

Abstract
Across biological scales, gene-regulatory networks employ autorepression (negative feedback) to maintain homeostasis and minimize failure from aberrant expression. Here, we present a proof of concept that disrupting transcriptional negative feedback dysregulates viral gene expression to therapeutically inhibit replication and confers a high evolutionary barrier to resistance. We find that nucleic-acid decoys mimicking cis-regulatory sites act as “feedback disruptors,” break homeostasis, and increase viral transcription factors to cytotoxic levels (termed “open-loop lethality”). Feedback disruptors against herpesviruses reduced viral replication >2-logs without activating innate immunity, showed sub-nM IC50, synergized with standard-of-care antivirals, and inhibited virus replication in mice. In contrast to approved antivirals where resistance rapidly emerged, no feedback-disruptor escape mutants evolved in long-term cultures. For SARS-CoV-2, disruption of a putative feedback circuit also generated open-loop lethality, reducing viral titers by >1-log. These results demonstrate that generating open-loop lethality, via negative-feedback disruption, may yield a class of antimicrobials with a high genetic barrier to resistance.
Keywords: feedback, autoregulatory circuit, transcriptional feedback, viral evolution, synthetic biology, nucleic acids
Graphical abstract
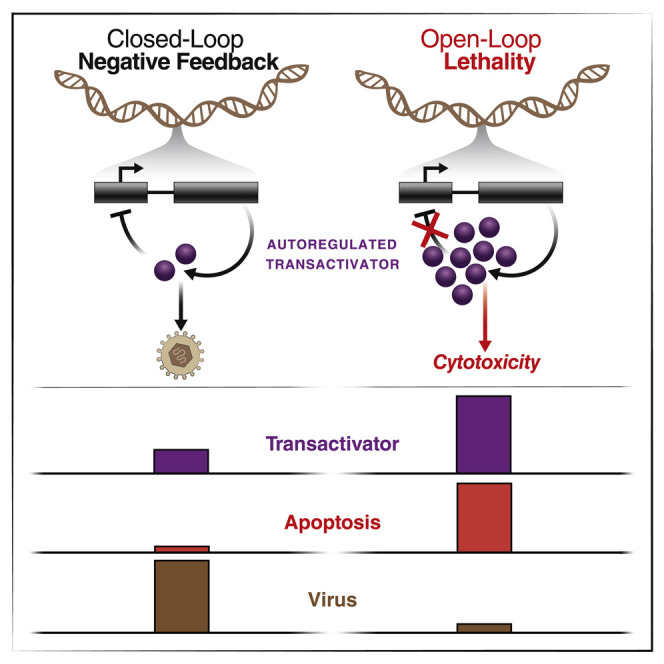
Interference with viral negative feedback loops results in overexpression of cytotoxic viral proteins and host cell death, presenting an approach to antiviral therapy with a high barrier to the evolution of resistance.
Introduction
The principle of feedback has been used since antiquity to regulate and limit intrinsic instability (Kenny, 1932). Early thermostats limited overheating; Watt’s centrifugal “governor” exploited feedback to limit steam engine volatility (Arago, 1840); and the “negative-feedback” amplifier (Black, 1934) curbed electrical fluctuations and led to modern control theory—which formalized negative feedback as a closed loop where a fraction of output is reversed in sign and returned as input to curtail a device “running away” (Bode, 1960; Nyquist, 1932). In biology, autorepression—also known as negative autoregulation or negative feedback (Alon, 2007; Jacob and Monod, 1961)—maintains homeostasis in systems ranging from bacterial transcription to mammalian fever response (Perez and Groisman, 2009; Roth et al., 2006).
In gene regulation, negative feedback is a recurrent motif (Ferrell, 2013) that optimizes a network’s responsiveness (Savageau, 1974) and enhances its robustness to perturbations (Austin et al., 2006; Becskei and Serrano, 2000; Hooshangi and Weiss, 2006; Nevozhay et al., 2009). A particularly tractable form of negative feedback is transcriptional autorepression—where a protein, the trans element, recognizes a sequence-specific DNA binding site, the cis element, on its own promoter. This cis-trans interaction, which is a protein-DNA interaction (PDI), often requires protein-protein interactions (PPIs), such as homo-multimerization of the protein. When the protein reaches sufficient concentrations, the PDI occurs with high probability, and promoter expression is sterically repressed. When protein levels drop, the PDI has a lower probability, and the promoter regains activity while the expression ensues, leading to homeostasis of protein levels.
Disrupting negative feedback, and associated homeostasis, detrimentally affects cells, with effects ranging from moderate to catastrophic. In bacteria, breaking negative-feedback circuits deoptimizes chemotaxis (Alon et al., 1999), whereas in fungi, breaking feedback degrades signal transduction (Yu et al., 2008). In mammals, breaking negative-feedback regulation of tumor necrosis factor α (TNF) or the tumor suppressor p53 induces programed cell death (Harris and Levine, 2005; Shelton et al., 1999; Van Antwerp et al., 1996), while breaking interferon negative feedback is toxic (Ivashkiv and Donlin, 2014). Given the essentiality of negative-feedback circuits in regulating microorganism and tumor physiology, disrupting feedback could be an attractive therapeutic strategy to generate a form of synthetic lethality that we hereafter call “open-loop lethality”—akin to disconnecting an engine’s governor to produce catastrophic failure.
Targeting feedback circuits could be particularly attractive from the perspective of resistance. The vast majority of traditional antimicrobials and antineoplastics target molecular moieties in which a small number of mutations can be sufficient to confer resistance. Many microorganisms and tumors exhibit high mutation rates [μ ∼10−3 mutations per genome per replication, (Drake, 1991; Jackson and Loeb, 1998)] such that the resulting genetic diversity often leads to the rapid emergence of resistant mutants that escape therapy (Goldberg et al., 2012; Lee et al., 2010; Meylan et al., 2018). In contrast, autorepression’s inherent input-output interactions suggest that disrupting transcriptional feedback, in theory, would have a high barrier to the evolution of resistance compared with traditional inhibitors. Specifically, resistance to feedback-disruption strategies would likely need to satisfy two criteria: (1) evolution of an alternate DNA-protein interaction via a coordinated set of mutations in both cis and trans elements to recapitulate a loop and (2) simultaneous maintenance of homeostasis (or, more importantly, fitness) for the intermediate open-loop variants. In other words, the protein (trans element) must evolve to recognize a new DNA binding sequence, and, concurrently, a new cognate DNA binding sequence (cis element) must also evolve. For DNA-protein interactions, a substantial body of literature has shown that altering binding specificity requires dozens of mutations (i.e., 18–50 single-base changes) distributed between both the DNA and the protein (Bogdanove et al., 2018; Takeuchi et al., 2014; Werther et al., 2017). From this perspective, targeting transcriptional feedback loops would mimic multidrug combination therapy, wherein multiple drugs simultaneously inhibit different viral targets—for a multidrug therapy requiring n mutations for resistance, escape mutants are predicted to arise at a rate ∼μ n, requiring substantially larger effective population sizes (i.e., N > μ −n). For feedback loops, the intermediate open-loop variants present in the population have a low probability of maintaining homeostasis, conferring decreased replicative fitness on these intermediate variants, thus presenting an added barrier. Consequently, feedback disruption could constitute a form of single-molecule combination therapy to limit resistance.
Here, we engineer DNA decoys that molecularly mimic cis-regulatory DNA sequences within feedback loops to competitively inhibit the DNA-protein interaction and thereby disrupt feedback-mediated homeostasis. As a proof of concept, we focus on a negative-feedback loop in the herpesvirus immediate early (IE) locus—which acts as a throttle or governor of viral replication—since new antivirals for drug-resistant herpesviruses are an important unmet medical need (Hofmann et al., 2020; Lurain and Chou, 2010; Piret and Boivin, 2014). Herpesviruses utilize transcriptional negative feedback to regulate coordinated gene-expression programs in which IE genes transactivate downstream viral genes driving the lytic viral replication cycle (Enquist and Leib, 2017; Mocarski et al., 2006; Shenk and Stinski, 2008; Weller and Coen, 2012). In the beta-herpesvirus cytomegalovirus (CMV)—which is a leading cause of birth defects and transplant failure—the 86-kDa IE protein (IE86; also known as IE2) is the major transcriptional transactivator and is indispensable. IE86 is also cytotoxic when dysregulated. Its homeostasis is regulated by a strong negative-feedback loop (Teng et al., 2012) mediated by IE86 binding as a high-order homo-multimer to a 14-base pair (bp) cis-repression sequence (crs) within its promoter, thereby autorepressing its transcription. Similarly, in the alpha-herpesvirus herpes simplex virus 1 (HSV-1), the IE175 protein (also known as ICP4), which plays a similar role in viral replication, is cytotoxic, and its homeostasis is regulated by a strong transcriptional negative-feedback loop (Chaturvedi et al., 2020; Paterson and Everett, 1988). Previous studies showed that genetic disruption of IE feedback via mutation of the crs increases IE protein levels past a cytotoxic threshold, generating a >100-fold reduction in viral replication (Isomura et al., 2008; Teng et al., 2012). In this study, we find that engineered DNA duplexes disrupt IE negative feedback, thereby increasing IE protein levels and generating open-loop lethality in infected cells. The effect contrasts with traditional transcription factor decoys—as feedback-disruptor duplexes increase rather than titrate levels of protein. We also show that the open-loop lethality strategy can be broadly generalized (e.g., to RNA sarbecoviruses such as SARS-CoV-2). Overall, the data show that targeting feedback circuitry to induce open-loop lethality yields a strong antiviral effect that carries a high barrier to the evolution of resistance and could enable escape-resistant therapeutic strategies.
Results
Theoretical considerations
Building off the concept of transcription factor decoys (Hecker and Wagner, 2017), we first asked whether molecular mimics of cis-regulatory DNA binding sites were theoretically capable of acting as competitive inhibitors in the context of feedback and specifically whether such decoys could break feedback to raise expression levels (Figure 1 A, bottom). We found that the established models of coupled negative-positive IE feedback (Teng et al., 2012; Vardi et al., 2018) predicted that DNA decoys could increase IE levels (Figure 1B). Mechanistically, the models showed that disrupting nonlinear negative-feedback induced a transition across a bistability (i.e., a separatrix) to a higher-expression state (Figure S1 A). Overall, the models indicated that DNA mimics could constitute feedback-disruptor (FD) molecules and specifically disrupt IE feedback.
Figure 1.

Theory and biochemical identification of feedback disruptors that induce open-loop lethality
(A) Schematics of the herpesvirus IE (IE86 and IE175) transcriptional negative-feedback circuits in the intact wild-type form (upper) and after disruption (lower) by putative feedback disruptors (FDs). When feedback is intact, IE proteins bind the cis-repression sequence (crs) in their respective IE promoters (cyan) and autorepress transcription. When feedback is disrupted (e.g., by titrating IE proteins with free crs-encoding DNA), the IE promoter is not autorepressed, and IE proteins reach cytotoxic levels.
(B) Computational modeling showing that FDs can effectively break feedback to increase IE protein levels.
(C) Probability of a drug-resistant CMV variant arising during the course of therapy. If only 1–2 mutations generate resistance (e.g., for GCV), the probability of resistance is 70%–100%. For three mutations, resistance becomes unlikely, and for >5 mutations, the probability becomes negligibly small. Uncertainties in probabilities (lines between diamonds) arise from the uncertainty in the mutation rate, drug efficacy, and dynamics of target cell proliferation.
(D) Top: schematic of different lengths of DNA duplexes tested for catalyzing homo-multimerization of the IE86 C terminus fragment protein (IE86C). Bottom: chromatographs of IE86C incubated with either a sequence-scrambled control or crs-containing DNA duplexes of differential lengths. A 28-bp crs-encoding duplex most efficiently titrates free IE86 protein from the 15 mL fraction into the 13 mL fraction (protein-DNA homomultimer fraction) with ∼98% of protein found in the 13-mL protein-DNA complex fraction in presence of the 28-bp duplex.
(E) Schematic of the minimal IE negative-feedback circuit (MIEP-IE86-IRES-GFP) stably expressed in ARPE-19 cells (“feedback-reporter cells”) and illustration of the effect of negative-feedback disruption on GFP fluorescence and cell viability.
(F) Representative flow cytometry plots of feedback-reporter cells after nucleofection with 28-bp DNA duplex encoding crs (FD86) or a scrambled 28-bp DNA duplex (FDScram). GFP expression analyzed at 48-h post-nucleofection; cell-death analyzed by Zombie Aqua at 96-h post-nucleofection. Mean ± 1 SD shown, n = 3 biological replicates. p values derived from Student’s t test: ∗∗∗∗ < 0.0005.
See also Figure S1.
Figure S1.

