

Abstract
Objective
The study objective was to determine whether overexpression of the mitochondrial antioxidant peroxidase, peroxiredoxin 3 (Prx3), reduces the severity of osteoarthritis (OA) in mice.
Methods
Age‐related OA (age 18 and 24 months) and OA induced by destabilization of the medial meniscus (DMM at age 6 months) were assessed in male mice that overexpress a human Prdx3 transgene encoding the Prx3 protein. Lox‐stop‐lox‐Prdx3 (iPrdx3) mice were crossed with aggrecan‐CreERT2 mice to produce iPrdx3AgCreERT2 or with Col2Cre to produce iPrdx3Col2Cre mice. Germline transgenics (Prdx3Tg) were also evaluated. Prx3 protein level was assessed by immunoblotting and functionally after induction of elevated mitochondrial hydrogen peroxide (H2O2) using menadione. Histological sections of stifle joints were scored for cartilage damage (Articular Cartilage Structure score [ACS]), osteophytes, and synovial hyperplasia and were evaluated by histomorphometry.
Results
Overexpression of Prx3 maintained mitochondrial membrane integrity and inhibited p38 phosphorylation in the presence of elevated H2O2. ACS scores of 18‐month‐old iPrdx3AgCreERT2 mice (mean ± SD, 4.88 ± 5.05) were significantly lower than age‐matched iPrdx3 controls (11.75 ± 6.34, P = 0.002) and trended lower in the 18‐month Prdx3Tg group (P = 0.14), whereas no significant differences between experimental and control groups at 24 months of age or in OA induced by DMM surgery were noted. Osteophyte scores trended lower in the 18‐month‐old Prdx3Tg group (P = 0.09) and at 24 months in the iPrdx3Col2Cre mice (P = 0.05). There were no significant group differences in synovial hyperplasia or histomorphometric measures.
Conclusion
Overexpression of the mitochondrial peroxidase Prx3 reduced the severity of age‐related OA, but not at advanced ages and not in DMM‐induced OA in younger mice.
INTRODUCTION
Reactive oxygen species (ROS) have been hypothesized to contribute to cartilage loss in osteoarthritis (OA), not only by causing oxidative damage to DNA, proteins, and lipids but also in their role as second messengers in catabolic signaling pathways that contribute to joint tissue destruction (1). The cellular ROS include hydrogen peroxide, superoxide, the hydroxyl radical, and peroxynitrite. A variety of cytosolic oxidases, such as the nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases, produce ROS, whereas generation of mitochondrial ROS is through the electron transfer chain and by mitochondrial oxidases, such as monoaminoxidase (2). There is evidence that increased mitochondrial ROS due to mitochondrial dysfunction contributes to the development of age‐related diseases, including OA (3). A role for mitochondrial ROS has been proposed for post‐traumatic OA as well. For example, inhibition of mitochondrial electron transport using intra‐articular injections of amobarbital reduced the severity of OA induced by intra‐articular fracture in pigs (4).
Hydrogen peroxide (H2O2) is the primary intracellular ROS responsible for the regulation of redox signaling because of its relative abundance and longer half‐life compared with other ROS (5). Levels of intracellular H2O2 are controlled by a family of six peroxiredoxins (Prx1‐6) that serve as peroxidases with peroxiredoxin 3 (Prx3) (encoded by the Prdx3 gene) controlling levels of mitochondrial H2O2 (5). We reported that normal cartilage from older adults and from OA joints both exhibited elevated levels of hyperoxidized Prx 1‐3, which occurs in the presence of excessive H2O2 and results in the inactivation of Prx function (6). The generation of excessive mitochondrial H2O2 in human chondrocytes in vitro resulted in hyperoxidation and inhibition of Prx3, which favored activation of the p38 mitogen activated protein (MAP) kinase over Akt signaling and resulted in increased catabolic signaling and cell death (6). The overexpression of catalase targeted to the mitochondria counteracted excessive mitochondrial H2O2, inhibited p38 activation, and promoted Akt activity and chondrocyte survival in vitro, whereas transgenic mice overexpressing catalase in the mitochondria (mitochondrial catalase [MCAT] mice) developed less severe age‐related OA (6). Likewise, adenoviral overexpression of mitochondrial Prx3 in chondrocytes was able to inhibit p38 activation and restore Akt signaling under conditions of excessive mitochondrial ROS (7). These results suggested that inhibition of excessive mitochondrial ROS may be a useful strategy to combat cartilage degradation in OA.
