction controlled mitochondrial dysfunction (see Figure 4E,4F). Thus, we hypothesized that BBR induces the apoptosis of A549 and PC9 cells via the ROS/ASK1/JNK pathway that promotes mitochondrial dysfunction. To test this hypothesis, an IHC analysis of the tumor tissues, was performed, and the results showed that BBR increased the levels of phosphorylation-ASK1, phosphorylation-JNK, and caspase-3. Additionally, consistent with observations in vitro, we found that BBR inhibited the activity of Bax in the tumors (see Figure 5).
Figure 5.

BBR induced NSCLC cell apoptosis via the activation of the ASK1/JNK pathway in vivo. (A) IHC analysis of the expression of phosphorylation-ASK1, phosphorylation-JNK, CL-caspase-3, and Bax in the tumor tissues treated with BBR (500 mg/kg). (B) The protein levels from (A) were quantified. ***P<0.001. BBR, berberine; ASK1, apoptosis signal-regulating kinase 1; JNK, c-jun-NH2-kinase; NSCLC, non-small cell lung cancer; IHC, immunohistochemical.
Discussion
BBR suppresses proliferation of NSCLC through diverse mechanisms including the inhibition of cancer cell invasion and metastasis, regulation of immune system, and the promotion of DNA damage (16). On the top of this, more researches reported BBR activate apoptosis of NSCLC. BBR upregulates tumor downregulates oncogenes (TNF-α, COX-2, MMP-2, and MMP-9) and suppressor genes (p21 and p53) mRNA and protein to enhance cell apoptosis (17). In addition, BBR can regulate the expression and interaction of HOTAIR and miR-34a-5p in NSCLC, which significantly inhibits epithelial-mesenchymal transition (EMT) and induces apoptosis (18). Moreover, BBR and icotinib synersiglically induce apoptosis by targeting ROS in epidermal growth factor receptor (EGFR)-resistant NSCLC cell lines (19). In a previous study, we showed that BBR-induced NSCLC apoptosis is related to the phosphorylation of JNK (20). To clarify the mechanisms upstream of JNK, we demonstrated that BBR effectively sustained the generation of the ROS and overproduction of ROS resulted in ASK1/JNK activation, which caused cell apoptosis in NSCLC cells.
ROS are important upstream molecules in the progression of cell death and survival. Thus, the induction of ROS production is an effective anti-cancer strategy. Some ROS-inducing agents, such as cisplatin, cyclophosphamide, and resveratrol (21), have been reported to induce apoptosis through deoxyribonucleic acid toxicity by the induction of intracellular ROS. For example, chlorpyrifos was found to trigger oxidative stress and induce apoptosis and necroptosis in fish liver cells by regulating the ROS/PTEN/PI3K/AKT axis (22). Additionally, increases in ROS also activate the ASK1/JNK (23), PI3K/AKT/mTOR (24), AMPK/p53 (25) and other apoptosis signaling pathways. There is extensive evidence that BBR promotes tumor cell apoptosis via the production of ROS in melanoma (26), breast cancer (27), and pancreatic cancer (28). Fan et al. found BBR increased ROS production in NSCLC cells (29); however, the mechanisms by which ROS exert their anti-tumor effect are not yet known. The ROS/ASK1/JNK signaling pathway is a possible mechanism for the BBR-dependent anti-apoptotic effect in NSCLC cells.
In this research, we demonstrate that BBR effectively inhibited the proliferation of A549 and PC9 cells in vitro by induced apoptosis. Annexin V and PI staining revealed that BBR was able to induce cellular apoptosis in the A549 and PC9 cells, and the effects of BBR on apoptosis-related genes in the A549 and PC9 cells were also analyzed. We found that BBR upregulated the expression of the apoptosis-promoting gene Bax while inhibiting the expression of the anti-apoptotic gene Bcl-2. The increased expression of Bax usually leads to increased mitochondrial membrane permeability, resulting in the release of pro-apoptotic factors, such as cytochrome c from mitochondria to cytosol, which initiates caspase cascade activation and promotes apoptosis progression (30). As a result, we also analyzed the cytochrome c protein levels in mitochondria and cytoplasm, and found that BBR treatment significantly promoted the transfer of cytochrome c from the mitochondria to cytoplasm, and this trend was positively correlated with drug concentration.