Simulations and in vitro analyses of FD DNA duplexes, related to Figure 1
(A) Numerical simulations of an experimentally validated ordinary differential equation (ODE) model of the major immediate early circuit of CMV (Teng et al., 2012) modified to include FDs as described in STAR Methods for three different FD dosages [0, 0.025, and 2.5] and heterogenous initial IE86 levels. All other parameters are kept constant.
(B) Parameter sensitivity analysis for the within-host CMV model of FD resistance. Two models of cell proliferation were tested across a range of parameter combinations (STAR Methods). Each “+” represents the predicted probability of resistance from a simulation using a particular parameter combination. Multiple overlapping “+”s (e.g., when the number of mutations n and the mutation rate μ are kept constant) indicate that the predicted risk of resistance is insensitive relative to other parameters (e.g., drug efficacy).
(C) Electrophoresis mobility shift assays (EMSA) verifying that the C terminus of IE86 specifically interacts with crs-containing DNA duplexes. Digoxigenin (DIG)-labeled DNA duplex probes of the crs and Δcrs sequences, as previously described (Asmar et al., 2004), were used for binding and detection. EMSA was performed after incubating DIG-labeled DNA probes with increasing concentrations of IE86 protein (0 μM to 20 μM) for 30 min at room temperature (lanes 1–6). To determine whether the IE86-crs interaction was sequence specific, either 20-fold of unlabeled crs DNA duplex (∗) was added (lane 7) or 20-fold unlabeled Δcrs DNA duplex (∗∗) was added (lanes 8 and 10).
(D) crs-encoding DNA duplexes ranging from 20 to 30 bp efficiently catalyze IE86 homomultimer formation. Size-exclusion chromatography of mixtures of DNA duplexes of different lengths (as indicated) with C terminus of IE86 protein. The IE86 homomultimer protein-DNA complex elutes at ∼13-mL column volume (red bar).
(E) Size-exclusion chromatography of maltose-binding protein (MBP) together with FD86 indicates that MBP does not oligomerize in the presence of FD86, thereby excluding an MBP-mediated protein-DNA interaction.
(F) Size-exclusion chromatography of purified full-length IE86 protein shows that FD86 efficiently catalyzes IE86 oligomerization, eluting at ∼11-mL column volume.
(G) Mock nucleofection of the IE86 feedback-reporter cell line.
(H) Top: quantitative (LICOR™) western blot analysis of IE86 from cell lysates of the IE86 feedback-reporter cell line at 2 days post-nucleofection of the crs-containing 28-bp DNA duplex; bottom: Coomassie blue loading control for western blot.
(I) Flow cytometry analysis of the IE86 feedback-reporter cell line after nucleofection with increasing doses of the crs-containing 28-bp DNA duplex and 100 μM of the sequence-scrambled 28-bp DNA duplex control analyzed at 3 days post-nucleofection (SSC, side scatter). We note that the efficiency of nucleofection is <100%, which manifests as an apparent bimodality.
(J) Size-exclusion chromatography of purified IE86C (N terminus tagged with MBP) incubated with the 28-bp crs-encoding DNA duplex containing one crs sequence (FD 1 ×) or two concatenated crs sequences (FD 2× -concat) for 30 min at room temperature (see Table S1 for FD sequences). Oligomerized fraction (% absorbance at ∼13mL fraction at OD280) was compared for both the samples.
(K) Flow cytometry analysis of IE86 feedback-reporter cells 2 days post-nucleofection with 25 μM of either FD 1 ×, or FD 2× -concat, or the control sequence-scrambled DNA duplex (FDScram).
(L) Quantification of feedback strength by gene-expression fluctuation (noise) analysis from single-cell time-lapse imaging. Each data point represents one cell. Feedback-reporter cells (n = 22), broken-feedback-reporter cells (Δcrs) (n = 42), and FD86-nucleofected feedback-reporter cells (25 μM, n = 49; 100 μM, n = 8) were imaged over 48 h. Single-cell imaging traces were analyzed by calculating the coefficient of variation (CV) and the mean of GFP expression for each individual cell. The expected noise obtained without disrupting feedback is given by the Poisson curve. Each dot represents one single-cell imaging trace.
(M) Phosphorothioation of feedback-disruptor DNA duplexes: flow cytometry dot plots of IE86 feedback-reporter cells 4 days post-nucleofection with 25 μM of either a 28-bp crs-encoding DNA duplex (FD86) containing four phosphorothioate bonds, a 28-bp sequence-scrambled DNA duplex (FDScram) also containing four phosphorothioate bonds, or a nonphosphorothioated version of FD86.
(N) Flow cytometry analysis of IE86 feedback-reporter cells 2 days post-nucleofection with 25 μM of a single-stranded 28mer crs-encoding oligonucleotide (ssFD86) containing four phosphorothioate bonds as compared with the sequenced-scrambled 28-bp DNA duplex also containing four phosphorothioate bonds (FDScram) in triplicate.
We next asked whether putative FD molecules could be beneficial in the context of mutational escape as compared with existing antiviral strategies. In viruses, the probability of escape is driven by pre-existing genetic diversity and selection (Coffin, 1995; Cudini et al., 2019; Drake and Hwang, 2005), leading to substantial levels of clinical resistance (Lurain and Chou, 2010; Piret and Boivin, 2014). For example, resistance to the CMV antiviral ganciclovir (GCV) and the HSV-1 antiviral acyclovir (ACV) is substantial. Single-base viral mutations are often sufficient for escape (Emery and Griffiths, 2000; Frobert et al., 2014) and arise rapidly (Drake and Hwang, 2005; Emery and Griffiths, 2000; Lu et al., 2002a; Schnipper and Crumpacker, 1980).
To estimate the likelihood of resistance to FD molecules, we built off previous computational models of GCV resistance (Emery and Griffiths, 2000) to calculate the likelihood that viruses with n mutations would emerge in vivo (Figures 1C and S1B). In agreement with clinical observations, the calculations showed that GCV-resistant mutants, which require a single mutation, arose with high probability (i.e., μ −1). In contrast, viruses with the requisite number of mutations needed to re-specify a DNA-protein interaction—predicted to be n > 18 based on protein-engineering literature (Bogdanove et al., 2018; Takeuchi et al., 2014; Werther et al., 2017)—emerged with a vanishingly small probability that scaled with ∼μ −n (Figures 1C and S1B). Parameter sensitivity analyses across different therapeutic efficacies, viral mutation rates, cell proliferation rates, and cell-death rates confirmed these results (Figure S1B).
Identification of a transcriptional feedback-disruptor candidate
Based on computational predictions of FD efficacy (Figures 1B and 1C), we set out to identify DNA duplexes that might act as IE FDs via binding of IE86 homomultimers. We developed an in vitro assay to quantify the efficiency of various linear DNA duplexes in catalyzing the formation of the IE86 protein-DNA complex (Figure 1D). First, the catalytically active C-terminal half of IE86 (Liu et al., 1991), termed IE86C, was bacterially expressed, purified via a maltose-binding protein (MBP) tag, and used to validate that IE86C could bind crs-encoding DNA duplexes in a sequence-specific manner (Figure S1C).
To determine the formation of the IE86C homomultimer on DNA, crs-encoding DNA duplexes of various lengths were assayed by size-exclusion chromatography. The assay showed that linear DNA duplexes between 21 and 34 bp catalyzed the formation of the predicted IE86C homomultimer (Teng et al., 2012) in complex with DNA, with 28-bp duplexes providing maximal binding (Figure 1D). Finer-grained analysis showed virtually no binding improvement for longer or shorter DNA duplexes from 24 to 32 bp (Figure S1D). Based on these data, the 28-bp DNA duplex was selected for further characterization. No homomultimer formation was observed for MBP with the 28-bp DNA or MBP alone (Figure S1E), and the 28-bp DNA also catalyzed homo-multimerization of full-length IE86 protein (Figure S1F).
To test whether the candidate 28-bp DNA duplexes disrupted feedback in cells, we used an ARPE-19 cell line stably transduced with a minimal IE86 negative-feedback reporter circuit (Teng et al., 2012) that expresses cDNA-encoded full-length IE86 and GFP from the IE86 promoter-enhancer. In these feedback-reporter cells, IE86 autorepresses the cassette, and an increase in GFP indicates disruption of autorepression and leads to cell death within 48–72 h (Figure 1E). To test whether duplexes disrupted autorepression and increased IE86 levels, 28-bp crs-encoding duplexes were transfected into feedback-reporter cells (Figure S1H). Next, flow cytometry analysis verified that increases in GFP correlated with increases in IE86; 28-bp duplexes led to dose-dependent feedback disruption (Figure S1I); and duplex concatemerization enhanced feedback disruption (Figures S1J and S1K).
To directly verify that FD duplexes disrupted negative feedback, we used an established assay that quantifies feedback strength from gene-expression fluctuations (noise) (Austin et al., 2006; Weinberger et al., 2008) based on longitudinal imaging traces for individual cells, thereby filtering out extrinsic cell-to-cell variability in FD transfection efficiency. The analysis showed that FD duplexes dramatically increased intrinsic IE86-GFP gene-expression noise, consistent with ablation of negative feedback (Figure S1L).
To enhance feedback disruption, we then stabilized the duplexes against cellular nuclease degradation by insertion of internal phosphorothioate bonds (Figure S1M)—a common approach to stabilize oligos against nucleases and improve cellular uptake (Seth et al., 2019; Stein and Castanotto, 2017). Transfection of the phosphorothioated 28-bp crs-encoding DNA duplexes disrupted feedback and led to substantial cell death, whereas the scrambled phosphorothioated 28-bp DNA duplex did not (Figures 1F and S1M). To verify that phosphorothioation did not inadvertently destabilize the duplex and stabilize single-stranded versions that might act as antisense oligos, we tested a phosphorothioated single-stranded non-duplex DNA oligonucleotide encoding the same 28-base crs sequence and found that it did not disrupt feedback (Figure S1N). These data indicated that crs-encoding phosphorothioated 28-bp DNA duplexes were candidate disruptors of IE86 feedback, and they are hereafter labeled FD86.
Disruption of IE feedback causes open-loop lethality via apoptosis, without activating innate immune or inflammatory pathways
To address the concern that FD duplexes might activate innate immune or inflammatory pathways via DNA sensors (Beutler, 2004; Lund et al., 2003), naive ARPE-19 cells—not expressing IE86—were transfected with FD86 or the scrambled control (FDScram). We observed no measurable cell death due to FD duplexes in naive cells (Figures 2A and 2B), consistent with double-stranded DNA sensors such as cGAS-STING requiring substantially longer duplexes than 28 bp for activation (Gantier, 2017; Luecke et al., 2017).
Figure 2.

Disruption of viral transcriptional feedback generates open-loop lethality without activating innate immune responses
(A and B) Cell viability analysis by flow cytometry after nucleofecting naive ARPE-19 cells with FD duplexes.
(C) RNA-seq heatmap of differentially enriched genes after nucleofecting feedback-reporter cells with FD86 or FDScram in biological duplicate. Six hundred and forty nine differentially enriched genes depicted; specific genes within apoptotic pathways listed.
(D) Pathway gene ontology (GO) enrichment analysis. The top ten most enriched pathways as analyzed in David are plotted.
(E and F) Apoptosis analysis by flow cytometry after nucleofecting feedback-reporter cells with FD duplexes, showing apoptosis by annexin V and TUNEL staining.
(G) qRT-PCR analysis of TLR9 expression 48 h after induction with a CpG-rich TLR agonist (ODN 2216) or FD duplexes. Error bars represent mean ± 1 SD, n = 3 biological replicates. p values derived from Student’s t test: ∗ < 0.05; ns, non-significant.
See also Figure S2.
Next, to determine the mechanism by which FD duplexes induced cell death in the presence of IE86, we used RNA-seq analysis of feedback-reporter cells transfected with FD86 or FDScram. Six hundered and forty nine differentially expressed genes were identified (Figure 2C), with apoptotic pathways prominently enriched (Figure 2D). Alternate ontology analysis confirmed the apoptotic pathway enrichment (Figures S2 A and S2B), and specific genes associated with apoptosis were identified (Figures 2C and 2D). To verify FD-induced apoptosis in IE86-expressing cells, assays for both early and late apoptotic markers were used (Figures 2E and 2F). Staining for annexin V (Figure 2E) and terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) (Figure 2F) showed that FD86 promoted apoptosis in the presence of IE86. Collectively, these results indicate that relatively short (28 bp) sequence-specific DNA duplexes can disrupt a viral feedback loop to generate a form of synthetic lethality that we termed “open-loop lethality.”
Figure S2.

RNA-seq analysis, related to Figure 2
(A) Heatmap showing the logCPM (log counts per million) values for clusters of differentially expressed genes from RNA-seq of cells nucleofected with either FD86 or FDScram (biological duplicates shown for each condition). The clusters are numbered on the left side of the plot, and the sample IDs are indicated at the bottom.
(B) Differentially expressed genes clustered by the logCPM values of FD and FDScram samples. Cluster 4 is a set of upregulated genes with enrichment of GO terms related to cell migration, endocytosis, and apoptosis. Analysis for other clusters not presented due to <100 genes. The y axis shows the enriched terms, and the x axis shows GeneRatio (the fraction of genes associated with a term that were among the genes in this cluster). The sizes of the data point represent the number of genes in the cluster associated with the respective term and the colors represent the FDR-adjusted p values. The apoptotic signaling pathway term is high in GeneRatio and Gene Count with an adjusted p value < 0.05.
(C) 25 genes enriched by FDScram treatment. The four genes that are also enriched in FD86-treated cells are marked with an asterisk.
(D) Specific genes with GO terms associated with inflammation that are enriched in cells overexpressing IE86 (FD86 treated).
To determine whether delivery of DNA per se contributed to the upregulation of innate immune or inflammatory pathways, we examined genes enriched by the 28bp FDScram relative to mock-transfected cells and found no enrichment of innate immune or interferon-stimulated pathways or upregulation of individual genes in inflammatory pathways (Figure S2C). Of the 649 genes enriched in FD86 treated cells, few were involved in inflammatory pathways (Figure S2D), with the exception of CD180, which downregulates TLRs (Yang et al., 2018), TGM2, EDNA2, and SCG2.
To verify that FD86 duplexes did not activate interferons, we examined the expression of TLR9, which is known to activate interferon in response to foreign DNA (Lund et al., 2003), and used a CpG-rich DNA oligonucleotide (ODN 2216) as a positive control for TLR9 activation (Krug et al., 2001). Unlike ODN 2216, FD duplexes did not significantly increase TLR9 expression (Figure 2G).
Overall, several pieces of evidence argue that FD duplexes do not act nonspecifically via activation innate immune pathways: (1) FD86 only induces cell death in IE86-expressing cells, and FDScram does not cause observable cell death in any context; (2) RNA-seq analysis finds no enrichment of innate immune or inflammatory pathways by FD duplexes; and (3) DNA sensors are not activated by FD duplexes. These data are broadly consistent with previous analyses showing that short dsDNA duplexes do not induce innate immune responses in cells and appear nontoxic in patients (Sen et al., 2012, 2009; Xi et al., 2005).
Feedback disruption inhibits viral replication via open-loop lethality
Next, we investigated disrupting IE86 feedback in the context of viral infection. We first verified that FD duplexes did not affect virus entry (Figures S3 A and S3B). Next, flow cytometry and microscopy analysis (Figures 3A and 3B) showed that FD86 generated a >10-fold increase in IE86 expression in cells infected with a CMV IE86-YFP fusion virus (Vardi et al., 2018). Dose-response analysis revealed that the FD86 50% inhibitory concentration (IC50) was 0.95 nM (Figure 3C) and that 25 μM was sufficient to achieve 100-fold inhibition of viral replication, consistent with IE feedback mutants (Teng et al., 2012). Subsequent analyses used the 25-μM concentration. The FD86 IC50 is lower than that of fomivirsen (IC50 = 30 nM), a 21-nt antisense DNA therapy (Mulamba et al., 1998), presumably due to the alternate mechanism of action of FD86.
Figure S3.

FD duplexes in the context of CMV infection, related to Figure 3
(A) Flow cytometry dot plots of naive or FD86-nucleofected ARPE-19 cells after infection with CMV TB40E-IE86-YFP (MOI = 0.1) showing no reduction in the percentage of infected IE86 expressing cells. Cells were infected 24 h after nucleofection with FD86 and were analyzed at 4 hpi (hours post infection).
(B) Viral entry assay. Fluorescent micrographs of HFF cells (nucleofected ± FD86), then infected with CMV TB40E-IE86-YFP, imaged at 2 hpi (MOI = 5), and stained for viral pp65 protein (red, 594 nm) and DAPI (blue, 405 nm).
(C) Schematic of the Δcrs CMV virus with a disrupted IE86-feedback circuit due to a 3-bp mutation in the crs.
(D) FD86 does not reduce Δcrs virus replication. Cells (human foreskin fibroblast, HFF) were nucleofected with 25 μM FD86 (or FDScram) and 24 h post-nucleofection infected with parent AD169 CMV virus or Δcrs AD169 CMV mutant virus (MOI = 0.1) and then titered (4 dpi for parent virus and 6 dpi for Δcrs virus due to its slower growth).
(E) FD86 does not increase IE86 levels in Δcrs infected cells. Fluorescent micrographs of HFF cells infected with Δcrs virus in the presence of FDScram or FD86 (6 dpi).
(F) FDs do not act through the cGAS-STING pathway. ARPE-19 (low cGAS-STING activity) and MRC-5 cells (high cGAS-STING activity) were nucleofected with 25 μM FD86 or mock and infected with CMV TB40E-IE86-YFP or AD169 (MOI = 0.1), respectively. Virus titers were assayed by TCID-50 at 4 dpi.
(G) FD86 interferes with replication of ganciclovir-resistant (GCVR) and foscarnet-resistant (FOSR) CMV strains. MRC-5 cells were nucleofected with 25 μM FD86 or FDScram and 24 h later were infected with either parent CMV AD169 (control) or GCVR or FOSR strains (CMV GDGrK17, CMV GDGrP53, CMV 759rD100-1, CMV PFArD100) at MOI = 0.1. Virus titers were assayed by TCID-50 at 4 dpi.
(H) Sequence homology for the crs of human CMV, murine CMV (mCMV), and rhesus CMV (RhCMV); green represents sequence homology, whereas red represents divergence.
(I) FDmCMV interferes with mCMV replication. NIH 3T3 mouse fibroblast cells were nucleofected with either 25 μM FDmCMV, FDScram, or mock nucleofected 24 h prior to mCMV infection at MOI = 0.1. At 4 dpi, virus titers were assayed by TCID-50 on 3T3 cells.
(J) FDRhCMV interferes with RhCMV replication. Telo-RF cells were nucleofected with either 25 μM FDRhCMV or (FDScram/mock) and infected with RhCMV (RhCMV 68.1 GFP) at MOI = 0.1. Virus titers were assayed by TCID-50 at 4 dpi.
(K) Live-dead analysis; left: flow cytometry analysis of ARPE-19 cells in the absence or presence of 100 μM FD175 stained with Zombie Aqua at 4 dpi; right: quantification of % Zombie Aqua positive cells from the flow plots.
(L) Live-dead analysis; left: Flow cytometry plots of ARPE-19 cells in the absence or presence of 100 μM FD86 + 10 μM of ganciclovir stained with Zombie Aqua at 4 dpi; right quantification of % Zombie Aqua positive cells from the flow plots.
Figure 3.