Much of the work that has implicated a role for mitochondrial ROS in OA has been performed in vitro or relies on indirect evidence, such as the demonstration of increased mitochondrial ROS in OA cartilage or evidence obtained using nonspecific antioxidants. Reduced OA severity in aged MCAT mice (6) provided in vivo evidence for mitochondrial H2O2 contributing to OA, but only a small number of mice were studied (n = 5 to 6 per group), and overexpressing catalase in the mitochondria has the limitation that catalase is normally found primarily in peroxisomes in the cytosol. Prx3, however, is located specifically in mitochondria where it serves as an important enzyme to catalyze H2O2 (5). Therefore, the objective of the current study was to determine the effects of transgenic overexpression of Prx3 on the development of age‐related OA and injury‐induced OA in vivo. Prx3 overexpression in mice has been shown to reduce levels of mitochondrial H2O2, resulting in improved glucose tolerance (8) as well as reduced age‐associated cognitive decline (9). A role for Prx3 in OA in vivo has not previously been investigated.
MATERIALS AND METHODS
Mouse studies
Animal studies were approved by the University of North Carolina Animal Care and Use Committee. Male mice on a C57BL/6 background were used. Inducible Prdx3 transgenic mice (iPrdx3) were generated with an iPrdx3 expression cassette in which loxP‐stop‐loxP was placed between the ubiquitous CAG promoter and human Prdx3 complementary DNA so that human Prx3 expression can be induced by removing the stop codon using Cre recombinase. We crossed these mice with aggrecan‐CreERT2 mice to produce (iPrdx3AgCreERT2) mice and treated them with tamoxifen as described (10) to induce transgene expression. Tamoxifen was administered at a dose of 40 μg/g by intraperitoneal injection at 4 months of age for the mice used in the destabilization of the medial meniscus (DMM) and aging experiments, repeated daily for 5 consecutive days. Additional tamoxifen doses were given over 3 days (Monday, Wednesday, Friday) at 12 months of age for the aging study. To control for any potential effects of tamoxifen, the control mice without the Cre driver (iPrdx3) also received tamoxifen. Conditional Prx3 transgenics were generated by crossing iPrdx3 mice with Col2‐Cre mice to produce iPrdx3Col2Cre mice. These latter mice were only used in the aging study at the 24‐month time point. Germline transgenics that overexpress human Prx3 globally (Prdx3Tg) were produced as previously described (8). Control mice were wild‐type littermates for the Prdx3Tg mice and iPrdx3 littermates for the iPrdx3AgCreERT2 and iPrdx3Col2Cre mice. The Prdx3Tg and iPrdx3 mice were generated at the Transgenic Core of the University of Michigan. The aggrecan‐CreERT2 mice were provided by Dr. Benoit de Crombrugghe (University of Texas MD Anderson Cancer Center), and the Col2‐Cre mice were provided by Dr. Di Chen (when he was on faculty at Rush University Medical Center, Chicago, Illinois). For genotyping, DNA isolated from ear punches was used in polymerase chain reaction. Primers for iPrdx3 were 5′‐GTT GTC GCA GTC TCA GTG GA ‐3′ and 5′‐GAC GCT CAA ATG CTT GAT GA ‐3′. Prdx3 was genotyped as previously described (8). Primers used to detect the intact nicotinamide nucleotide transhydrogenase (NNT1) allele were the following: 5′‐GAC CAA TGC CAT CTC AGG TT=3′ and 5′‐AAG GGC CGA CAC ATT CTA TG=3′.
DMM and sham control surgeries were performed on the right knee as previously described (11) at 6 months of age in separate groups of mice. For the aging study, mice were aged to 18 and 24 months. A total of 251 mice were used for this study. Exact numbers of mice used for each experiment are presented in the results and ranged from 12 to 18 per experimental group. Numbers per group were based on a previously published power analysis from a similar study of male C57BL/6 DMM and aging mice (10).