Next, we found that BBR-induced apoptosis was dependent on the dose-dependent phosphorylation of the ASK1 and JNK. ASK1/JNK signaling is indeed a key regulator of apoptosis (31), and we found that the JNK inhibitor SP600125 was able to prevent BBR-induced NSCLC cell apoptosis. This is consistent with the previous findings for colon cancer cells that BBR-induced, dose-dependent induction of apoptosis was accompanied by sustained phosphorylation of JNK and the generation of the ROS, what’s more, the induction of apoptosis was alleviated by inhibitors specific for JNK (SP6000125) and the scavenger of ROS (NAC) reversed BBR-induced apoptosis effects via inhibition of JNK expression (10).We also measured the level of intracellular ROS in A549 and PC9 cells after BBR treatment, and found that BBR significantly increased ROS levels. The pretreatment of NSCLC cells with NAC was sufficient to suppress ROS generation and associated apoptosis following BBR treatment, indicating that such ROS production is necessary for programmed cell death induction. These findings are consistent with those generated using prostate cancer cells that showed that NAC treatment prevented BBR-induced ROS generation in PC-3 cells, thereby interfering with their apoptosis (9). In fact, our observation supported our initial hypothesis that NAC-mediated ROS ablation would be sufficient to largely ablate BBR-induced ASK1/JNK phosphorylation and associated cell death (see Figure 6). Similarly, Xie et al. previously found that BBR induced breast cancer cell death via the ROS/ASK1/JNK pathway (27). Thus, these results indicate that treatment with BBR induced the apoptosis of NSCLC cells via the ROS/ASK1/JNK pathway.
Figure 6.
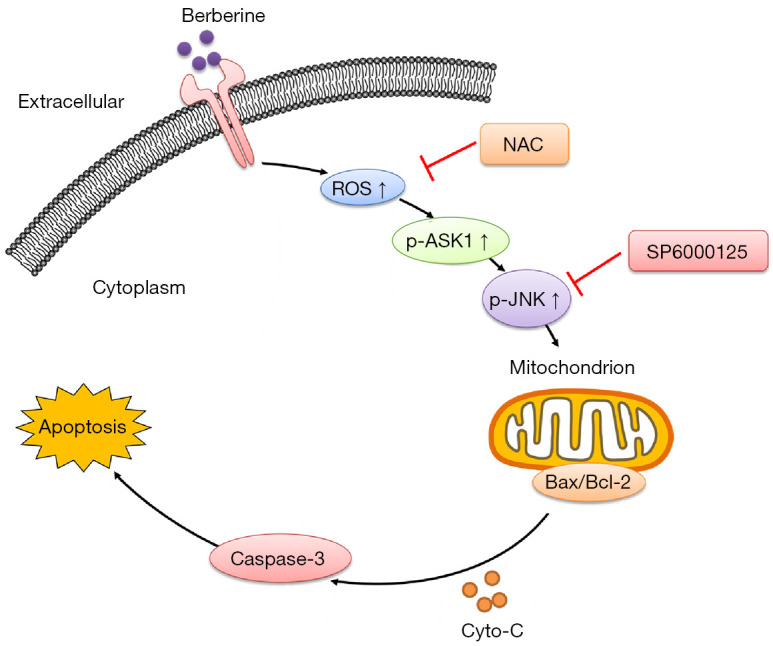
An overview of the BBR-induced apoptotic death of NSCLC cells. BBR-induced ROS generation promotes ASK1/JNK phosphorylation, which in turn suppresses Bcl-2 family proteins, driving a loss of MMP, the release of mitochondrial cytochrome c, and the consequent caspase-dependent apoptosis pathway. NAC, N-acetyl cysteine; ROS, reactive oxygen species; ASK1, apoptosis signal-regulating kinase 1; JNK, c-jun-NH2-kinase; BBR, berberine; NSCLC, non-small cell lung cancer; MMP, mitochondrial membrane potential.