Open-loop lethality generates an antiviral effect against diverse herpesviruses
(A) Flow cytometry analysis showing that feedback disruption generates IE86 overexpression in CMV-infected cells. ARPE-19 cells were nucleofected with 25 μM FD86 or FDScram, then 24 h later infected with CMV (TB40E) encoding an IE86-YFP fusion (MOI = 0.1), and analyzed at 2 days postinfection (dpi).
(B) Micrographs of YFP fluorescence in ARPE-19 cells at 24 h postinfection with TB40E-IE-YFP treated with FD86 or FDScram (MOI = 1.0). Scale bar, 200 μm.
(C) FD86 dose-response curve and corresponding IC50 value. ARPE-19 cells were nucleofected with FD86 at the concentration specified, infected with TB40E-IE86-YFP virus (MOI = 0.1), and virus titered 4 dpi (error bars represent mean ± 1 SD, n = 3 biological replicates).
(D) Antiviral effect of FD86 on CMV. ARPE-19 cells were nucleofected with 25 μM FD86 (or mock/FDScram), and 24-h post-nucleofection, cells were infected with TB40E-IE86-YFP virus at different MOIs (0.1, 0.5, 1.0, 2.0), and titered at 4 dpi (error bars represent mean ± 1 sd, n = 3 biological replicates).
(E) Apoptosis induction in CMV-infected cells: ARPE-19 cells were nucleofected with 25 μM FD86 (or mock/FDScram), 24 h later infected with TB40E-IE86-YFP virus (MOI = 1), and at 48 hpi stained for annexin V and analyzed by flow cytometry.
(F) Feedback disruption leads to IE175 overexpression in HSV-1-infected cells. ARPE-19 cells were nucleofected with 25 μM FD175 or FDScram (or mock), then 24 h later infected with HSV-1 (17syn+ strain) encoding an IE175-YFP (MOI = 0.1), and analyzed at 2 dpi.
(G) Micrographs of YFP fluoresence in Vero cells 12 h postinfection with 17syn+ IE175-YFP (MOI = 1.0) and treated with FD175 or FDScram. Scale bar, 200 μm.
(H) FD175 dose-response curve and corresponding IC50 values. Titers were calculated on Vero cells nucleofected with FD175 at the concentration indicated, infected with HSV-1 IE175-YFP (17syn+ strain, MOI = 0.1) 24 h later, and titered 2 dpi (error bars represent mean ± 1 sd, n = 3 biological replicates).
(I) Antiviral effect of FD175 on HSV-1. Vero cells were nucleofected with 25 μM FD175 (or mock/FDScram), and 24 h post-nucleofection, cells were infected with HSV-1 (17syn+ strain) at different MOIs (0.1, 0.5, 1.0, 2.0), and titered at 4 dpi (error bars represent mean ± 1 SD, n = 3 biological replicates).
(J) Ferroptosis induction in HSV-infected Vero cells. Cells were nucleofected with 25 μM FD175 (or mock/FDScram) and then 24 h later, infected with HSV-1 (17syn+ strain) IE175-YFP (MOI = 1), and at 24-hpi cells, were harvested, stained with a ferroptosis marker (BODIPY C11), and analyzed by flow cytometry. p values derived from Student’s t test: ns, non-significant, ∗∗ < 0.01.
See also Figures S3, S4, and S5.
To confirm that FD-induced cell death was due to disruption of IE86 feedback, we used a previously characterized mutant virus (Teng et al., 2012), which lacks IE86 negative feedback due to a three-base mutation in the crs and exhibits lower viral fitness (Figure S3C). As expected, FD86 did not reduce replication or significantly increase IE86 expression in this mutant (Figures S3D and S3E). These data confirm that FD-mediated viral inhibition results from specific disruption of IE86 autorepression, resulting in IE86 overexpression and open-loop lethality.
Given the unique mechanism of action of open-loop lethality, we hypothesized that—in contrast to existing antivirals—FD86 would be robust to the multiplicity of infection (MOI), as increased virus would deliver more cytotoxic IE86 substrate. As predicted, inhibition of viral replication was robust across MOIs from 0.1 to 2.0 (Figure 3D).
Importantly, FDScram did not exhibit antiviral effects, consistent with the open-loop lethality model, and this supports that FDs do not act via nonspecific activation of innate DNA sensors. To further verify that FD activity was independent of cGAS-STING, we tested the effect of FDs in MRC-5 cells, which exhibit high cGAS-STING expression (Meade et al., 2019) relative to ARPE-19 cells (Tigano et al., 2021) and observed little difference in FD antiviral effects and no effect of mock transfection in either setting (Figure S3F). Moreover, the possibility that high concentrations of FD86 were cytotoxic was ruled out by assaying cell death in uninfected ARPE-19s under 100 μM FD86, both in the presence and in the absence of GCV (Figures S3K and S3L).
To be sure that the antiviral effects were not specific to virus strain (TB40E) or cell type (ARPE-19 and MRC-5) used, we tested CMV AD169 in human foreskin fibroblasts and in six different GCV-resistant CMV strains. FD86 inhibited virus by 100-fold in all cell types and strains tested (Figure S3G). We also designed FD duplexes for murine CMV (mCMV, FD88) and rhesus CMV (RhCMV, FDRhCMV) (Figure S3H; Table S1) and observed comparable reductions in mCMV and RhCMV viral titers (Figures S3I and S3J).
To determine whether viral inhibition resulted from IE86 open-loop lethality via apoptosis, apoptosis markers were assayed in CMV-infected cells. The assay confirmed that FD86 treatment increased apoptosis in infected cells, whereas treatment with FDScram did not (Figures 3E and S5E). We also employed a cell-death rescue assay based on small-molecule inhibitors of various cell-death pathways. The rescue assay indicated that IE86 feedback disruption activated a number of cell-death pathways—not unexpected given crosstalk (Hong et al., 2017)—but with apoptosis inhibition yielding the most pronounced rescue (Figures S5A and S5B). These data indicate that FD86 induces open-loop lethality via premature apoptosis in CMV-infected cells to reduce viral titers.
Figure S5.

FD duplexes induce cytotoxicity by crosstalk between cell-death pathways, related to Figure 3
(A and B) (A) Bar graph quantification and (B) flow cytometry plots of the cell-death “rescue” assay. FD86 induces apoptosis in CMV-infected cells. ARPE-19 cells were nucleofected with 25 μM FD86 (or mock/FDScram) and at 24 h later infected with CMV (TB40E-IE86-YFP, MOI = 0.1) in the presence of the indicated cell-death inhibitors: auto (autophagy inhibitor); apo (apoptosis inhibitor); nec (necroptosis inhibitor); fer (ferroptosis inhibitor). Cells were harvested at 48 hpi, stained for dead cells with Zombie Aqua (BioLegend, Cat# 423101), and analyzed by flow cytometry. Experiment performed in three biological replicates. p values were calculated using two-way ANOVA followed by Tukey’s multiple comparisons test.
(C and D) (C) Bar graph quantification and (D) flow cytometry plots of the cell-death “rescue” assay. FD175 induces ferroptosis in HSV-infected cells. Vero cells were nucleofected with 25μM FD175 (or mock/FDScram), then 24 h later infected with HSV-1 (17syn+ strain) IE175-YFP virus (MOI = 0.1) in the presence of the indicated cell-death inhibitors: auto (autophagy inhibitor); apo (apoptosis inhibitor); nec (necroptosis inhibitor); and fer (ferroptosis inhibitor). Cells were harvested at 24 hpi, stained for dead cells with Zombie Aqua (BioLegend, Cat# 423101), and analyzed by flow cytometry. Experiment performed in three biological replicates. p values were calculated using two-way ANOVA followed by Tukey’s multiple comparisons test.
(E) Triplicate repeats of FD-treated CMV-infected ARPE-19 cells stained with apoptosis marker. ARPE-19 cells were nucleofected with 25 μM FD86 (or mock/FDScram); 24 h later, cells were infected with CMV (TB40E) IE86-YFP virus (MOI = 1) and at 48 h post infection stained with annexin V.
(F) Triplicate repeats of HSV-1 infected Vero cells stained for ferroptosis (BODIPY C11) in the presence of FDs. Vero cells, which were nucleofected with 25 μM FD175 (or mock/FDScram), were infected with HSV-1 IE175-YFP (17syn+ strain) at MOI = 1 at 24 h post-nucleofection. At 24 hpi, cells were harvested and stained.
Open-loop lethality is generalizable to other herpesvirus circuits
As FD duplexes inhibit diverse CMVs (β herpesviruses), and as HSV-1 (an α herpesvirus) also encodes a feedback circuit to maintain homeostasis of its cytotoxic transactivator protein IE175 (Chaturvedi et al., 2020), we hypothesized that HSV-1 FDs could be developed. To test this, the 14-bp crs within FD86 was replaced with a 15-bp sequence taken from the IE175 promoter to generate the FD175 candidate, a 29-bp DNA duplex. FD175 did not inhibit viral entry of HSV-1 (17syn+ strain) (Figures S4 A and S4B), but it disrupted IE175 autorepression and increased IE175 expression >10-fold compared with a scrambled DNA duplex (Figures 3F and 3G).
Figure S4.

FD duplexes in the context of HSV-1 infection, related to Figure 3
(A) Flow cytometry dot plots of naive or FD175-nucleofected ARPE-19 cells after infection with HSV-1 strain 17syn+ IE175-YFP (MOI = 0.1). Cells were infected 24 h post-nucleofection with FD175 and analyzed at 4 hpi. The uninfected control is the same as in Figure S3A.
(B) Fluorescent micrographs of Vero cells (nucleofected ± FD175), then infected with HSV-1 17syn+ IE175-YFP, imaged at 1 hpi (MOI = 20), and stained for ICP5 protein (red, 594 nm) and DAPI (blue, 405 nm).
(C) Flow cytometry live-dead analysis using Zombie Aqua of uninfected (naive) Vero cells (i.e., control) showing gating for dead cells. Dead cell gate was drawn by comparing with HSV-1-infected Vero cells in Figure S5D (left-most column). Percentage of dead cells is <1%.
(D) Uninfected Vero cells nucleofected with FDScram or FD175 (mean of biological triplicates shown); nucleofection of FD175 does not generate significant increases in dead cells compared with nucleofection of FDScram control.
(E) FD175 interferes with HSV-1 (KOS strain) replication. Vero cells were nucleofected with either 25 μM FD175 (or mock/FDScram) and 24 h later were infected with HSV-1 (KOS strain) at MOI = 0.1, then virus quantified by TCID-50 at 4 dpi.
(F and G) Absence of nonspecific antiviral effects. (F) Vero cells were nucleofected with 25 μM FD86 or FDScram and infected with HSV-1 (17syn+ strain IE175-YFP virus, MOI = 0.1), followed by quantification of virus titer 4 dpi by TCID50. (G) ARPE-19 cells were nucleofected with 25 μM FD86 or FDScram and infected with CMV (TB40E-IE86-YFP MOI = 0.1), followed by quantification of virus titer 4 dpi by TCID50.
(H) Left: flow cytometry plots of naive Vero cells or in the presence of 100 μM FD175 stained with Zombie Aqua stain at 4 dpi; right: quantification of % Zombie Aqua positive cells from the flow plots.
(I) Left: flow cytometry plots of naive Vero cells or in the presence of 100 μM FD175 + 10μM of acyclovir stained with Zombie Aqua stain at 4 dpi; right: quantification of % Zombie Aqua positive cells from the flow plots.
Similar to FD86, FD175 exhibited a low IC50 (0.06 nM) and inhibited HSV-1 replication ∼100-fold across a broad range of MOIs (Figures 3H–3I); it did not induce cell death in uninfected cells (Figures S4C, S4D, S4H, and S4I). A cell-death rescue assay found that IE175 feedback disruption activated diverse cell-death pathways, including apoptosis, with ferroptosis dominating (Figures S5 C and S5D)—consistent with significant crosstalk between ferroptosis and apoptosis (Hong et al., 2017). A direct assay for ferroptosis in HSV-infected cells confirmed that IE175 open-loop lethality is mediated via ferroptosis (Figures 3J, S5C, S5D, and S5F). Similar antiviral effects of FD175 were observed for a second HSV-1 strain (KOS) (Figure S4E). Overall, the results indicate that the FD-mediated antiviral strategy may be generalizable to other herpesvirus circuits.
Open-loop lethality exhibits a high genetic barrier to the evolution of resistance
To test the hypothesis that feedback disruption presents a high barrier for the evolution of resistance, both phenotypic and genotypic resistance assays were used. The phenotypic resistance assay employed a continuous-culture system (Figure 4 A). To benchmark the FD86 antiviral effect and assess the emergence of antiviral resistance, we compared against fomivirsen (antisense DNA), to which resistance develops rapidly (Mulamba et al., 1998). Fomivirsen initially reduced viral titer, but resistance emerged within 3–4 rounds of infection, as evidenced by a positive slope in the viral titer (Figures 4B and S6A). In contrast, FD86 reduced CMV titers in the initial round by 100-fold and then steadily to below the limit of detection by day 52, with no evidence of CMV resistance observed (Figure 4B). To verify virus clearance, continuous-culture supernatants were sub-cultured in the absence of FDs, and consistent with the data from the parent continuous culture, virus was below the limit of detection in these subcultures from day 52 onward (Figure S6 B).
Figure 4.

Open-loop lethality exhibits a high genetic barrier to the evolution of resistance
(A) Schematic of the continuous-culture experiment. ARPE-19 cells (±FD) were infected, and at 4 dpi, supernatant was collected and was used to infect naive ARPE-19 cells (±FD) until day 60.
(B) Continuous-culture titers for CMV (TB40E-IE86-YFP) in the presence of FD86 (red), fomivirsen (blue), or mock treatment (gray). Fomivirsen resistance (positive slope of the titering dynamics) was observed beginning at day 12 (Figure S6A).
(C) Continuous culture for HSV-1 (17syn+ IE175-YFP) in the presence of 25 μM FD175 (red) or mock treatment (gray). ACV resistance (positive slope of the titering dynamics) is observed beginning at day 4 (Figure S6D). All plots show mean ± 1 SD, n = 3 biological replicates.
See also Figure S6.
Figure S6.

Evolution of resistance to existing antivirals; FD duplexes do not induce cell death in uninfected bystander cells, related to Figures 4 and 5
(A) Outgrowth of fomivirsen-resistant virus in continuous culture. Viral titers from rounds 3–5 of continuous cultures ±25 μM fomivirsen (no dose escalation was used). Positive slope in titers in presence of fomivirsen is observed beginning at round 3 of infection.
(B) Verification that FD86 decreases CMV titers below detection; subculturing of supernatants from continuous culture. We used the established TCID50 calculation described previously (Reed, 1938). This calculation computes both the TCID50/ml and a single-well detection limit from the initial dilution and titration factor. Because there are multiple replicates (in this case eight replicate wells) and the statistics are Boolean, it is possible to reliably calculate TCID50/ml values below the single-well detection limit for the entire row of eight wells.
(C) Fitness recovery assay: analysis of FD86-treated continuous cultures upon removal of FD86. Supernatants from continuous cultures on indicated days were cultured on ARPE-19 cells ± FD86. Titers recover 1.5–2 logs upon removal of FD86.
(D) HSV-1 resistance to acyclovir (ACV). Viral titers of ARPE-19 cells infected with HSV-1 (strain 17syn+ IE175-YFP; MOI = 1) ±100μM ACV (no dose escalation used) and supernatant transferred every 2 days over three consecutive rounds of infection. Virus titers were assayed by TCID-50 every 2 days post transfer; a positive slope in the titers (i.e., outgrowth of ACV-resistant virus) is evident despite 100 μM ACV.
(E and F) Continuous cultures of AD169 either without FD86, with 25 μM FD86, or with FD86 dose escalation.
(G and H) Continuous cultures of Δcrs AD169 either without FD86, with 25 μM FD86, or with FD86 dose escalation.
(I) No evidence of escape through increasing expression of UL38 or vMIA. qRT-PCR of UL38 and vMIA (normalized to beta-actin) from the AD169 continuous cultures with FD86 dose escalation. Significance tests were carried out using a one-way ANOVA with Dunnet’s test for multiple comparisons.
(J) The AD169 strain sustains greater fitness than the Δcrs AD169 strain does over multiple weeks of FD86 treatment in continuous culture. Data points from day 8 to 36 in (E) and (G) were compared. Significance was determined by Student’s t test, ∗∗ < 0.01.
(K) UL38 overexpression does not rescue CMV from FD-induced open-loop lethality. MRC5 cells were nucleofected with a UL38 expression vector (and either mock/FDScram/FD86), then 24 h later, infected with CMV and titered at 4 dpi.
(L) Flow cytometry viability analysis of uninfected neighboring “bystander” cells. ARPE-19 cells were nucleofected with 25 μM FD86, and 24 h post-nucleofection, cells were infected with CMV TB40E-IE86-YFP (MOI = 0.5). At 2 hpi, cells were washed twice in PBS to remove any attached virus and co-cultured with naive mCherry-expressing ARPE-19 cells (“bystanders”). Cells were analyzed by flow cytometry at 36 hpi (i.e., once FD-mediated cytotoxicity was present but prior to virus release).
To be sure that viral inhibition and the lack of resistance could not be explained by selection for a reduced-fitness viral variant (e.g., that spontaneously arose early on), we analyzed culture supernatants after removal of FDs (i.e., a fitness recovery assay) to determine whether viral titers rebounded. Removal of FDs resulted in a 1.5–2 log recovery in viral titers (Figure S6C), indicating that a sustained antiviral effect of FDs was responsible for reducing viral titers. Thus, these fitness recovery data do not support the alternative hypothesis that lack of resistance is explained by a reduced-fitness variant that emerges early and is gradually diluted out.
These results were confirmed using a genotypic resistance assay. Sequence analysis detected only transient single-nucleotide polymorphisms in the IE-protein regions responsible for DNA binding (Table S2). There were no mutations found in the 500-bp region surrounding the promoter crs sequence (Table S3), and sequencing of samples from the recovery assay did not reveal any consensus mutations (Table S4), indicating that no resistant variants emerged.
We also tested for the emergence of antiviral resistance to FD175 in HSV-1 continuous cultures and benchmarked against ACV (Figure 4C). Consistent with previous reports (Coen and Schaffer, 1980; Schnipper and Crumpacker, 1980), we found HSV-1 resistance to ACV emerged within two rounds (Figure S6D), whereas FD175 steadily reduced HSV-1 titers to below detection by day 40, with no evidence of increasing titers or resistance (Figure 4C).
As TB40E infections in ARPE-19 cells are thought to be largely cell associated, we repeated the continuous-culture assays using HCMV AD169, which is thought to produce more cell-free virus, in MRC5 cells. Similar viral yield reduction and clearance of virus were observed with AD169 in MRC5 cultures (Figure S6E).
To select for potential FD-resistant variants, we used an established dose-escalation approach (Biron et al., 1986; Cihlar et al., 1998; Lurain et al., 1994). Despite initiating the dose escalation at FD concentrations far below the IC50 and maintaining the infected cultures at sub-IC50 doses for weeks, no evidence of resistance to FDs was detected, and virus was cleared shortly after the IC50 dose was reached (Figure S6F). Parallel dose-escalation studies performed on the Δcrs mutant virus also could not detect evidence of resistance (Figures S6G, S6H, and S6J). Notably, the fitness of the Δcrs mutant virus was significantly lower than that of the wild-type virus under FD86 treatment across days 8–36 (Figure S6J), indicating that the Δcrs virus does not meet the criterion for a resistant mutant, i.e., its fitness is not higher than the wild type under antiviral selective pressure.
To test the hypothesis that overexpression of viral anti-apoptotic genes could overcome FD-mediated cytotoxicity and generate resistance, we examined long-term continuous cultures for expression increases in anti-apoptotic viral genes (UL38 and vMIA) (Figure S6I), and no significant increase in UL38 or vMIA expression could be detected. To further examine whether anti-apoptotic genes could overcome the FD antiviral effect, we ectopically expressed UL38 resulting in 5-fold overexpression, but this did not reduce the FD antiviral effect (Figure S6K). These data on the inability of anti-apoptotic genes to overcome the FD antiviral effect appear consistent with the extensive crosstalk between different cell-death pathways and the combinatorial mechanism of FD-induced lethality (Figures S5A and S5B). Overall, these data support the hypothesis that open-loop lethality confers a high barrier to the evolution of resistance.
Open-loop lethality does not result in bystander killing, and it has combinatorial capacity
A potential concern with a therapeutic mechanism that overexpresses a cytotoxic protein is that these proteins will be released from dying cells and be cytotoxic to uninfected bystander cells. To test this hypothesis, infected cultures were examined during the first round of infection, as bystander death would generate death in excess of the percentage of cells infected. However, the proportion of dead cells was always lower than the input virus MOI (Figures 3E and 3J). To further test bystander killing, we developed a bystander co-culture assay (Figure 5 A) that showed significant FD-mediated apoptosis in infected (IE86-YFP positive) cells but not in bystander cells (Figures 5B and S6L). These data are consistent with findings that during regulated apoptotic and ferroptotic cell death, plasma-membrane integrity is not disrupted, and cells do not release their inner contents (Yu et al., 2017).
Figure 5.