Immunoblotting and immunofluorescence
Immunoblotting of cartilage lysates from mouse femoral caps was performed as described (6) to verify the level of Prx3 expression using an antibody that recognizes human and mouse Prx3 (Abcam ab73349). Immunofluorescence experiments were performed on mouse chondrocytes isolated from wild‐type, Prdx3Tg, iPrdx3, and iPrdx3‐AgCre femoral caps. Freshly isolated, unpassaged cells were cultured in DMEM/F12 containing 10% FBS on 24‐well glass bottom plates (Cellvis P24‐1.5P) for 72 hours in room air oxygen. To induce oxidative stress, cells were treated with 25 μM menadione in culture medium for 18 hours. Mitochondrial staining was performed with 200 nM MitoTracker Deep Red FM (Invitrogen M22426) in culture medium for 30 minutes at 37°C and then visualized via fluorescence microscopy on an EVOS M5000 Imaging System (ThermoFisher). Fluorescent images were analyzed and quantified using CellProfiler. Menadione‐treated mouse chondrocytes were also evaluated by measuring p38 and extracellular‐signal regulated kinase (ERK) phosphorylation as previously described (6).
Histological evaluation
Mice were euthanized at 10 weeks after DMM surgery or at 18 months and 24 months of age for the aging study. Mouse stifle joints were fixed, processed, sectioned, and stained, and mid‐coronal sections were scored for cartilage damage (Articular Cartilage Structure [ACS] 0‐12), osteophytes (0‐3), and synovial hyperplasia (0‐3) in the medial and lateral tibial plateaus as previously described in detail (12). These sites were chosen based on our previous studies demonstrating that the great majority of lesions occur on the tibial plateaus in the DMM model and in aging studies, with DMM mice demonstrating more severe medial disease and aging mice more severe lateral disease (12). Scores from the two sites were summed. Histomorphometric measurements, including the thickness and area of articular cartilage, calcified cartilage, and subchondral bone, were performed as described (12). All measures were made by observers for whom experimental groups were anonymized.
Statistical analysis
GraphPad Prism 8.12 was used for data analysis and to generate graphs, with histomorphometry analysis performed in RStudio (version 1.3.1093). Results are presented as mean ± SD. Mann‐Whitney nonparametric testing was used to compare sham and DMM operated mice (separately for each group) to judge the effectiveness of DMM surgery in inducing OA and then to compare the DMM mice from the experimental group to their appropriate controls. For the aging study, the Mann‐Whitney test was used to compare each experimental group to its appropriate control group. Histomorphometric and cell culture experiments were analyzed using Welch t tests.
RESULTS
Expression of functional human Prx3 in mouse transgenics
Immunoblotting of femoral cap lysates confirmed increased Prx3 protein expression in the Prdx3Tg, iPrdx3AgCreERT2, and iPrdx3Col2Cre mice compared with their controls (Supplementary Figure S1). To determine whether the overexpressed Prx3 was functional, mitochondrial membrane integrity was evaluated using Mitotracker Deep Red in chondrocytes under conditions of menadione‐induced oxidative stress. Menadione treatment resulted in loss of mitochondrial membrane integrity in chondrocytes from control mice (wild type, iPrdx3) but not in chondrocytes isolated from Prx3 overexpressing mice (Prdx3Tg and iPrdx3AgCre), indicating that the overexpressed Prx3 was able to reduce levels of mitochondrial H2O2 (Figure 1A and B). Menadione‐induced phosphorylation of p38, but not ERK, was also reduced in chondrocytes from Prdx3Tg mice compared with wild‐type controls (Figure 1C and D). This is consistent with our previous study (6, 7) in which menadione‐induced phosphorylation of p38, but not ERK, was inhibited in human chondrocytes overexpressing Prx3 by adenoviral transduction.
Figure 1.