In conclusion, the results of the present study indicated that BBR mediated the generation of ROS, which activated the ASK1/JNK pathway, and subsequently resulted in loss of MMP, and eventually triggered A549 and PC9 cell apoptosis. Based on our findings, we proposed a model by which BBR induced apoptosis in NSCLC via the ROS/ASK1/JNK pathways, which provides evidence of the therapeutic potential of BBR for NSCLC patients.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was financially supported by the National Natural Science Foundation of China (Nos. 31770131 and 81473469), the Shanghai Shen Kang Hospital Development Center Plan (No. SHDC12018119), the International Cooperation Project of the Belt and Road (No. 20400750600), and the Shanghai Municipal Commission of Health and Family Plan (No. 201840056).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Animal experiments were performed under a project license (No. SHDSYY-2015-3298) granted by the Ethics Committee of Shanghai Tenth People’s Hospital (Tenth People’s Hospital Affiliated to Tongji University) and in compliance with the institutional guidelines for the care and use of animals.
Reporting Checklist: The authors have completed the ARRIVE and MDAR reporting checklists. Available at https://atm.amegroups.com/article/view/10.21037/atm-22-1298/rc
Data Sharing Statement: Available at https://atm.amegroups.com/article/view/10.21037/atm-22-1298/dss
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://atm.amegroups.com/article/view/10.21037/atm-22-1298/coif). The authors have no conflicts of interest to declare.
(English Language Editor: L. Huleatt)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7-30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2.Meador CB, Hata AN. Acquired resistance to targeted therapies in NSCLC: Updates and evolving insights. Pharmacol Ther 2020;210:107522. 10.1016/j.pharmthera.2020.107522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Liu Y, Du X, et al. The Anti-Cancer Mechanisms of Berberine: A Review. Cancer Manag Res 2020;12:695-702. 10.2147/CMAR.S242329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Huang X, Tao C, et al. Berberine chloride suppresses non-small cell lung cancer by deregulating Sin3A/TOP2B pathway in vitro and in vivo. Cancer Chemother Pharmacol 2020;86:151-61. 10.1007/s00280-020-04050-y [DOI] [PubMed] [Google Scholar]
- 5.Li J, Liu F, Jiang S, et al. Berberine hydrochloride inhibits cell proliferation and promotes apoptosis of non-small cell lung cancer via the suppression of the MMP2 and Bcl-2/Bax signaling pathways. Oncol Lett 2018;15:7409-14. 10.3892/ol.2018.8249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar R, Awasthi M, Sharma A, et al. Berberine induces dose-dependent quiescence and apoptosis in A549 cancer cells by modulating cell cyclins and inflammation independent of mTOR pathway. Life Sci 2020;244:117346. 10.1016/j.lfs.2020.117346 [DOI] [PubMed] [Google Scholar]
- 7.Lu JJ, Fu L, Tang Z, et al. Melatonin inhibits AP-2β/hTERT, NF-κB/COX-2 and Akt/ERK and activates caspase/Cyto C signaling to enhance the antitumor activity of berberine in lung cancer cells. Oncotarget 2016;7:2985-3001. 10.18632/oncotarget.6407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang R, Qiao H, Chen S, et al. Berberine reverses lapatinib resistance of HER2-positive breast cancer cells by increasing the level of ROS. Cancer Biol Ther 2016;17:925-34. 10.1080/15384047.2016.1210728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meeran SM, Katiyar S, Katiyar SK. Berberine-induced apoptosis in human prostate cancer cells is initiated by reactive oxygen species generation. Toxicol Appl Pharmacol 2008;229:33-43. 10.1016/j.taap.2007.12.027 [DOI] [PubMed] [Google Scholar]
- 10.Hsu WH, Hsieh YS, Kuo HC, et al. Berberine induces apoptosis in SW620 human colonic carcinoma cells through generation of reactive oxygen species and activation of JNK/p38 MAPK and FasL. Arch Toxicol 2007;81:719-28. 10.1007/s00204-006-0169-y [DOI] [PubMed] [Google Scholar]
- 11.Call JA, Eckhardt SG, Camidge DR. Targeted manipulation of apoptosis in cancer treatment. Lancet Oncol 2008;9:1002-11. 10.1016/S1470-2045(08)70209-2 [DOI] [PubMed] [Google Scholar]
- 12.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016;1863:2977-92. 10.1016/j.bbamcr.2016.09.012 [DOI] [PubMed] [Google Scholar]