Open-loop lethality does not lead to bystander killing and enables combinatorial therapeutic approaches
(A and B) Schematic of bystander cell-death assay and flow cytometry analysis.
(C) Virus titer for CMV in the presence of FDScram, GCV (10 μM), FD86, or FD86 with GCV in HFF cells at 4 dpi.
(D) Virus titer for HSV-1 in the presence of FDScram, ACV (10 μM), FD175, or FD175 with ACV in Vero cells at 2 dpi.
(E) Multiplexed feedback disruption can inhibit CMV and HSV-1 replication. Inset: schematic of the mixed-infection experiment. Main panel: qPCR analysis of CMV and HSV-1 viral genomes in a mixed-infection experiment at 4 dpi. All error bars show mean ± 1 SD, n = 3 biological replicates.
See also Figure S6.
Given the lack of bystander cytotoxicity, we next asked whether FDs could be combined with conventional antivirals, such as GCV or ACV (Figures 5C and 5D). FDs generated reductions in viral titer comparable with GCV and ACV, and, strikingly, combinations of FD86+GCV and FD175+ACV generated additive reductions in viral titer of ∼4-logs, consistent with FDs acting via a mechanism distinct from that of GCV and ACV. Critically, no cytotoxic effects were observed from FD86+GCV or FD175+ACV in uninfected cells (Figure S3, Figure S4L and S4I).
Given that herpesvirus infections of unknown etiology are a significant clinical problem (Cunningham, 2011; Elia et al, 2016), we tested whether FDs could be multiplexed. Cells were cotransfected with FD86 and FD175, or with FDScram alone as a control, and infected with both CMV and HSV-1 (MOI = 0.1). qPCR analysis showed that the relative genomic copy number of both CMV and HSV-1 was effectively reduced by the multiplex treatment (Figure 5E). Moreover, consistent with the feedback disruption mechanism, the antiviral effects of FD86 and FD175 were each specific to their cognate viruses (Figures S4F and S4G).
Overall, the results argue that FD duplexes have the potential for use in combination with standard-of-care therapies to enhance the antiviral effect, and with each other to address infections of unknown etiology.
Open-loop lethality inhibits viral replication in murine models
To test whether open-loop lethality could generate an antiviral effect in vivo, we first employed an established murine model of HSV-1 corneal infection (Lahmidi et al., 2017) (Figure 6 A). As predicted, FD175 treatment resulted in a significant reduction in the percentage of HSV-1 infected cells in the mouse cornea (Figures 6B and 6C), reduced viral titers in mouse corneas by 150-fold (Figure 6D), and reduced viral genome replication by about 2 logs (Figure 6E).
Figure 6.

In vivo antiviral efficacy of open-loop lethality
(A) Schematic of the HSV-1 corneal infection model in mice. Mice undergo corneal debridement followed by infection with HSV-1 (17syn+ IE175-YFP). At 6 hpi, FD175, FDScram, or PBS was topically applied to the cornea. Corneas were harvested at 2 dpi for analysis.
(B) YFP fluorescence images of corneas after harvesting (nuclei stained with DAPI). Scale bars, 100 μm.
(C) Quantification of HSV-1 YFP expressing cells in corneas. Five corneas imaged per sample.
(D) HSV-1 viral titers from infected corneas 2 days post-treatment with PBS, FDScram, or FD175. Each data point represents a pooling of corneas (i.e., nine corneas per treatment).
(E) HSV-1 viral genomic DNA quantification by qPCR 2 days post-treatment. Each data point represents a pooling of corneas (i.e., nine corneas per treatment).
(F) Schematic of the mCMV systemic infection model in mice. Mice were infected with 105 PFU mCMV and injected intraperitoneally with FD88 (n = 5) on gold nanoparticles or a vehicle control (n = 5) at 3 h pre- and 18 h post infection. At 5 dpi, liver and spleen were harvested for analysis.
(G) Quantification of viral IE88 and GpB mRNA by qRT-PCR in FD88-treated or vehicle control-treated samples.
(H) qPCR quantification of mCMV replication by DNA genome copy number in FD88 or vehicle-treated samples.
(I) Infectious virus titer quantified by TCID-50 from animals treated with FD88 or vehicle control. p values derived from Student’s t test: ∗<0.05, ∗∗<0.01, ∗∗∗<0.001.
Next, to test whether FDs reached concentrations sufficient to suppress systemic infection in vivo, we used the mCMV systemic infection model and delivered FDs that disrupt mCMV IE3 (IE88) feedback (termed FD88) via nanoparticles (Figure 6F). Mice infected with mCMV (polyclonal Smith strain) and treated with FD88 exhibited a significant increase in viral IE88 gene expression (Figure 6G), reduced viral titers (Figure 6G), reduced viral genome replication (Figure 6H), and reduced infectious viral particles in the liver and spleen (Figure 6I). Strikingly, FD88-treated animals exhibited significant protection from body-weight loss compared with infected control animals (Figure 6J). Together, these results show proof of concept that feedback disruption leading to open-loop lethality can generate a systemic antiviral effect.
Open-loop lethality in an RNA virus; disrupting SARS-CoV-2 autorepression inhibits replication
To test whether the open-loop lethality concept could be generalized to inhibit other viruses, we examined a predicted autorepression circuit in the beta coronavirus SARS-CoV-2 (Li et al., 2021b). The putative circuit involves the non-structural protein NSP15 interacting with transcriptional regulatory sequences (TRSs) on viral genomic RNA (gRNA) to inhibit gRNA replication, transcription, and downstream translation of viral proteins (Figure 7 A). Since several SARS-CoV-2 proteins are known to induce apoptosis (Li et al., 2021a), we hypothesized that disrupting autorepression would cause the accumulation of these viral proteins, leading to open-loop lethality and reduced virus production. Thus, we tested whether excess TRS RNA decoys acted as FDs. Short 24-nt RNAs containing a tetramer repeat of the TRS (termed FDTRS) were engineered, and to ensure that these short RNAs did not activate innate immune responses, a size-matched scrambled FD RNA (FDScram) was used as a negative control. Cells were transfected with FDTRS RNAs and infected with SARS-CoV-2 isolate WA-1 (MOI = 0.05), and the effects on replication were examined as described (Chaturvedi et al., 2021). As predicted, during the first viral replication cycle (∼8 h), FDTRS RNAs induced a significant increase in both sub-genomic and genomic viral RNAs, including NSP15, ORF1a, and Spike RNA (Figures 7B and S7A). Since Spike protein is associated with increased apoptosis, we examined early and late apoptosis markers, annexin V and TUNEL, and found significant increases in infected cells treated with FDTRS RNAs compared with controls (Figures 7C, 7D and S7B). Increased apoptosis was associated with a >1-log reduction in SARS-CoV-2 replication (Figure 7E). These data validate the existence of a predicted negative-feedback circuit in SARS-CoV-2 and show that disrupting this circuit generates open-loop lethality leading to an antiviral effect in an RNA virus.
Figure 7.

Open-loop lethality generates an antiviral effect against SARS-CoV-2
(A) Schematic of the putative SARS-CoV-2 transcriptional negative-feedback circuit. Nsp15 proteins, which are transcribed from a sub-genomic RNA, cleave genomic RNAs (gRNAs) at the transcriptional regulatory sequences (TRS), thereby suppressing genomic and sub-genomic RNA levels. Feedback can be disrupted by supplying excess TRS encoding RNAs (FDTRS) as decoys to titrate nsp15, thereby leading to the accumulation of genomic RNAs and increased synthesis of non-structural, structural, and accessory proteins, including cytotoxic viral proteins above cytotoxic levels.
(B) Vero cells were nucleofected with 25 μM FDTRS or FDScram, followed by infection with SARS-CoV-2 (WA-1 strain, MOI = 0.05) and quantification of nsp15, ORF1a, and Spike RNA by qRT-PCR within the first round of replication (i.e., 8 hpi).
(C) Apoptosis analysis at 12 hpi by qRT-PCR for annexin V.
(D) Apoptosis analysis at 48 hpi by TUNEL assay.
(E) Virus titer for SARS-CoV-2 after treatment with either FDTRS or FDScram. p values derived from Student’s t test: ∗<0.05, ∗∗<0.01, ∗∗∗<0.001.
See also Figure S7.
Figure S7.