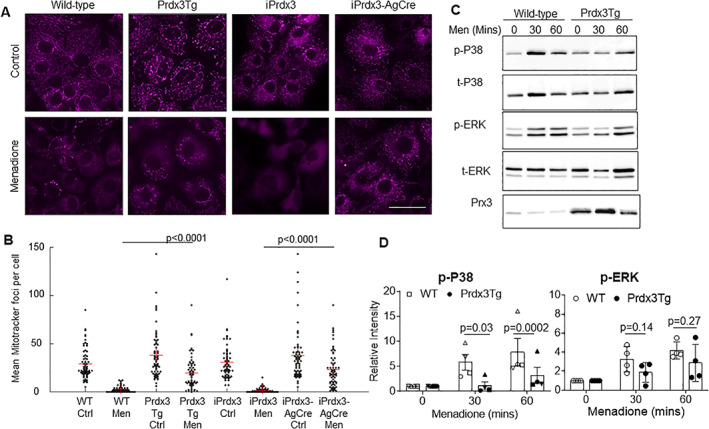
Expression of the Prdx3 transgene in chondrocytes reduces the effects of mitochondrial H2O2. (A) Mitochondrial membrane staining of chondrocytes derived from wild‐type, Prdx3Tg, iPrdx3, and iPrdx3AgCre mice treated with 0.1% DMSO (Control) or 25 μM menadione for 18 hours. Scale bar = 50 μm. (B) Quantification of mean mitochondrial foci per cell from 65 randomly sampled cells from each treatment group. Results are representative of three independent experiments. (C) Immunoblot analysis of p38 and ERK phosphorylation in menadione‐treated chondrocytes from WT and Prdx3Tg mice. Immunoblot shown is representative of n = 4 experiments. (D) Densitometric analysis of immunoblots (n = 4) demonstrating reduced p38 but not ERK phosphorylation in Prdx3Tg mice compared with WT controls. Results are the relative intensity of phospho‐p38 to total p38 and phospho‐ERK to total ERK normalized to time 0.
Mice overexpressing Prx3 are not protected from DMM‐induced OA
The DMM model was used to determine whether overexpression of human Prx3 in 6‐month‐old mice was able to reduce the severity of surgically induced OA. All groups of mice that underwent DMM surgery exhibited significantly more cartilage damage (ACS grade) and osteophyte formation at 10 weeks after surgery, a time point when the disease is moderately severe, compared with sham‐operated groups (Figure 2). There was no difference between the various Prdx3 transgenic lines and their controls. Minimal synovial hyperplasia, which is typical of the DMM model, was present and did not differ among the groups. There were also no differences among the DMM groups in histomorphometric measures (Supplementary Table S1).
Figure 2.
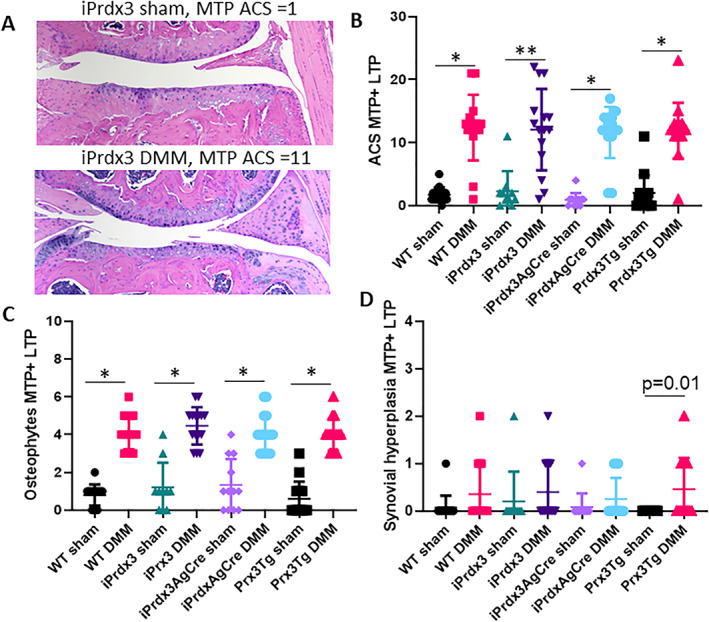
Effects of Prx3 protein overexpression on DMM‐induced OA in mice. Six‐month‐old Prx3‐overexpressing mice and their controls underwent sham or DMM surgery to induce OA. Mice were evaluated for OA by histology at 10 weeks after surgery. (A) Representative H&E images of the MTP from a sham and DMM mouse (10×). (B) ACS grades for the MTP+LTP. (C) Osteophyte scores for the MTP+LTP. (D) Synovial hyperplasia scores for the MTP+LTP. Results are the mean ± SD. WT controls, sham and DMM n = 15; iPrdx3 controls, sham n = 12 and DMM n = 14; iPrdx3AgCre sham n = 13 and DMM n = 15; Prxd3Tg sham and DMM n = 15. *P < 0.0001, **P = 0.0002. ACS, Articular Cartilage Structure; DMM, destabilization of the medial meniscus; H&E, hematoxylin–eosin; LTP, lateral tibial plateau; MTP, medial tibial plateau; MTP + LTP, sum of the medial and lateral tibial plateaus; OA, osteoarthritis; Prx3, peroxiredoxin 3; Prx3Tg, germline transgenics that overexpress human Prx3 globally; WT, wild‐type.