- 13.Ma J, Zhao D, Lu H, et al. Apoptosis Signal-Regulating Kinase 1 (ASK1) Activation is Involved in Silver Nanoparticles Induced Apoptosis of A549 Lung Cancer Cell Line. J Biomed Nanotechnol 2017;13:349-54. 10.1166/jbn.2017.2359 [DOI] [PubMed] [Google Scholar]
- 14.Matsuzawa A, Nishitoh H, Tobiume K, et al. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal 2002;4:415-25. 10.1089/15230860260196218 [DOI] [PubMed] [Google Scholar]
- 15.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001;2:222-8. 10.1093/embo-reports/kve046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Meng X, Wu D, et al. A Natural Isoquinoline Alkaloid With Antitumor Activity: Studies of the Biological Activities of Berberine. Front Pharmacol 2019;10:9. 10.3389/fphar.2019.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalaiarasi A, Anusha C, Sankar R, et al. Plant Isoquinoline Alkaloid Berberine Exhibits Chromatin Remodeling by Modulation of Histone Deacetylase To Induce Growth Arrest and Apoptosis in the A549 Cell Line. J Agric Food Chem 2016;64:9542-50. 10.1021/acs.jafc.6b04453 [DOI] [PubMed] [Google Scholar]
- 18.Zheng F, Li J, Ma C, et al. Novel regulation of miR-34a-5p and HOTAIR by the combination of berberine and gefitinib leading to inhibition of EMT in human lung cancer. J Cell Mol Med 2020;24:5578-92. 10.1111/jcmm.15214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen P, Dai CH, Shi ZH, et al. Synergistic inhibitory effect of berberine and icotinib on non-small cell lung cancer cells via inducing autophagic cell death and apoptosis. Apoptosis 2021;26:639-56. 10.1007/s10495-021-01694-w [DOI] [PubMed] [Google Scholar]
- 20.Chen QQ, Shi JM, Ding Z, et al. Berberine induces apoptosis in non-small-cell lung cancer cells by upregulating miR-19a targeting tissue factor. Cancer Manag Res 2019;11:9005-15. 10.2147/CMAR.S207677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldman EH, Chen L, Fu H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3-3 dissociation. J Biol Chem 2004;279:10442-9. 10.1074/jbc.M311129200 [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Wang L, Shi X, et al. Chlorpyrifos induces the apoptosis and necroptosis of L8824 cells through the ROS/PTEN/PI3K/AKT axis. J Hazard Mater 2020;398:122905. 10.1016/j.jhazmat.2020.122905 [DOI] [PubMed] [Google Scholar]
- 23.Huang M, Li X, Jia S, et al. Bisphenol AF induces apoptosis via estrogen receptor beta (ERβ) and ROS-ASK1-JNK MAPK pathway in human granulosa cell line KGN. Environ Pollut 2021;270:116051. 10.1016/j.envpol.2020.116051 [DOI] [PubMed] [Google Scholar]
- 24.Fang S, Wan X, Zou X, et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis 2021;12:88. 10.1038/s41419-020-03357-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S, Xu A, Gao Y, et al. Graphene oxide exacerbates dextran sodium sulfate-induced colitis via ROS/AMPK/p53 signaling to mediate apoptosis. J Nanobiotechnology 2021;19:85. 10.1186/s12951-021-00832-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Gong Q, Song C, et al. Berberine-photodynamic therapy sensitizes melanoma cells to cisplatin-induced apoptosis through ROS-mediated P38 MAPK pathways. Toxicol Appl Pharmacol 2021;418:115484. 10.1016/j.taap.2021.115484 [DOI] [PubMed] [Google Scholar]
- 27.Xie J, Xu Y, Huang X, et al. Berberine-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species generation and mitochondrial-related apoptotic pathway. Tumour Biol 2015;36:1279-88. 10.1007/s13277-014-2754-7 [DOI] [PubMed] [Google Scholar]
- 28.Park SH, Sung JH, Kim EJ, et al. Berberine induces apoptosis via ROS generation in PANC-1 and MIA-PaCa2 pancreatic cell lines. Braz J Med Biol Res 2015;48:111-9. 10.1590/1414-431x20144293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan XX, Leung EL, Xie Y, et al. Suppression of Lipogenesis via Reactive Oxygen Species-AMPK Signaling for Treating Malignant and Proliferative Diseases. Antioxid Redox Signal 2018;28:339-57. 10.1089/ars.2017.7090 [DOI] [PubMed] [Google Scholar]
- 30.Burke PJ. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017;3:857-70. 10.1016/j.trecan.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue J, López JM. Understanding MAPK Signaling Pathways in Apoptosis. Int J Mol Sci 2020;21:2346. 10.3390/ijms21072346 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as