FDTRS decoys in the context of SARS-CoV-2 infection, related to Figure 7
(A) qRT-PCR analysis for ORF1a expression fold-change at 8, 12, and 24 hpi; Vero cells were nucleofected with 25 μM FDTRS or FDScram and then infected with SARS-CoV-2 (WA-1 strain, MOI = 0.05).
(B) Raw flow cytometry dot plots for apoptosis analysis at 48 hpi by TUNEL assay; Vero cells were nucleofected with 25 μM FDTRS or FDScram and then infected with SARS-CoV-2 (WA-1 strain, MOI = 0.05).
Discussion
Generalizability of open-loop lethality
Overall, these data show that open-loop lethality can be generated by decoys that competitively inhibit DNA- or RNA-protein interactions to disrupt negative-feedback circuits. FD decoys increase rather than decrease (i.e., titrate) transcription factor levels, thereby breaking homeostasis. This molecular mechanism of action—a form of synthetic lethality—does not function via innate immune mechanisms and limits open-loop lethality only to cells encoding the targeted circuit. Feedback disruption generates antiviral open-loop lethality against multiple herpesviruses, confers a high barrier to the evolution of resistance, and enables feedback-disruption strategies to synergize with conventional therapeutics (Figure 4, Figure 5, Figure 6, Figure 7). While this study focused on viral feedback circuits, in principle, open-loop lethality may be a viable therapeutic strategy against autoregulatory circuits in other microbes or neoplasms.
From the perspective of gene-regulatory networks, the feedback circuits described herein have interlinked positive and negative feedback. This coupled-feedback architecture amplifies the effects of open-loop lethality by creating a bistable system where FDs cause a transition across the separatrix to a new cytotoxic steady state (Figures 3E and 3J). Notably, negative-feedback-“linearizer” circuits also exhibit increased circuit output under titration of feedback regulators (Nevozhay et al., 2009), suggesting that the FD approach could apply broadly to negative feedback.
From the antiviral perspective, open-loop lethality could overcome a current treatment challenge: the reduction in antiviral efficacy that accompanies high-viremic loads for many conventional antiviral therapies, which increases the potential for the outgrowth of resistant mutants (Åsberg et al., 2016). However, for open-loop lethality strategies, higher MOIs benefit the drug efficacy, as high MOIs deliver more potentially cytotoxic protein to the cell. In terms of resistance, viral regulatory loci appear to have lower genetic variability than viral enzymes or receptors do (Cudini et al., 2019; Lu et al., 2002b; Renzette et al., 2015), supporting that such circuits are essential (i.e., attractive therapeutic targets).
A number of viral proteins are known to be proapoptotic and tightly regulated, including the HIV-1 Vpr protein, the HBV HBx protein, and PB1-F2 from influenza (Boya et al., 2004; Zamarin et al., 2005), and inflammatory cytokines are also known to generate cytotoxicity when dysregulated (Cauwels and Brouckaert, 2007). Recent studies showing RNA-mediated negative feedback on transcription via condensates (Henninger et al., 2021) or small regulatory RNAs (Hoyos et al., 2020) suggest generalized targets for feedback disruption. Other studies indicate that feedback regulation in signaling pathways may be targetable (Longo et al., 2013), and certain “integral” feedback architectures (Aoki et al., 2019) could be particularly attractive targets for disruption due to their exceptionally tight control characteristics.
While delivery of nucleic acids remains a challenge, the FD decoys reported here are short, and significant clinical advances have been made with the recent FDA approval of antisense and exon-skipping oligonucleotide therapies delivered via nanoparticles (Adams et al., 2018, Kanasty et al., 2013, Khvorova and Watts, 2017) and COVID mRNA vaccines. Such nucleic-acid therapies are generally thought to offer increased specificity with the potential for fewer off-target effects than small molecules.
Limitations of the study
As with any theoretical framework, our mathematical model for the evolution of resistance is limited by several assumptions. For tractability, the model considers point mutations as the mechanism of evolutionary adaptation and does not explicitly include gene amplification, recombination, or selection. Here, we discuss the potential of these alternate mechanisms, as well as others, to generate resistance.
There is a well-established body of literature demonstrating that the selection of pre-existing mutants is the most likely mechanism for the emergence of resistance (Emery and Griffiths, 2000; Luria and Delbruck, 1943; Ribeiro and Bonhoeffer, 2000). For example, the Δcrs mutant could be a potential starting candidate to select for resistance to FDs, assuming IE2 could evolve to bind selectively to the mutated crs. However, no resistance was detected in Δcrs continuous cultures (Figures S6G and S6H), suggesting that selection is unlikely even from this starting point. More importantly, the Δcrs virus has significantly lower fitness than the FD-treated wild type has (Figure S6J), implying that the Δcrs mutant would be outcompeted prior to selection.
Recombination is a major contributor to within-host viral genetic diversity (Coen and Schaffer, 1980; Cudini et al., 2019; Fu et al., 2002; Thiry et al., 2005; Wildy, 1955) and another mechanism for the evolution of resistance. However, recombinant-driven resistance typically requires extant resistant strains circulating within the population to serve as evolutionary substrates, and as discussed above, the mechanisms for pregenerating such resistant mutants are unclear.
An alternate scheme could be for the virus to escape open-loop lethality by evolving more rapid viral maturation kinetics. For herpesviruses, this appears unlikely for several reasons. First, the IE proteins are detectable by 2–3 h post infection (hpi) (Teng et al., 2012), and IE2-induced apoptosis is detectable by 24 hpi, whereas the viral life cycle is ∼96 h. Thus, the virus would likely need to accelerate its life cycle by many days (∼4 ×) to complete maturation prior to apoptosis. In fact, this putative viral acceleration scheme contravenes the empirical data for the Δ crs mutant, which replicates with slower kinetics given its reduced MIEP activity (Teng et al., 2012). This deceleration results in a substantial fitness loss compared with the wild-type virus, and previous attempts to rescue/accelerate the Δ crs mutant generated further reductions in fitness (i.e., a 4-log reduction in titer). Indeed, dynamical systems theory predicts that mutants with accelerated kinetics will in fact be more sensitive to open-loop lethality, rather than less sensitive.
Gene amplification can generate antiviral drug resistance, but it typically manifests rapidly within several passages (Bayer et al., 2018; Erlandson et al., 2014; Slabaugh et al., 1988), whereas the long-term continuous-culture data herein do not manifest phenotypic resistance or genetic duplication (Tables S2 and S3). A potential explanation is that amplification or duplication often overcomes inhibitors (e.g., molecular enzyme poisons), whereas for FDs, IE amplification would likely enhance open-loop lethality. While amplification of anti-apoptotic genes could overcome open-loop lethality, 5× overexpression of anti-apoptotic proteins was insufficient (Figures S6I and S6K), perhaps due to the extensive crosstalk between multiple cell-death pathways activated by feedback disruption (Figures S5A–S5D).
Altogether, these data suggest that the open-loop lethality strategy may present a high barrier for the evolution of resistance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| goat anti-mouse Alexa 594 secondary antibody | Thermo Fisher Scientific | Cat# A-11005, RRID:AB_2534073) |
| mouse anti-pp65 primary antibody, clone 8F5 | Nowak et al. (1984) | N/A |
| mouse anti-ICP5 primary antibody | Abcam | Cat# ab6508, RRID:AB_305530 |
| IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Cat# 926–32210, RRID:AB_621842 |
| mouse anti-IE86 primary antivody, clone 8B1.2 (ASR) | Millipore | Cat #MAB810, RRID:AB_95254 |
| Bacterial and virus strains | ||
| CMV TB40E-IE86-YFP | Vardi et al. (2018) | N/A |
| HSV-1 17syn+ IE175-YFP | Everett et al. (2003) | N/A |
| mCMV strain K181 | Morley et al. (2002) | N/A |
| mCMV strain Smith | ATCC | Cat #CRL-1404 |
| CMV AD169 | HIV Reagents Program | ARP-1910 |
| CMV AD169Δcrs | Teng and Bolovan-Fritts et al, Cell 2012 | N/A |
| CMV GDGrK17 | HIV Reagents Program | ARP-1669 |
| CMV GDGrP53 | HIV Reagents Program | ARP-1670 |
| CMV 759rD100-1 | HIV Reagents Program | ARP-1912 |
| CMV PFArD100 | HIV Reagents Program | ARP-1666 |
| Rhesus CMV 68.1 EGFP virus | Chang and Barry (2003) | N/A |
| BL21 competent E. coli cells | New England Biolabs Inc | Cat # C2530H |
| SARS-CoV-2 isolate (USA-WA1/2020) | SARS-CoV-2 isolate (USAWA1/2020) was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS Related Coronavirus 2, Isolate USAWA1/2020. |
NR-52281 |
| Chemicals, peptides, and recombinant proteins | ||
| BODIPY 581/591 C11 Lipid Peroxidation Sensor | Invitrogen | D3861 |
| 3-methyl adenine authophagy inhibitor | Sigma Aldrich | M9281 |
| NLRP3 inhibitor, CAS 256373-96-3 | Sigma Aldrich | Cat # 5381200001 |
| Ferrostatin-1 | Sigma Aldrich | Cat #SML0583 |
| collagenase I | Sigma-Aldrich | Cat #C0130 |
| Ganciclovir | Sigma-Aldrich | Cat #G2536 |
| Acyclovir | Sigma-Aldrich | Cat #A0220000 |
| Valproic Acid | Calbiochem | Cat # 1069-66-5 |
| Critical commercial assays | ||
| RNAeasy RNA extraction kit | Qiagen | 74104 |
| Ovation RNA-Seq System v2 kit | NuGEN | 7102 |
| Ovation Ultralow Library System V2 | NuGEN | 0344NB |
| Tunel Assay Apoptosis Detection kit | Biotium | 30074 |
| QuantiTet Reverse Transcription kit | QIAGEN | 205311 |
| Fast SYBR Green Master Mix | Applied Biosystems | #4385612 |
| DNeasy Blood and tissue kit | Qiagen | 69504 |
| Zombie Aqua Fixable Viability kit | BioLegend | Cat # 423101 |
| Annexin V Apoptosis Detection Kit | BioLegend | Cat #640932 |
| Experimental models: Cell lines | ||
| ARPE-19 cells | ATCC | Cat #CRL-2302 |
| MRC-5 fibroblasts | ATCC | Cat #CCL-171 |
| Telo-RF cells | Kirchoff et al. (2002) | N/A |
| NIH/3T3 mouse fibroblast | ATCC | Cat #CRL-1658 |
| ARPE-19 stably expressing the MIEP-IE86-GFP minimal circuit | Teng et al. (2012) | N/A |
| Vero-E6 cells | ATCC | Cat #CRL-1586 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | Jackson lab | # 000664 |
| BALB/c mice | Jackson lab | #000651 |
| Oligonucleotides | ||
| ODN 2216 | Table S1 | N/A |
| Feedback disruptors and Fomivirsen | Table S1 | N/A |
| Recombinant DNA | ||
| pMALcXS | Macias and Stinski (1993) | N/A |
| pMAL-cX5 vector | New England Biolabs Inc | Cat # N8108 |
| IE86-FL-pMAL-cX5 vector | This paper | N/A |
| pUL38 expression vector | Terhune et al. (2007) | N/A |
| Software and algorithms | ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
| MATLAB | Math works, Inc | https://www.mathworks.com/products/matlab.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Leor S. Weinberger (leor.weinberger@gladstone.ucsf.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer agreement.
Experimental model and subject details
Animals
All animal experiments were conducted in accordance with procedures approved by the UCSF Institutional Animal Care and Use Committee. For HSV-1 infection studies, experiments were performed with 6- to 10-week old male and female sibling Black 6 mice. Breeding pairs were purchased from The Jackson Laboratory (C57BL/6J mice, Stock No: 000664) and maintained under pathogen-free conditions in the UCSF barrier facility. For mCMV infection studies, experiments were performed using 28- to 30-week old mice; specifically, 6-week old male BALB/c mice (Jackson Laboratory, Stock No: 000651) were maintained under pathogen-free conditions in the UCSF barrier facility until they were 28- to 30-week-old.
Viruses
The CMV TB40E-IE86-YFP virus was previously described (Vardi et al., 2018) and CMV AD169 (Cat #ARP-1910), GDGrK17 (Cat #ARP-1669), CMV GDGrP53 (Cat #ARP-1670), CMV 759rD100-1 (Cat #ARP-1912), CMV PFArD100 (Cat #ARP-1666) were obtained through the NIH AIDS Reagent Program. mCMV strain K181 (Morley et al., 2002) was kindly provided by Lewis Lanier (UCSF) and Rhesus CMV 68.1 EGFP virus (Chang and Barry, 2003) was kindly provided by Peter Barry, UC-Davis. mCMV strain Smith was obtained from ATCC (Cat #CRL-1404). The clinical strain of HSV-1 17syn+ IE175-YFP (Everett et al., 2003) was kindly provided by Roger Everett, MRC Virology Unit, Glasgow, Scotland that was passaged originally from a clinical isolate (Brown et al., 1973). SARS-CoV-2 isolate (USA-WA1/2020) was obtained from Biodefense and Emerging Infections (BEI) Resources. SARS-CoV-2 live virus experiments were performed at the Gladstone Institutes in a Biosafety Level 3 (BSL3) containment facility under an approved Biosafety Use Authorization from UCSF and in compliance with UCSF guidelines and procedures.
Cells
ARPE-19 cells (ATCC, Cat #CRL-2302) were routinely maintained in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium (DMEM)/F-12 (Mediatech Inc.) with 10% fetal bovine serum (FBS) (HyClone) and 50 U/ml Penicillin-Streptomycin (Mediatech Inc.) at 37°C and 5% CO2 in a humified incubator. The ARPE-19 feedback-reporter cell line stably expresses a MIEP-IE86-IRES-GFP minimal cassette and has been previously described (Teng et al., 2012); to potentiate responsiveness, these cells are cultured in presence of Valproic Acid (VPA) 24 hours prior to perturbation (e.g. transfection). The broken feedback-reporter cell line expressing MIEP(Δcrs)-IE86-IRES-GFP (Teng et al 2012) was prepared by freshly transducing ARPE-19 cells with the reporter construct. MRC-5 fibroblasts (ATCC, Cat #CCL-171), NIH/3T3 mouse fibroblast (ATCC, Cat #CRL-1658), Telo-RF (Kirchoff et al., 2002) and Vero-E6 (Vero C1008, ATCC Cat #CRL-1586) were maintained in DMEM with 10% FBS and 50 U/ml Penicillin and Streptomycin (Mediatech Inc.). Fomivirsen (Table S1), Ganciclovir (Sigma-Aldrich, Cat #G2536) and Acyclovir (Sigma-Aldrich, Cat #A0220000) were added to media at the indicated concentrations following virus inoculate removal.
Method details
Feedback disruptor mathematical modeling and numerical simulations
An experimentally validated ODE model of the CMV IE86 negative feedback circuit (Teng et al., 2012; Vardi et al., 2018) was modified to include state variables for free IE86 protein (IE86), crs feedback-disruptor (FD) DNA duplexes, and the IE86–FD DNA-protein interaction complex (Complex):