Effects of Prx3 overexpression on age‐related OA
In contrast to DMM‐induced OA, the ACS grades of the 18‐month‐old mice were significantly lower in the iPrdx3AgCreERT2 group (4.88 ± 5.05, mean ± SD) than the age‐matched iPrdx3 control group (11.75 ± 6.34, P = 0.002), whereas a trend toward a significantly lower ACS grade was noted in the 18‐month Prdx3Tg group (P = 0.14), which also exhibited a trend (P = 0.09) in lower osteophyte scores (Figure 3A‐C). The Prdx3Tg mice had the lowest ACS scores of any group (3.40 ± 5.26), but one outlier and the variability in the littermate wild‐type control group (6.78 ± 7.75) reduced statistical significance. There were no differences in histomorphometric measures (Supplementary Table S2). At 24 months of age, there were no significant differences in ACS scores among the groups (Figure 3E), with a modest decrease in osteophyte scores (Figure 3F) noted in the iPrdx3Col2Cre mice (2.62 ± 1.12) compared with the iPrdx3 controls (3.69 ± 1.55, P = 0.05). No differences in synovial hyperplasia scores were noted at either age (Figure 3D and G), and there were no differences in histomorphometric measures (Supplementary Table S3).
Figure 3.
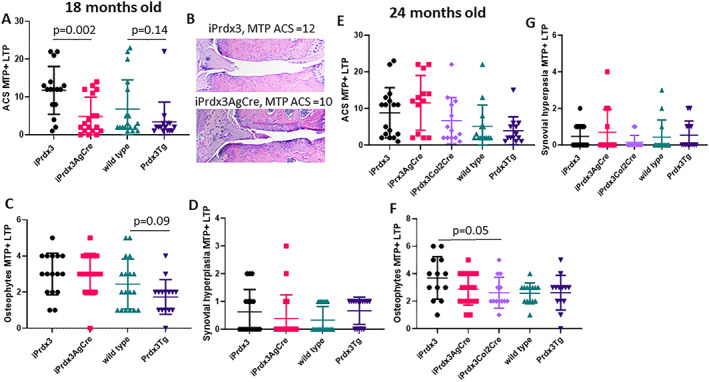
Effects of Prx3 protein overexpression on age‐related OA in mice. Mice at 18 months of age (A‐D) and 24 months of age (E‐G) were evaluated by histology using the same measures shown in Figure 2. Results are the mean ± SD. iPrdx3 controls, n = 18 at 18 months and n = 14 at 24 months; iPrdx3AgCre n = 18 at 18 months and n = 13 at 24 months; iPrdx3Col2Cre n = 13 at 24 months; wild‐type controls, n = 18 at 18 months and n = 14 at 24 months; Prxd3Tg n = 15 at 18 months and n = 13 at 24 months. ACS, Articular Cartilage Structure; LTP, lateral tibial plateau; MTP, medial tibial plateau; MTP + LTP, sum of the medial and lateral tibial plateaus; OA, osteoarthritis; Prx3, peroxiredoxin 3; Prx3Tg, germline transgenics that overexpress human Prx3 globally.
Because the Prx3 transgenic mice used in this study had originally been generated on a mixed C57BL/6J;SJL background, we genotyped the mice to determine whether any strain carried a deletion of the NNT gene that has been reported in C57BL/6J mice. Loss of NNT has been demonstrated to alter mitochondrial redox balance (13) that could potentially affect the results of our study. The iPrdx3 mice either with or without AgCreERT2 did not carry the NNT deletion, whereas the germline Prdx3Tg colony and their wild‐type control littermates did carry it (Supplementary Figure S2). Because we used iPrdx3 littermates as controls for iPrdx3AgCreERT2 and iPrdx3Col2Cre mice and wild‐type littermates as controls for the Prdx3Tg mice differences in expression of NNT did not explain differences in the results.