where represents the basal IE86 expression rate, represents the IE86 feedback gain constant, represents the IE86 cooperativity index for crs binding (hill coefficient), represents the IE86 cooperativity index for transcriptional activation of the MIEP upstream of the crs (hill coefficient), represents a Michaelis constant for IE86 negative feedback, represents a Michaelis constant for IE86 negative feedback, represents a Michaelis constant for FD binding, represents the per-capita IE86 protein degradation rate, represents the Complex association/formation rate, represents the Complex dissociation/fragmentation rate. In this model, the IE86 can oligomerize ( ∼ 6)22 to bind either the crs in its own promoter (MIEP)—thereby mediating negative feedback and down-regulating its own expression rate—or can bind the FD DNA duplex, thereby sequestering IE86 that might otherwise downregulate its own expression via negative feedback. For both binding events, IE86 can transition between free and FD-bound states but the negative feedback bound state is not explicitly modeled for parsimony and to not introduce unnecessary parameters. Dimeric IE86 ( ∼ 2) can promiscuously bind promoters to positively regulate transcription (Lukac et al., 1994; Nishimura et al., 2017), likely including the MIEP itself upstream of the crs. Because both the positive and negative feedback operate through regulation of the MIEP, their effects were multiplied to obtain a net transcriptional rate scaled by , as done previously in the viral context (Teng et al., 2012). Based on the phosphorothioation studies, the DNA duplexes were assumed to be stable (i.e., not degrade) over the course of the simulation time. Under this assumption, there are only two independent state variables, since Complex(t) + FD(t) is equal to the initial FD supplied to the system.
The ODEs were numerically solved using MATLAB after specifying the initial FD dose. Parameters were chosen by first fixing the Hill coefficients = 6 and = 2, then searching parameter space to obtain qualitative agreement with experiments. For Figure S1A, , , , , , , and . The initial FD doses were 0, 0.025, or 2.5. To approximate a range of IE86 expression across a population of cells sorted for circuit activity, initial IE86 levels were randomly drawn from a cut Gaussian distribution where below 3 were excluded.
Within-host CMV model for calculation of probability of resistance to FDs
To calculate the probability of mutants arising during infection, a within-host model for CMV infection, based on a previous model widely used for chronic viral infections (Perelson, 2002) was used. The ODEs describing within-host infection dynamics are:
| (Equation 1) |
This model considers the dynamics of target cells (T), cells infected but not yet producing virus, i.e. in the eclipse phase (E), productively infected cells (I) and virus (V). Target cells are generated at rate , and die at per capita rate . They can be infected by virus interacting with target cells at rate , leading to productively infected cells (I). Productively infected cells die at a per capita rate in the absence of FD, and produce viruses at rate p per cell. Under FD treatment, the death rate increases by because of the cytotoxic effect of FD-induced open-loop lethality. Viruses are cleared at per capita rate c. models the efficacy of a therapy.
From Equation 1, one can calculate the within-host reproductive number, , using the next-generation matrix method (Diekmann et al., 2010), in the absence of treatment:
| (Equation 2) |
where T 0 is the target cell concentration in an uninfected individual.
CMV infects a range of host cells, including T lymphocytes, epithelial cells, endothelial cells and hematopoietic cells (Rice et al., 1984; Sinzger et al., 2008; Vanarsdall and Johnson, 2012). Here, the net per capita rate of change, i.e., the combined per capita rate of proliferation and death of target cells (), is assumed to be -0.04/day, a rate estimated for human T lymphocytes (Mohri et al., 2001). Because it is assumed that there is a source of new target cells, p-d must be negative since the source rate plus proliferation must match the death rate. It is further assumed that the infection-free target cell concentration is 106/mL. Then assuming the system is at steady state pre-infection 104/mL/day, the infected cell half-life is set to 1 day, corresponding to a death rate 0.69/day (Emery et al., 1999). The average eclipse period of an infected cell, 1/k, is set to 1 day (Emery et al., 2002). Furthermore, the clearance rate of CMV, c, was set to be 23/day, matching the rate estimated for HIV and HCV (Guedj et al., 2013; Ramratnam et al., 1999).
The values of the other parameters in the model are estimated using the parameter values above and previously reported clinical data. First, the doubling time of the plasma CMV load was estimated to be 1.5 days in 18 bone marrow transplant recipients (Emery et al., 1999), which translates to an exponential growth rate of 0.46/day. This rate, , corresponds to the dominant eigenvalue of the Jacobian matrix of Equation 1 at the infection free equilibrium. Then, is related to r and other parameters by the equation
| (Equation 3) |
Substituting the values of k, r, c and produces 2.48.
From Equation 1, the set-point viral load in the absence of treatment, , is:
| (Equation 4) |
A typical CMV set-point viral load is 105 copies/mL in patients with CMV retinitis (Emery et al., 1999). Then, is calculated as:
Finally, from Equation 2, p is calculated as
Note that the initial density of target cells T 0 is not well established and that changes in the estimate of T 0 will change the estimate of p without affecting the estimates of other parameters.
The values of the parameters in the model are summarized in Table S6.
Calculation of probability of resistance mutants appearing in a host
The probability of resistant mutants appearing in a host is calculated using simulation of the ODE model above. Viral load before initiation of therapy was assumed to be at steady-state, and therapy assumed to have 90% efficacy (i.e., ), starting at time 0. The model was then numerically solved to calculate the total number of preexisting viral particles and viral particles produced during the treatment via the following equation:
| (Equation 5) |
where the virus is assumed to distribute through extracellular body fluid in a 70 kg person where the volume of the extracellular body fluid, , is assumed to be 15,000 mL.
The life-cycle of CMV involves replication of CMV genome in the nucleus. Mutations can be generated during replication of viral DNA with the mutation rate per site per generation estimated in the range of 2∗10-7 (Renzette et al., 2015) to 2∗10-8 (Drake and Hwang, 2005). Given the size of CMV genome (2.4∗105 bp), the mutation rate per genome, , takes values between 4.8∗10-3 and 4.8∗10-2, which assumes that mutation at any site in the genome can lead to resistance whereas, in reality only a fraction of sites (e.g., in the IE locus) generate resistance mutations. Therefore, this calculated risk of resistance represents an overestimate.
For FD intervention, where the genetic barrier to resistance is relatively high, the probability of generating resistance, , can be approximated as
| (Equation 6) |
where is the number of mutations needed to generate resistance (with n set to 3, 5, 10, or 15 in the analysis below).
The calculations for the probability of FD resistance take into consideration parameter value uncertainties including the mutation rate , (varied between 0.5, 1 or 2/day), and (varied between values of 0.8, 0.9 or 0.99). The combination of all possible choices of parameters (including the 4 choices for n) leads to 2∗3∗3∗4=72 simulations (results of these 72 simulations are shown in Figure S1B). Note that because the probability of resistance changes monotonically with changes in each of these parameters, using extreme values for these parameters in the simulation is sufficient to derive the upper and the lower bounds of the probability of resistance (shown in Figure 1C).
The probability of resistance appears to be largely determined by and n (Figure S1M), and is relatively insensitive to changes in the efficacy of the therapy , and the rate of increased cytotoxicity , as long as the therapy maintains efficacy and the viral load declines exponentially under therapy.
For additional sensitivity analysis of the model’s functional form, a second within-host model for CMV infection that uses logistic proliferation of target cells was examined. The ODEs describing the model are:
| (Equation 7) |
where is the maximum proliferation rate and T 0 is the concentration of the target cells in the absence of infection, and T 0=106 /mL. The expressions for R0 and the initial growth rate r remain the same as the expressions in the basic model (Equations 2 and 3, respectively). The set-point viral load in the absence of treatment, , can be calculated by setting the ODE system (Equation 7) to steady state and solve the resulting equations. The expression for is:
| (Equation 8) |
As mentioned above, Emery et al. estimated that a typical set-point viral load is 105 copies/mL (Emery et al., 1999). Then, is calculated from Equation 8 as:
| (Equation 9) |
All other parameters are set the same as in the basic model.
Numerical simulations of the logistic proliferation model, show that the predicted probabilities of resistance are very similar to the predictions of the basic model (Figure S1B). The upper and lower bounds shown (Figure 1C) are based on the predicted probabilities of resistance using all the parameter combinations from both the basic model and the logistic proliferation model.
Double-stranded FD DNA-duplex preparation
FD86, FD175, FD88, FDRhCMV were made by annealing sequence specific oligonucleotides (see Table S1 for sequences). Briefly, DNA oligonucleotides were obtained from Integrated DNA Technologies and resuspended in annealing buffer (100mM Potassium acetate; 30mM HEPES, pH 7.5). Oligonucleotides of complementary sequences were mixed in equimolar amount, heated to 95°C for 2 minutes, and gradually cooled to 25°C over the period of 45 minutes in a S1000 Thermocycler (Bio-Rad) and stored at –20°C.
Protein expression, EMSA and DNA binding assays
BL21 competent E. coli cells (New England Biolabs Inc, Cat # C2530H) were transformed with a pMALcXS plasmid encoding the C-terminus part of IE86 fused to the maltose binding protein, as described (Macias and Stinski, 1993), and induced with isopropyl b-D thiogalactoside (IPTG) in 1L luria broth (Thermofisher Scientific) containing ampicillin (Sigma-Aldrich). Purification for C-terminus IE86 protein: Cells were pelleted at 10,000 RPM for 30 minutes at 4°C and resuspended in 40mL lysis buffer (20mM HEPES, pH 7.4, 1M NaCl, 1mM EDTA, 1mM DTT, Roche protease inhibitor cocktail). Lysozyme was added at a final concentration of 1mg/ml and cells were incubated on ice for 30 minutes, followed by addition of 1mM PMSF and sonication on ice (sonicator at 40% amplitude, 10 seconds on, 30 seconds off-6 repeats). 1mM MgSO4, 0.1mg/ml DNAse (Sigma-Aldrich) and 25U/ml Benzonase (Sigma-Aldrich, Cat #E1014) were then added, cells further incubated on ice for 15-30 minutes, centrifuged at 12,000 rpm for 30 minutes at 4°C and the supernatant incubated with amylose resins (Sigma-Aldrich, St Louis, MO) for 2 hours at 4°C with gentle agitation. Batch bound resin was poured into a column and washed with 10 column volumes of lysis buffer, and eluted in elution buffer (20mM HEPES, pH 7.4°C, 250mM NaCl, 10mM Maltose, 1mM EDTA and 1mM DTT). The eluted protein was passed through a Superose 6 column (GE Healthcare Life Sciences) using gel filtration buffer (20mM HEPES, pH 7.4, 250mM NaCl and 1mM DTT) and used for EMSA or for binding assays. Cloning and purification of full length IE86 protein: Full length IE86 protein was codon optimized and cloned in pMAL-cX5 vector (New England Biolabs Inc., Cat # N8108). Cells were transformed in BL-21 cells and IE86-FL-pMAL-cX5 transformed cells were grown in 1L luria broth (Thermofisher Scientific) containing ampicillin (Sigma-Aldrich) and induced for 1 hour with IPTG (Sigma-Aldrich, Cat #I6758). Cells were pelleted and frozen in liquid nitrogen and stored at -80o freezer until used. Pellet was resuspended in 40mL lysis buffer (20mM HEPES, pH 7.4, 10% glycerol, 0.2M MgSO4, 1M NaCl, 1mM EDTA, 1mM DTT, Roche protease inhibitor cocktail). Lysozyme was added at a final concentration of 1mg/ml and cells were incubated on ice for 30 minutes, followed by addition of 1mM PMSF and sonication on ice (sonicator at 40% amplitude, 10 seconds on, 30 seconds off-6 repeats). 1mM MgSO4, 0.1mg/ml DNAse (Sigma-Aldrich) and 25U/ml Benzonase (Sigma- Aldrich) were then added, cells further incubated on ice for 15–30 minutes, centrifuged at 12,000 rpm for 30 minutes at 4°C and the supernatant incubated with amylose resins (Sigma-Aldrich) for 2 hours at 4°C with gentle agitation. Batch bound resin was poured into a column and washed with 10 column volumes of lysis buffer, and eluted in elution buffer (20mM HEPES, pH 7.4°C, 10% glycerol, 0.2M MgSO4, 250mM NaCl, 10mM Maltose, 1mM EDTA and 1mM DTT). The eluted protein was passed through a Superose 6 column (GE Healthcare Life Sciences) using gel filtration buffer (20mM HEPES, pH 7.4, 250mM NaCl 10% glycerol, 0.2M MgSO4, and 1mM DTT) and used for binding assays. Binding assays were performed by incubating duplexes of various lengths (as labeled in the figures) with purified MBP-IE86 or MBP-IE-FL for 30 minutes at room temperature and running on Superpose-6 column in the presence of gel filtration buffer and monitored oligomerized form of MBP-IE86 (at 13ml column volume on FPLC) in the presence of different sizes of FD86. To verify the sequence specific interaction between crs and MBP-IE86, EMSA was performed for crs and Δcrs oligonucleotides (Asmar et al., 2004). DNA oligonucleotides were obtained from Integrated DNA Technologies (San Jose, CA) (see Table S1 for sequences) and resuspended in annealing buffer (100mM Potassium acetate; 30mM HEPES, pH 7.5). Oligonucleotides of complementary sequences were mixed in equimolar amount, heated to 95°C for 2 minutes, and gradually cooled to 25°C over 45 minutes in a S1000 Thermocycler (Bio-Rad) and stored at –20oC. Briefly, oligonucleotides were incubated with the C-terminus-IE86 protein for 30 minutes in the presence of 1X binding buffer (10 mM HEPES-NaOH [pH 8.0], 50 mM KCl, 100 mM EDTA, and 5% glycerol, and subjected to electrophoresis on 1% agarose gel prepared in 1X TAE (Tris-acetate EDTA) buffer at 10V at 4°C. The gel was stained with ethidium bromide and imaged.
Feedback quantification by single-cell fluctuation (noise) analysis
ARPE-19 cells carrying the MIEP-IE86-IRES-GFP feedback reporter (Teng et al., 2012) were nucleofected with FD86 and seeded onto 8-chamber imaging slides (μ-Slide 8 Well Glass Bottom, Ibidi, Cat # 80826) using 40,000 cells in 200 μL of media supplemented with 1 mM valproic acid. We note that the efficiency of nucleofection is less than 100%. As a control, "naive" feedback reporter cells were sorted for moderate GFP (i.e., higher background circuit activity) to increase the GFP signal for imaging, allowed to recover over several passages, and 20,000 cells were seeded per chamber in 200 μL media. For the broken-feedback reporter, ARPE-19 cells were freshly transduced with the MIEP(Δcrs)-IE86-IRES-GFP reporter construct (Teng et al., 2012). Imaging started 3 d post-seeding. Cells were imaged for 24 h on a FluoView 3000 confocal laser scanning microscope (Olympus) equipped with a 40.0×/1.25 N.A. silicon oil immersion objective (UPLSAPO40XS, Olympus) and 488nm laser (OBIS). Fields of view were acquired every 15 minutes, at 0.1554 μm xy resolution and a z resolution of 2 μm, with a stack of three z-slices separated by 2 μm. The z-slices were maximally projected in FIJI and movies were manually analyzed to extract a median GFP fluorescence at each frame, producing a timeseries for each cell (Weinberger et al., 2008). The mean and coefficient of variance (CV=σ/μ) were computed from each timeseries.
Quantifying open-loop lethality, IC50, and viral yield reduction in CMV
To measure increases in IE86 expression by flow cytometry, ARPE-19 cells were nucleofected with FD86 or FDScram at 25 μM (or mock). Then 24 h later, the cells were infected with CMV (TB40E) encoding an IE86-YFP fusion (TB40E-IE86-YFP) at MOI=0.1, harvested, and analyzed by flow at 2 dpi. To visualize IE86 expression by fluorescence microscopy, ARPE-19 cells were nucleofected with FD86 or FDScram at 25 μM. Then 24 h later, the cells were infected with TB40E-IE86-YFP at MOI=1.0, and imaged at 1 dpi, since the immediate-early genes are already expressed by this early time point. To perform an FD86 dose-response analysis and calculate an IC50 against viral titer, ARPE-19 cells were nucleofected with FD86 at increasing concentrations ranging from 10-5 nM to 105 nM. Then 24 h later, the cells were infected with TB40E-IE86-YFP (MOI=0.1). At 4 dpi (one replication cycle), virus was harvested by subjecting the cells to three freeze-thaw cycles, then 10-fold serial dilution titrations on naive ARPE-19 cells was performed with eight replicates per dilution in 96-well plates. Titration results were converted to PFU/ml using median tissue culture infectious dose (TCID-50) (Reed, 1938). The detection limit shown is the single-well detection limit. To measure CMV yield reduction due to open-loop lethality, ARPE-19 cells were nucleofected with FD86 or FDScram at 25 μM (or mock). Then 24 h later, the cells were infected with a TB40E-IE86-YFP at increasing MOIs (0.1, 0.5, 1.0, 2.0), and virus was titered at 4 dpi. To quantify apoptosis induction in CMV-infected cells, ARPE-19 cells were nucleofected with FD86 or FDScram at 25 μM (or mock). Then 24 h later, the cells were infected with TB40E-IE86-YFP (MOI=1), and cells were harvested and stained for Annexin V at 2 dpi, followed by flow cytometry. See also Figures 3A–3E. Similar protocols were used to nucleofect MRC-5 and HFF cells prior to infection and titration.
Quantifying open-loop lethality, IC50, and viral yield reduction in HSV-1
To measure increases in IE175 expression by flow cytometry, ARPE-19 cells were nucleofected with FD175 or FDScram at 25 μM (or mock). Then 24 h later, the cells were infected with HSV-1 (17syn+ strain) encoding IE175-YFP at MOI=0.1, harvested, and analyzed by flow at 2 dpi. To visualize IE175 expression by fluorescence microscopy, ARPE-19 cells were nucleofected with FD175 or FDScram at 25 μM. Then 24 h later, the cells were infected with HSV-1 IE175-YFP (17syn+ strain) at MOI=1.0, and imaged at 12 hpi, since the immediate-early genes are already expressed by this time point. To perform an FD175 dose-response analysis and calculate an IC50 against viral titer, Vero cells were nucleofected with FD175 at increasing concentrations ranging from 10-5 nM to 105 nM. Then 24 h later, the cells were infected with HSV-1 IE175-YFP (17syn+ strain) at MOI=0.1. At 2 dpi (one replication cycle), virus was harvested by subjecting the cells to three freeze-thaw cycles, then 10-fold serial dilution titrations on naive ARPE-19 cells was performed with eight replicates per dilution in 96-well plates. Titration results were converted to PFU/ml using median tissue culture infectious dose (TCID-50) (Reed, 1938). The detection limit shown is the single-well detection limit. To measure HSV-1 yield reduction due to open-loop lethality, Vero cells were nucleofected with FD175 or FDScram at 25 μM (or mock). Then 24 h later, the cells were infected with a HSV-1 IE175-YFP (17syn+ strain) at increasing MOIs (0.1, 0.5, 1.0, 2.0), and virus was titered at 4 dpi. To quantify ferroptosis induction in HSV-1 infected cells, Vero cells were nucleofected with FD175 or FDScram at 25 μM (or mock). Then 24 h later, the cells were infected with HSV-1 IE175-YFP (17syn+ strain) at MOI=1. At 1 dpi, cells were harvested and stained with a ferroptosis marker (BODIPY C11), followed by flow cytometry. See also Figures 3F–3J.
UL38 overexpression
MRC-5 cells were nucleofected with 2 μg of UL38 expression plasmid (kindly provided by Scott S. Terhune, (Terhune et al., 2007)) or mock 24 h before infection followed by transfection of 25μM of FD86 or FDScram (using lipofectamine 3000) at 12 h before infection. Cells were infected with AD169 virus (MOI=0.1) and harvested at 4 dpi to quantify UL38 expression and virus titer. For UL38 expression quantification, cells were lysed in TRIzol LS (cat#10296010, Invitrogen), using 0.75ml TRIzol LS per 1 million cells. RNA was extracted using the Direct-zol RNA extraction kit (cat#R2070T, Zymo Research Inc.), DNAse treated using RNAse-free DNAse I (cat#EN0521, Thermofisher Scientific), reverse transcribed using SuperScript II Reverse transcriptase using oligo d(T) primers (cat#12574026, Thermofisher Scientific), and cDNA was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR) using SYBR green PCR master mix (cat#4309155, Thermofisher Scientific) with primers specific to UL38. For quantification of virus titer, cells were subjected to three freeze-thaws followed by centrifugation at 10,000 rpm for 10 minutes, supernatant was harvested and virus titer was quantified by TCID-50.
RNA-seq analysis
Total RNA was isolated from feedback-reporter cells in biological duplicate and purified using RNAeasy RNA extraction kit, Qiagen. To simplify gene ontology analysis, cells were not pre-cultured in VPA prior to FD transfection. RNA-seq libraries were prepared with Ovation RNA-Seq System v2 kit (NuGEN, Cat #1702). Total RNA (100ng) was reverse transcribed to synthesize the first-strand cDNA using a combination of random hexamers and a poly-T chimeric primer. RNA template was partially degraded by heating and the second strand cDNA was synthesized using DNA polymerase. The double-stranded cDNA was amplified using single primer isothermal amplification (SPIA—a linear cDNA amplification process in which RNase H degrades RNA in DNA/RNA heteroduplex at the 5′-end of the double-stranded DNA, after which the SPIA primer binds to the cDNA and the polymerase starts replication at the 3′-end of the primer by displacement of the existing forward strand). Random hexamers were used to amplify the second-strand cDNA linearly. The cDNA was fragmented with the Covaris S-series System according to the manufacturer’s instructions to produce fragmented DNA with a fragment size in the desired range. Finally, libraries were generated (using seven cycles of amplification) from the SPIA amplified cDNA using the Ovation Ultralow Library System V2 (NuGEN, Cat #0344NB). RNA-seq libraries were analyzed by Bioanalyzer and quantified by qPCR (KAPA). High-throughput sequencing was performed on one lane of a HiSeq 4000, single-read 50bp (Illumina) and completed libraries were sequenced on a NOVAseq 4000. Sequencing reads were pseudo-aligned with kallisto (Bray et al., 2016). Kallisto count function quantified transcript abundance for subsequent differential expression analysis with DESeq2 (Love et al., 2014) in RStudio. All genes with less than 200 total reads across all samples were filtered out. The DESeq2 results function directly compared two sample types (i.e. 28 bp DNA vs Scramble), creating a table of genes with corresponding log2 fold change in expression, p-value and adjusted p-value. Genes with an adjusted p-value less than 0.05 and log2 (fold change) either greater or less than 1 were extracted to generate lists of significantly up- and down-regulated genes and uploaded to DAVID Bioinformatics for Functional GO analysis to identify enriched biological processes (Huang da et al., 2009). Pathways were identified and ranked by fold enrichment. For follow-up functional-enrichment analysis, the package DESeq2 computed normalized counts per million (CPM) and mapped fragments from the raw count matrix using the robust median ratio method. Next, CPM values were log transformed (base 2) after adding a small number (0.01) to prevent logarithms of 0. This yielded a matrix of logCPM values for the four samples, which was clustered using hopach with cosine angle distance metric (Pollard and van der Laan, 2002) yielding nine clusters for the differential genes, whose logCPM values were plotted as a heatmap after setting the mean for each gene to zero. Enrichment analyses was performed to identify gene ontology (GO) terms for the clusters that had more than 100 genes with available Entrez IDs using the enrichGO function from the clusterProfiler package to search for biological process sub-ontologies and assessed the significance of enrichment of a GO term relative to a background set of all the genes detected (Yu et al., 2012). Terms with FDR-adjusted p<0.05 were considered to be significantly enriched.
Cell death and TLR9 analysis in feedback-reporter cells
To measure cell death caused merely by nucleofection of DNA duplexes, naive ARPE-19 cells were nucleofected with 25 μM FD86 or FDScram, then 48 hours post-nucleofection, cells were harvested, stained using Zombie Aqua, and analyzed by flow cytometry in biological triplicate. To measure differential gene expression by RNA-seq, total RNA from feedback-reporter cells was harvested at 72 hours post nucleofection in biological duplicate and processed as described in "RNA-seq analysis".
To measure cell death caused by open-loop lethality, ARPE-19 feedback-reporter cells or naïve cells were nucleofected with the FD86 or FDScram and 72 h later cells were trypsinized, stained, and analyzed by flow cytometry for apoptosis markers (Annexin V Apoptosis Detection Kit, Biolegend, Cat # 640932) or by using the TUNEL Assay Apoptosis Detection kit (Biotium, Cat #30074) according to the manufacturer’s instructions.
TLR9 expression analysis was performed on ARPE-19 cells 48h after nucleofection with FD86 or addition of 1 μM ODN 2216 (Integrated DNA Technologies, Inc) to the cells. Total cell RNA was extracted using an RNEASY RNA isolation kit (Qiagen, Cat #74104) and reverse transcribed using a QuantiTet Reverse Transcription kit (Qiagen; Cat #205311). Relative quantification of genomic DNA or cDNA was performed on a 7900HT Fast Real-Time PCR System (ThermoFisher Scientific, Cat #4329003) using sequence specific primers (Table S1) and Fast SYBR Green Master Mix (Applied Biosystems, Cat #4385612).
Western blot analysis
106 ARPE-19 cells were centrifuged at 2,000g for 10 min at 4°C and resuspended in PBS (Sigma- Aldrich). Cells were pelleted, resuspended in ice-cold RIPA lysis buffer (Sigma-Aldrich, Cat #R0278) by vortexing and incubated at 4°C for 30 min. The lysed samples were then pelleted at 13,000 rpm for 20 min at 4°C and supernatants were assayed by western blot as previously described (Vardi et al., 2018). Briefly, 20μg of cell lysate total protein was added to 1x loading buffer (100mM Tris-HCl (pH6.8), 200mM DTT, 4% SDS, 0.1% Bromophenol blue, 20% glycerol), and boiled for 10 minutes at 95°C. The lysed samples and precision plus kaleidoscope pre-stained protein marker (Bio-Rad, Cat # 1610375) were then loaded on 10% Mini-PROTEAN TGX precast protein gel (Bio-Rad) in duplicate and ran at 90V for 2 h in Tris-glycine running buffer (925mM Tris, 250mM Glycine and 0.1% SDS). One gel was stained for two h at room temperature with coomassie blue stain (1g coomassie brilliant blue (Bio-Rad), Methanol (50%[v/v]), Glacial acetic acid (10%[v/v]), final volume to 1L with Milli-Q H2O, followed by destaining (40% methanol, 7% acetic acid in final volume of 1L in Milli-Q H2O. The gel was blotted on a PVDF membrane using a semi-dry transfer unit (Trans-Blot Semi-Dry Electrophotetic transfer cell, Bio-Rad) at 25V for 45 minutes. The membrane was blocked with Odyssey blocking buffer (LI-COR Biosciences, Cat #927) for 2 h at room temperature with gentle agitation. IE86 protein was detected by a 2 h incubation of the membrane with an anti-IE86 antibody (1:100 dilution, Millipore, Cat #MAB810, RRID:AB_95254) in Odyssey blocking buffer at room temperature followed by three 5-minute wash steps (1x PBS + 0.01% Tween-20). The membrane was then incubated with a IRDye 800CW Goat anti-Mouse IgG Secondary Antibody (1:20,000 dilution, LI-COR Biosciences Cat #926-32210, RRID:AB_621842) for 1 h in the dark, washed three times in wash buffer (1x PBS + 0.01% Tween-20) and imaged on an Odyssey system (LI-COR Biosciences).
Herpesvirus qPCR, flow cytometry and fluorescence microscopy
Genomic DNA from CMV or HSV-1 infected ARPE-19 cells, or mouse corneal tissue dissociated with collagenase I for 30 minutes, was extracted using DNeasy Blood and tissue kit (Qiagen, Cat #69504). Flow cytometry was performed on BD FACS-Calibur (BD, Biosciences) at the Gladstone Flow Cytometry Core and analyzed using FlowJo™ (FlowJo, LLC). Imaging of CMV and HSV-1 cell entry was performed as follows: HFFs and Vero cells grown on glass coverslips and infected with CMV TB40E IE86-YFP (MOI=5 imaged at 2h post-infection) or HSV-1 17syn+ IE175-YFP (MOI=20 imaged at 1h post-infection), respectively. Cells were fixed with 4% formaldehyde, permeabilized with 0.1% Triton-X100 for 15min and blocked in normal goat serum for 1h. HFFs were incubated with a mouse anti-pp65 primary antibody (Nowak et al., 1984) (1:200 dilution, clone 8F5, gift from the Shenk lab) while Vero cells were incubated with a mouse anti-ICP5 primary antibody (1:200, Abcam, Cat #ab6508, RRID:AB_305530). HFFs and Vero cells were then incubated with a goat anti-mouse Alexa 594 secondary antibody (1:250, Thermo Fisher Scientific, Cat #A-11005, RRID:AB_2534073), mounted and imaged on a Zeiss Axiovert inverted fluorescence microscope (Carl Zeiss) with a 100X oil-immersion objective.
For analysis of cell-death pathways, cells were infected with respective virus at MOI=0.1, at 48 hpi, cells were trypsinized, stained with Zombie Aqua Fixable Viability kit (BioLegend, Cat # 423101), and subjected to flow cytometry. Cells were stained for apoptosis marker (Annexin V Apoptosis Detection Kit, Biolegend, Cat #640932, cells harvested at 48 hpi) and ferroptosis markers (BODIPY 581/591 C11, Thermofisher Scientific, Cat #D3861, cells harvested at 24 hpi) and subjected to flow cytometry. Analysis of cell-death pathways was performed using cell-death pathway inhibitors and staining for death-pathway markers. Briefly, cells were nucleofected with the FD86, FD175, or FDScram (25μM each) or mock nucleofected in the presence of one of the following inhibitors: auto= autophagy inhibitor, 3-methyladenine authophagy inhibitor, Sigma-Aldrich, Cat #M9281; apo= apoptosis inhibitor, NLRP3 inhibitor, CAS 256373-96-3, Sigma-Aldrich, Cat # 5381200001); fer= ferroptosis inhibitor, Ferrostatin-1, Sigma-Aldrich, Cat #SML0583; nec=necroptosis inhibitor, Necrostatin-1, Sigma-Aldrich, Cat #4311-88-0).
Continuous culture
For the CMV continuous cultures, ARPE-19 or MRC5 cells were nucleofected with 25 μM FD86 or mock nucleofected and at 24 h post-nucleofection, infected with either CMV TB40E-IE86-YFP, CMV AD169 IE2-YFP, or CMV Δcrs IE2-YFP virus (Teng et al., 2012) at an MOI=0.1. Then, at 4 dpi, and every 4 days thereafter until day 60, the culture supernatant was collected and transferred to naïve ARPE-19 cells +/- FD86. For dose-escalation studies, naïve cells not exposed to FD86 were infected and increasing FD86 doses delivered every 4 days according the following dose scheme: 0.1 nM, 0.2 nM, 0.5 nM, 0.75 nM, 1 nM, 10 nM, 100 nM, 1 μM, 5 μM, 10 μM, 25 μM. For Fomivirsen analysis, 25 μM Fomivirsen was added at 1 hpi. Supernatants were also titered on naïve ARPE-19 cells by TCID-50 as described above. For the HSV-1 continuous culture: ARPE-19 cells were nucleofected with 25 μM FD175 or mock nucleofected and then infected with HSV-1 (17syn+ IE175-YFP) (MOI=0.1). At 4 dpi and then every 4 days thereafter until day 40, supernatants were transferred to infect naïve ARPE-19 cells (+/- FD175). Supernatants were also used for TCID-50 analysis on naïve ARPE-19 cells as described above. To assay for the putative reduced-fitness viral variant, continuous culture supernatants were collected on days 16, 32 and 48 and used to infect ARPE-19 cells in the presence/absence of FD86, and supernatants were collected for titering on naive ARPE-19 cells at 4 dpi.
Bystander assays
ARPE-19 cells were nucleofected with FD86, then 24h later infected with CMV (TB40E-IE86-YFP). At 2 dpi, prior to release of progeny virus, cells were co-cultured with naïve mCherry-labeled cells (‘bystanders’). At 36h after co-culture, cells were harvested and analyzed by flow cytometry. This timeline was chosen to ensure sufficient time for FD-mediated cytotoxicity to manifest but be shorter than the 96-hour replication cycle for virus release so as not to be confounded by 2nd round virus infection.
Combinatorial therapeutic approaches and multiplexed feedback disruption
To demonstrate combinatorial effects of FD86 with the standard-of-care CMV antiviral ganciclovir (GCV), HFF cells were nucleofected with 25 μM FD86, FDScram, or mock. Then, cells were either cultured with 10 μM GCV alone or in combination with FD86 nucleofection, and infected with TB40E-IE86-YFP. Virus was titered at 4 dpi. To demonstrate combinatorial effects of FD175 with the standard-of-care HSV-1 antiviral acyclovir (ACV), Vero cells were nucleofected with 25 μM FD175, FDScram, or mock. Then, cells were either cultured with 10 μM ACV alone or in combination with FD175 nucleofection, and infected with HSV-1 (17syn+ IE175-YFP). Virus was titered at 2 dpi.
To test the efficacy of multiplexed feedback disruption in a mixed-infection setting, ARPE-19 cells were nucleofected with equimolar amounts of FD86 and FD175 or with FDScram, then were co-infected with CMV (TB40E-IE86-YFP) and HSV-1 (17syn+ IE175-YFP) at MOI=0.1. After 4 dpi, DNA was harvested and qPCR for CMV and HSV-1 viral genomes was performed using primers specific for CMV and HSV-1.
HSV-1 corneal infection assays in mice
All experiments were performed with 6- to 10-week-old male and female sibling Black 6 mice. Breeding pairs were purchased from The Jackson Laboratory (C57BL/6J mice, Stock No: 000664) and maintained under pathogen-free conditions in the UCSF barrier facility. All animal experiments were conducted in accordance with procedures approved by the UCSF Institutional Animal Care and Use Committee. Corneal epithelial debridement was performed on mice as previously described (Chan and Werb, 2015). Briefly, mice were anesthetized by isofluorane inhalation (Abbott Laboratories). The central part of the epithelium was removed down to the basement membrane using an Algerbrush II (Katena Products, Inc., Cat # K2-4900). 5 μl of HSV-1 17syn+ YFP-IE175 (105 PFU) were immediately applied to the debrided cornea. 6 h later, mice were anesthetized again and 5 μl of 25 μM FD175, FDScram or PBS was topically applied to the cornea for 5 min. At 2 dpi, eyes were enucleated and the corneas were dissected to remove the lens, iris, and retina. Four incisions were made equal distances apart to aid in flattening the corneas. Fresh corneas were counterstained using 0.5 μg/ml DAPI, mounted on slides with Fluoro-gel (Electron Microscopy Sciences, Cat #17985-10) and imaged on a Zeiss Axiovert Confocal microscope. Fluorescent microscopy of corneas infected with HSV-1 was performed on a Zeiss Axiovert fluorescent microscope for DAPI (excitation at 345nm) and YFP (excitation at 514 nm). Images were analyzed using ImageJ software (Schneider et al., 2012). Image quantification of YFP was based on a dual-thresholding for DAPI to generate image masks (first a size thresholding for DAPI signal >10 μm2 then an intensity threshold for DAPI intensity > 100 a.u.). YFP intensity was then quantified within the mask, for YFP particles > 2 μm2. Corneas were dissociated using collagenase I (Sigma-Aldrich, St Louis, MO), total DNA was extracted using DNeasy Blood & Tissue kit (Qiagen, Cat #69504), and subjected to qPCR using Fast SYBR green master mix (Applied Biosystems, Cat #4385612), analyzed on a 7900HT Fast Real-Time PCR System (Thermofisher Scientific, Cat #4329003). Titration of virus in corneas was performed by dissociating corneal tissues using collagenase I (Sigma-Aldrich, Cat #C0130) for 30 minutes, subjecting them to three freeze-thaw cycles and using them for TCID-50 analysis.
mCMV systemic infection assays in mice
All animal experiments were conducted in accordance with procedures approved by the UCSF Institutional Animal Care and Use Committee. 6-week old male BALB/c mice (Jackson Laboratory) were maintained under pathogen-free conditions in the UCSF barrier facility until they were 28- to 30-week-old, followed by infection and treatment as follows. FD nanoparticles were formulated as previously described (Zhang et al., 2012). Mice were administered 500 μl of FD88 nanoparticle formulation (final concentration 100 μM) (n=5) or vehicle control (n=5) via intraperitoneal (IP) injection at 3 h pre- and 18 h post-infection. Mice were infected with 105 PFU of mCMV (Smith Strain) via IP route, weighed at day 0 and 5, and liver and spleen harvested on day 5 and homogenized. Total DNA was isolated using PCI method, and RNA was extracted using Trizol and Zymo direct-zol RNA extraction kit. Infectious virus titer was quantified by TCID-50. To quantify relative mCMV expression levels, total RNA was analyzed by qRT-PCR using primers for GpB (glycoprotein B) and IE88 normalized to beta-actin. To quantify mCMV replication, genomic copy number was assayed by qPCR analysis of genomic DNA using primers for IE88 and normalized to beta-actin.
SARS-CoV-2 analyses
All live virus experiments were performed at the Gladstone Institutes in a Biosafety Level 3 (BSL3) containment facility under an approved Biosafety Use Authorization from UCSF and in compliance with UCSF guidelines and procedures. Vero-E6 cells were nucleofected with 25 μM FDTRS or FDScram, at 24-h post transfection, cells were infected with SARS-CoV-2 (WA-1 strain) at MOI=0.05. Cells were harvested in Trizol LS (cat# 10296010, Invitrogen) or in 10% formalin at 8 h, 12 h, 24 h and 48 h post infection. RNA was extracted from Trizol treated cells using Direct-zol RNA extraction kit (cat#R2070T, Zymo Research Inc.), DNAse treated using RNAse free DNase-1 (cat#EN0521, Thermofisher Scientific), reverse transcribed for cDNA preparation using SuperScript II Reverse Transcriptase with oligo d(T) primers (cat#12574026, Thermofisher Scientific), and analyzed by quantitative real-time polymerase chain reaction (qRT-PCR) analysis using SYBR green PCR master mix (cat#4309155, Thermofisher Scientific) with sequence specific primers. All qRT-PCR samples were normalized using beta-actin primers. Cells were harvested in formalin, subjected to TUNEL assay and analyzed by flow cytometry. Infectious SARS-CoV-2 titers were quantified by plaque assay on Vero-E6 cells as described previously (Chaturvedi et al., 2021). Briefly, Vero-E6 cells were plated as a confluent monolayer in 12-well plates 24h before performing the plaque assay, on the day of plaque assay, media was aspirated, cells were layered with 250 μl of diluted virus in modified DMEM media (DMEM, 2% FBS, L-glut, P/S), incubated for 1h at 37°C and gently rocked every 15 minutes. After incubation, 2ml of overlay media (1.2% Avicel in 1XMEM) was added to each well. At 3 dpi, overlay media was aspirated, monolayer was washed with PBS, fixed with 10% formalin for 1 h, and stained with 0.1% crystal violet for 10 minutes, and washed with cell culture grade water. Plaques were manually counted to calculate PFU/ml.
Quantification and statistical analysis
Statistical differences were determined by using the two-tailed unpaired Student's t test, and 1-way/2-way ANOVA followed by Tukey’s multiple comparisons test (GraphPad Prism), unless otherwise noted. A p-value less than 0.05 was considered statistically significant: ∗<0.05, ∗∗<0.01, ∗∗∗<0.001, ∗∗∗∗<0.0001, ns: not significant. Statistical details of experiments can be found in the Figure Legends and results section.
Acknowledgments