DISCUSSION
These results demonstrate that transgenic overexpression of the mitochondrial antioxidant protein Prx3, an important enzyme for catalysis of mitochondrial H2O2, can decrease age‐related OA, particularly cartilage damage, in 18‐month‐old mice. However, Prx3 overexpression was not sufficient to reduce the severity of OA at more advanced ages or in young adult mice with DMM‐induced OA. These findings provide partial support for the hypothesis that increased mitochondrial H2O2 is a significant contributor to the development of age‐related OA. The findings do not rule out a role for mitochondrial dysfunction and increased mitochondrial H2O2 in injury‐induced OA or OA at the most advanced ages. Besides Prx3, there are additional mechanisms by which H2O2 is metabolized in the mitochondria that may be at play, such as reduction by glutathione and glutathione peroxidase (14). Furthermore, mechanisms by which mitochondrial dysfunction contributes to OA exist that are independent of elevated mitochondrial H2O2, such as changes in energy metabolism and ATP production (reviewed in [3]).
Overexpression of the mitochondrial antioxidant Prx3 might have been more effective in the aging model than the DMM model because aging is associated with a lower antioxidant capacity (reviewed in [1]), which would not be expected in the young adult mice used for the DMM study. Because of the difficulty in measuring H2O2 in vivo, it is not known whether equivalent amounts of mitochondrial H2O2 are produced in these models. There are several possible reasons why Prx3 overexpression was not as effective in the mice at 24 months of age. It is possible that mitochondrial production of H2O2 continues to rise with age such that the increased level of transgenic Prx3 expression was insufficient to counter higher levels of mitochondrial H2O2 that are capable of inactivating Prx3 at the more advanced age. Also, an age‐related decline in NADPH levels could negatively affect Prx3 function. The recycling of oxidized Prxs back to their reduced state requires the activity of the NADPH‐dependent thioredoxin reductase (15). Cellular deficiency of NADPH can result in accumulation of unresolved Prxs not able to serve their antioxidant function.
Transgenic Prx3 overexpression has been successfully employed to examine the role of mitochondrial H2O2 in other age‐related conditions, including myocardial infarction (16) and cognitive decline (9). We did not use a Prx3 knock‐out model for comparison to Prx3 overexpression because past studies have shown that Prx3 knock‐out mice exhibit skeletal muscle damage and metabolic abnormalities by 10 months of age that would confound the results of an OA study (17).
There are limitations to our study. ROS other than H2O2 may contribute to OA, such as the hydroxyl radical and peroxynitrite, that may not be altered by overexpression of Prxs. Only male mice were evaluated, and measures of pain behavior were not included. Because Prx3 is localized to the mitochondria, the present study cannot rule out the involvement of cytosolic H2O2 in injury‐induced or age‐related OA. Further studies are needed to directly compare the inhibition of cytosolic H2O2 with mitochondrial H2O2 in OA models and to examine the role of additional antioxidant systems capable of reducing H2O2.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Loeser had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Loeser, Coryell, Gopalakrishnan, Ran, Carlson.
Acquisition of data
Loeser, Coryell, Armstrong, Collins, Gopalakrishnan, McDermott.
Analysis and interpretation of data
Loeser, Coryell, Armstrong, Gopalakrishnan, Ran, Carlson.
Supporting information
Figure S1. Prx3 levels in cartilage from wild‐type and Prdx3 transgenics.
Figure S2. Presence of the nicotinamide nucleotide transhydrogenase (NNT) gene in the mouse colonies.
Table S1. Histomorphometric analysis of DMM mice with transgenic Prx3 overexpression.
Table S2. Histomorphometric analysis of 18‐month‐old aged mice with transgenic Prx3 overexpression.
Table S3. Histomorphometric analysis of 24‐month‐old aged mice with transgenic Prx3 overexpression.
ACKNOWLEDGMENTS
We thank Kathryn Kelley for technical assistance and Katalin Kovacs and Brea Pool for preparation of histologic sections.
Supported by the National Institute on Aging (R01‐AG‐044034), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (T32‐AR‐050938), and the Office of the Director of the National Institutes of Health (T35‐0D‐011118).
The authors have nothing to disclose of relevance to this manuscript.
REFERENCES
- 1. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med 2019;132:73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiol Rev 2014;94:909‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blanco FJ, Rego I, Ruiz‐Romero C. The role of mitochondria in osteoarthritis. Nature Rev Rheumatol 2011;7:161‐9. [DOI] [PubMed] [Google Scholar]
- 4. Coleman MC, Goetz JE, Brouillette MJ, Seol D, Willey MC, Petersen EB, et al. Targeting mitochondrial responses to intra‐articular fracture to prevent posttraumatic osteoarthritis. Science Trans Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rhee SG, Woo HA, Kang D. The role of peroxiredoxins in the transduction of H2O2 signals. Antioxid Redox Signal 2018;28:537‐57. [DOI] [PubMed] [Google Scholar]
- 6. Collins JA, Wood ST, Nelson KJ, Rowe MA, Carlson CS, Chubinskaya S, et al. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro‐survival signalling in aging chondrocytes. J Biol Chem 2016;291:6641‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins JA, Wood ST, Bolduc JA, Nurmalasari NPD, Chubinskaya S, Poole LB, et al. Differential peroxiredoxin hyperoxidation regulates MAP kinase signaling in human articular chondrocytes. Free Radic Biol Med 2019;134:139‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L, Na R, Gu M, Salmon AB, Liu Y, Liang H, et al. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell 2008;7:866‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen L, Na R, Ran Q. Enhanced defense against mitochondrial hydrogen peroxide attenuates age‐associated cognition decline. Neurobiol Aging 2014;35:2552‐61. [DOI] [PubMed] [Google Scholar]
- 10. Loeser RF, Kelley KL, Armstrong A, Collins JA, Diekman BO, Carlson CS. Deletion of JNK enhances senescence in joint tissues and increases the severity of age‐related osteoarthritis in mice. Arthritis Rheumatol 2020;72:1679‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loeser RF, Olex A, McNulty MA, Carlson CS, Callahan M, Ferguson C, et al. Microarray analysis reveals age‐related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum 2012;64:705‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Armstrong AR, Carlson CS, Rendahl AK, Loeser RF. Optimization of histologic grading schemes in spontaneous and surgically‐induced murine models of osteoarthritis. Osteoarthritis Cartilage 2021;29:536‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leskov I, Neville A, Shen X, Pardue S, Kevil CG, Granger DN, et al. Nicotinamide nucleotide transhydrogenase activity impacts mitochondrial redox balance and the development of hypertension in mice. J Am Soc Hypertens 2017;11:110‐21. [DOI] [PubMed] [Google Scholar]
- 14. Marí M, De Gregorio E, De Dios C, Roca‐Agujetas V, Cucarull B, Tutusaus A, et al. Mitochondrial glutathione: recent insights and role in disease. Antioxidants (Basel) 2020;9:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Veal EA, Underwood ZE, Tomalin LE, Morgan BA, Pillay CS. Hyperoxidation of peroxiredoxins: gain or loss of function?. Antioxid Redox Signal 2018;28:574‐90. [DOI] [PubMed] [Google Scholar]
- 16. Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, et al. Overexpression of mitochondrial peroxiredoxin‐3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006;113:1779‐86. [DOI] [PubMed] [Google Scholar]
- 17. Zhang YG, Wang L, Kaifu T, Li J, Li X, Li L. Featured article: accelerated decline of physical strength in peroxiredoxin‐3 knockout mice. Exp Biol Med (Maywood) 2016;241:1395‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Prx3 levels in cartilage from wild‐type and Prdx3 transgenics.
Figure S2. Presence of the nicotinamide nucleotide transhydrogenase (NNT) gene in the mouse colonies.
Table S1. Histomorphometric analysis of DMM mice with transgenic Prx3 overexpression.
Table S2. Histomorphometric analysis of 18‐month‐old aged mice with transgenic Prx3 overexpression.
Table S3. Histomorphometric analysis of 24‐month‐old aged mice with transgenic Prx3 overexpression.