We dedicate the manuscript to the memory of Cynthia Bolovan-Fritts, who made this work possible. We thank L. Lanier, P. Barry, R. Everett, and S. Terhune for providing mCMV strain K181, RhCMV 68.1 EGFP, HSV-1 17syn+ IE175-YFP, and UL38 expression plasmid. We thank R. Rodick, E. Ingerman, M. Ott, J.J. Miranda, S. Zheng, M. Cavrois, N. Raman, and the Weinberger lab for discussions and K. Claiborn for reviewing the manuscript. We acknowledge Gladstone Flow Cytometry Core, funded through NIH P30 AI027763 and the Pendelton Charitable Trust. M.F.C. acknowledges NIH (R01 EY022739, NIH-NEI EY002162), Research to Prevent Blindness, and That Man May See Research Grant. Portions of this work were done under the auspices of the US DOE under contract 89233218CNA000001 and supported by NIH grants R01-OD001095 and R01-AI028433 (to A.S.P.). This work was supported by the Bowes Distinguished Professorship, the Pew Scholars in the Biomedical Sciences Program, NIH Director’s New Innovator Award OD006677 and Pioneer Award OD17181 programs (to L.S.W.).
Author contributions
S.C. and L.S.W. conceived and designed the study. S.C., M.P., M.W., D.R.R., G.C., A.J.K., K.D., J.G., and M.F.C., designed and performed the experiments and curated the data. M.P., N.V., L.S.W., R.K., and A.S.P. performed the mathematical modeling. S.C., M.P., M.W., and L.S.W. wrote the paper.
Declaration of interests
Competing interests: L.S.W. is an inventor of “Compositions and methods of use thereof for identifying antiviral agents” (US patent no. US10106817B2). L.S.W. and S.C. are inventors of “Compositions for herpesvirus transcriptional feedback circuit disruption and uses thereof” (US patent no. US2021/0395746A1).
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science and one or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: May 12, 2022
Corrected online: April 3, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.04.022.
Supplemental information
Extended FD86 treatment over 60 days does not appear to generate dominant mutations. Sanger sequencing of IE86 (Exon 5) from virus in the continuous culture experiment in the presence of FD86 (Figure 5B). Viral DNA was extracted, IE86 exon 5 was PCR amplified (see Table S1 for primer sequences), Sanger sequenced, and aligned with the IE86 sequence for homology analysis. Despite apparent transient SNPs (e.g., at nucleotide position 640), no mutations “sweep” the population or are maintained in the consensus sequence over time.
Sequencing analysis of the CMV promoter region from the continuous culture experiment in the presence of FD86 (Figure 5B). Viral DNA was extracted and 250bp upstream and downstream of the crs was amplified by PCR (see Table S1 for primer sequences), Sanger sequenced, and then aligned with the viral promoter sequence for homology analysis. No acquired consensus mutations are observed over 14 replication cycles.
Sequencing analysis of IE86 (Exon 5) using virus obtained by culturing FD86-treated continuous culture supernatants (days 16, 32, and 48) in the presence or absence of FD86 (Figure S5C). Viral DNA was extracted, IE86 exon 5 was PCR amplified (see Table S1 for primer sequences), Sanger sequenced, and aligned with the IE86 sequence for homology analysis. No acquired consensus mutations are observed.
Sequencing analysis of the CMV promoter region using virus obtained by culturing FD86-treated continuous culture supernatants (days 16, 32, and 48) in the presence or absence of FD86 (Figure S5C). Viral DNA was extracted and 250-bp upstream and downstream of the crs was amplified by PCR (see Table S1 for primer sequences), Sanger sequenced and aligned with the viral promoter sequence for homology analysis. No acquired consensus mutations are observed.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. Complete model details for simulations are provided in this paper, and this paper does not report original code.
References
- Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.-C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- Alon U. Chapman & Hall/CRC; 2007. An introduction to systems biology: Design principles of biological circuits. [Google Scholar]
- Alon U., Surette M.G., Barkai N., Leibler S. Robustness in bacterial chemotaxis. Nature. 1999;397:168–171. doi: 10.1038/16483. [DOI] [PubMed] [Google Scholar]
- Aoki S.K., Lillacci G., Gupta A., Baumschlager A., Schweingruber D., Khammash M. A universal biomolecular integral feedback controller for robust perfect adaptation. Nature. 2019;570:533–537. doi: 10.1038/s41586-019-1321-1. [DOI] [PubMed] [Google Scholar]
- Arago, M. (1840). History of the steam engine, with a reply to the Criticisms to which the first publication of the article gave rise. Blasco de garay 1615, Solomon de Caus 1629. Sir Samuel Morland. Branca 1663. The Marquis of Worcester 1683. Denis Papin 1698, A.A. Lieut, and U.S.N. Harwood, translators 120 years B. C. Hiero of Alexandria 1543, p. 1690 and 1695. Captain Savery. (1705). Newcomen, Cawley, and Savery. Journal of the Franklin Institute, of the State of Pennsylvania, for the Promotion of the Mechanic Arts; Devoted to Mechanical and Physical Science, Civil Engineering, the Arts and Manufactures, and the Recording of American and Other Patent Inventions (1828-1851) 25, 3.
- Åsberg A., Humar A., Rollag H., Jardine A.G., Kumar D., Aukrust P., Ueland T., Bignamini A.A., Hartmann A. Lessons learned from a randomized study of oral valganciclovir versus parenteral ganciclovir treatment of cytomegalovirus disease in solid organ transplant recipients: The VICTOR trial. Clin. Infect. Dis. 2016;62:1154–1160. doi: 10.1093/cid/ciw084. [DOI] [PubMed] [Google Scholar]
- Asmar J., Wiebusch L., Truss M., Hagemeier C. The putative zinc finger of the human cytomegalovirus IE2 86-kilodalton protein is dispensable for DNA binding and autorepression, thereby demarcating a concise core domain in the C terminus of the protein. J. Virol. 2004;78:11853–11864. doi: 10.1128/JVI.78.21.11853-11864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin D.W., Allen M.S., McCollum J.M., Dar R.D., Wilgus J.R., Sayler G.S., Samatova N.F., Cox C.D., Simpson M.L. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–611. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- Bayer A., Brennan G., Geballe A.P. Adaptation by copy number variation in monopartite viruses. Curr. Opin. Virol. 2018;33:7–12. doi: 10.1016/j.coviro.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becskei A., Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Biron K.K., Fyfe J.A., Stanat S.C., Leslie L.K., Sorrell J.B., Lambe C.U., Coen D.M. A human cytomegalovirus mutant resistant to the nucleoside analog 9-([2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine (BW B759U) induces reduced levels of BW B759U triphosphate. Proc. Natl. Acad. Sci. USA. 1986;83:8769–8773. doi: 10.1073/pnas.83.22.8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black H.S. Stabilized feedback amplifiers. Bell Syst. Tech. J. 1934;13:1–18. [Google Scholar]
- Bode H. Feedback: The history of an idea. Selected papers on mathematical trends in control theory. 1960:106–123. [Google Scholar]
- Bogdanove A.J., Bohm A., Miller J.C., Morgan R.D., Stoddard B.L. Engineering altered protein-DNA recognition specificity. Nucleic Acids Res. 2018;46:4845–4871. doi: 10.1093/nar/gky289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P., Pauleau A.L., Poncet D., Gonzalez-Polo R.A., Zamzami N., Kroemer G. Viral proteins targeting mitochondria: Controlling cell death. Biochim. Biophys. Acta. 2004;1659:178–189. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brown S.M., Ritchie D.A., Subak-Sharpe J.H. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J. Gen. Virol. 1973;18:329–346. doi: 10.1099/0022-1317-18-3-329. [DOI] [PubMed] [Google Scholar]
- Cauwels A., Brouckaert P. Survival of TNF toxicity: dependence on caspases and NO. Arch. Biochem. Biophys. 2007;462:132–139. doi: 10.1016/j.abb.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Chan M.F., Werb Z. Animal models of corneal injury. Bio Protoc. 2015;5:e1516. doi: 10.21769/bioprotoc.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W.L., Barry P.A. Cloning of the full-length rhesus cytomegalovirus genome as an infectious and self-excisable bacterial artificial chromosome for analysis of viral pathogenesis. J. Virol. 2003;77:5073–5083. doi: 10.1128/JVI.77.9.5073-5083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi S., Engel R., Weinberger L. The HSV-1 ICP4 transcriptional auto-repression circuit functions as a transcriptional ‘accelerator’circuit. Front. Cell. Infect. Microbiol. 2020;10:265. doi: 10.3389/fcimb.2020.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi S., Vasen G., Pablo M., Chen X., Beutler N., Kumar A., Tanner E., Illouz S., Rahgoshay D., Burnett J., et al. Identification of a therapeutic interfering particle-A single-dose SARS-CoV-2 antiviral intervention with a high barrier to resistance. Cell. 2021;184:6022–6036. doi: 10.1016/j.cell.2021.11.004. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihlar T., Fuller M.D., Mulato A.S., Cherrington J.M. A point mutation in the human cytomegalovirus DNA polymerase gene selected in vitro by cidofovir confers a slow replication phenotype in cell culture. Virology. 1998;248:382–393. doi: 10.1006/viro.1998.9299. [DOI] [PubMed] [Google Scholar]
- Coen D.M., Schaffer P.A. Two distinct loci confer resistance to acycloguanosine in herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA. 1980;77:2265–2269. doi: 10.1073/pnas.77.4.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- Cudini J., Roy S., Houldcroft C.J., Bryant J.M., Depledge D.P., Tutill H., Veys P., Williams R., Worth A.J.J., Tamuri A.U., et al. Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. Proc. Natl. Acad. Sci. USA. 2019;116:5693–5698. doi: 10.1073/pnas.1818130116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham E.T. Cytomegalovirus: Ophthalmic perspectives on a pervasive pathogen. Expert Rev. Ophthalmol. 2011;6:489–491. [Google Scholar]
- Diekmann O., Heesterbeek J.A., Roberts M.G. The construction of next-generation matrices for compartmental epidemic models. J. R. Soc. Interface. 2010;7:873–885. doi: 10.1098/rsif.2009.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA. 1991;88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J.W., Hwang C.B. On the mutation rate of herpes simplex virus type 1. Genetics. 2005;170:969–970. doi: 10.1534/genetics.104.040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia, M.H., J.J., and Gaudio, P.A. (2016). Cytomegalovirus anterior uveitis in immunocompetent patients. In EyeNet Magazine, pp, 37–38.
- Emery V.C., Cope A.V., Bowen E.F., Gor D., Griffiths P.D. The dynamics of human cytomegalovirus replication in vivo. J. Exp. Med. 1999;190:177–182. doi: 10.1084/jem.190.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery V.C., Griffiths P.D. Prediction of cytomegalovirus load and resistance patterns after antiviral chemotherapy. Proc. Natl. Acad. Sci. USA. 2000;97:8039–8044. doi: 10.1073/pnas.140123497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery V.C., Hassan-Walker A.F., Burroughs A.K., Griffiths P.D. Human cytomegalovirus (HCMV) replication dynamics in HCMV-naive and -experienced immunocompromised hosts. J. Infect. Dis. 2002;185:1723–1728. doi: 10.1086/340653. [DOI] [PubMed] [Google Scholar]
- Enquist L.W., Leib D.A. Intrinsic and innate defenses of neurons: detente with the herpesviruses. J. Virol. 2017;91:e01200. doi: 10.1128/JVI.01200-16. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandson K.J., Cotter C.A., Charity J.C., Martens C., Fischer E.R., Ricklefs S.M., Porcella S.F., Moss B. Duplication of the A17L locus of vaccinia virus provides an alternate route to rifampin resistance. J. Virol. 2014;88:11576–11585. doi: 10.1128/JVI.00618-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D., Sourvinos G., Orr A. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 2003;77:3680–3689. doi: 10.1128/JVI.77.6.3680-3689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell J.E., Jr. Feedback loops and reciprocal regulation: Recurring motifs in the systems biology of the cell cycle. Curr. Opin. Cell Biol. 2013;25:676–686. doi: 10.1016/j.ceb.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frobert E., Burrel S., Ducastelle-Lepretre S., Billaud G., Ader F., Casalegno J.S., Nave V., Boutolleau D., Michallet M., Lina B., Morfin F. Resistance of herpes simplex viruses to acyclovir: An update from a ten-year survey in France. Antiviral Res. 2014;111:36–41. doi: 10.1016/j.antiviral.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Fu X., Wang H., Zhang X. High-frequency intermolecular homologous recombination during herpes simplex virus-mediated plasmid DNA replication. J. Virol. 2002;76:5866–5874. doi: 10.1128/JVI.76.12.5866-5874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier M.P. Length does matter for cGAS. EMBO Rep. 2017;18:1675–1676. doi: 10.15252/embr.201744773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg D.E., Siliciano R.F., Jacobs W.R., Jr. Outwitting evolution: Fighting drug-resistant TB, malaria, and HIV. Cell. 2012;148:1271–1283. doi: 10.1016/j.cell.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj J., Dahari H., Rong L., Sansone N.D., Nettles R.E., Cotler S.J., Layden T.J., Uprichard S.L., Perelson A.S. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc. Natl. Acad. Sci. USA. 2013;110:3991–3996. doi: 10.1073/pnas.1203110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S.L., Levine A.J. The p53 pathway: Positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Hecker M., Wagner A.H. Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 2017;144:29–34. doi: 10.1016/j.bcp.2017.06.122. [DOI] [PubMed] [Google Scholar]
- Henninger J.E., Oksuz O., Shrinivas K., Sagi I., LeRoy G., Zheng M.M., Andrews J.O., Zamudio A.V., Lazaris C., Hannett N.M., et al. RNA-mediated feedback control of transcriptional condensates. Cell. 2021;184:207–225.e24. doi: 10.1016/j.cell.2020.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann E., Sidler D., Dahdal S., Bittel P., Suter-Riniker F., Manuel O., Walti L.N., Hirzel C. Emergence of letermovir resistance in solid organ transplant recipients with ganciclovir resistant cytomegalovirus infection: A case series and review of the literature. Transpl Infect. Disorders. 2020;23:e13515. doi: 10.1111/tid.13515. [DOI] [PubMed] [Google Scholar]
- Hong S.H., Lee D.H., Lee Y.S., Jo M.J., Jeong Y.A., Kwon W.T., Choudry H.A., Bartlett D.L., Lee Y.J. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget. 2017;8:115164–115178. doi: 10.18632/oncotarget.23046. 29383150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshangi S., Weiss R. The effect of negative feedback on noise propagation in transcriptional gene networks. Chaos. 2006;16:026108. doi: 10.1063/1.2208927. [DOI] [PubMed] [Google Scholar]
- Hoyos M., Huber M., Förstner K.U., Papenfort K. Gene autoregulation by 3′ UTR-derived bacterial small RNAs. eLife. 2020;9:e58836. doi: 10.7554/eLife.58836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura H., Stinski M.F., Kudoh A., Nakayama S., Murata T., Sato Y., Iwahori S., Tsurumi T. A cis element between the TATA Box and the transcription start site of the major immediate-early promoter of human cytomegalovirus determines efficiency of viral replication. J. Virol. 2008;82:849–858. doi: 10.1128/JVI.01593-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A.L., Loeb L.A. The mutation rate and cancer. Genetics. 1998;148:1483–1490. doi: 10.1093/genetics/148.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F., Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- Kenny E.J.A. The date of Ctesibius. The Class. Q. 1932;26:190–192. [Google Scholar]
- Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchoff V., Wong S., St J.S., Pari G.S. Generation of a life-expanded rhesus monkey fibroblast cell line for the growth of rhesus rhadinovirus (RRV) Arch Virol. 2002;147:321–333. doi: 10.1007/s705-002-8322-9. [DOI] [PubMed] [Google Scholar]
- Krug A., Rothenfusser S., Hornung V., Jahrsdörfer B., Blackwell S., Ballas Z.K., Endres S., Krieg A.M., Hartmann G. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur. J. Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Lahmidi S., Yousefi M., Dridi S., Duplay P., Pearson A. Dok-1 and Dok-2 are required to maintain herpes simplex virus 1-specific CD8(+) T cells in a murine model of ocular infection. J. Virol. 2017;91 doi: 10.1128/JVI.02297-16. e02297-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.H., Molla M.N., Cantor C.R., Collins J.J. Bacterial charity work leads to population-wide resistance. Nature. 2010;467:82–85. doi: 10.1038/nature09354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Li J., Wang P.H., Yang N., Huang J., Ou J., Xu T., Zhao X., Liu T., Huang X., et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2021;1867:166260. doi: 10.1016/j.bbadis.2021.166260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Cheng Z., Wang F., Chang J., Zhao Q., Zhou H., Liu C., Ruan J., Duan G., Gao S. A negative feedback model to explain regulation of SARS-CoV-2 replication and transcription. Front. Genet. 2021;12:641445. doi: 10.3389/fgene.2021.641445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Hermiston T.W., Stinski M.F. A cis-acting element in the major immediate-early (IE) promoter of human cytomegalovirus is required for negative regulation by IE2. J. Virol. 1991;65:897–903. doi: 10.1128/jvi.65.2.897-903.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo D.M., Selimkhanov J., Kearns J.D., Hasty J., Hoffmann A., Tsimring L.S. Dual delayed feedback provides sensitivity and robustness to the NF-kappaB signaling module. PLOS Comput. Biol. 2013;9:e1003112. doi: 10.1371/journal.pcbi.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q., Hwang Y.T., Hwang C.B. Detection of mutations within the thymidine kinase gene of herpes simplex virus type 1 by denaturing gradient gel electrophoresis. J. Virol. Methods. 2002;99:1–7. doi: 10.1016/s0166-0934(01)00388-3. [DOI] [PubMed] [Google Scholar]
- Lu Q., Hwang Y.T., Hwang C.B. Mutation spectra of herpes simplex virus type 1 thymidine kinase mutants. J. Virol. 2002;76:5822–5828. doi: 10.1128/JVI.76.11.5822-5828.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke S., Holleufer A., Christensen M.H., Jønsson K.L., Boni G.A., Sørensen L.K., Johannsen M., Jakobsen M.R., Hartmann R., Paludan S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18:1707–1715. doi: 10.15252/embr.201744017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac D.M., Manuppello J.R., Alwine J.C. Transcriptional activation by the human cytomegalovirus immediate-early proteins: Requirements for simple promoter structures and interactions with multiple components of the transcription complex. J. Virol. 1994;68:5184–5193. doi: 10.1128/jvi.68.8.5184-5193.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J., Sato A., Akira S., Medzhitov R., Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurain N.S., Chou S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010;23:689–712. doi: 10.1128/CMR.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurain N.S., Spafford L.E., Thompson K.D. Mutation in the UL97 open reading frame of human cytomegalovirus strains resistant to ganciclovir. J. Virol. 1994;68:4427–4431. doi: 10.1128/jvi.68.7.4427-4431.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria S.E., Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias M.P., Stinski M.F. An in vitro system for human cytomegalovirus immediate early 2 protein (IE2)-mediated site-dependent repression of transcription and direct binding of IE2 to the major immediate early promoter. Proc. Natl. Acad. Sci. U. S. A. 1993;90:707–711. doi: 10.1073/pnas.90.2.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade N., King M., Munger J., Walsh D. mTOR dysregulation by vaccinia virus F17 controls multiple processes with varying roles in infection. J. Virol. 2019;93 doi: 10.1128/JVI.00784-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan S., Andrews I.W., Collins J.J. Targeting antibiotic tolerance, pathogen by pathogen. Cell. 2018;172:1228–1238. doi: 10.1016/j.cell.2018.01.037. [DOI] [PubMed] [Google Scholar]
- Mocarski E.S., Shenk T., Pass R.F. In: Fields’ virology. Knipe D.M., editor. Lippincott Williams & Wilkins; 2006. Cytomegaloviruses; pp. 2708–2772. [Google Scholar]
- Mohri H., Perelson A.S., Tung K., Ribeiro R.M., Ramratnam B., Markowitz M., Kost R., Hurley A., Weinberger L., Cesar D., et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 2001;194:1277–1287. doi: 10.1084/jem.194.9.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley P.J., Ertl P., Sweet C. Immunisation of Balb/c mice with severely attenuated murine cytomegalovirus mutants induces protective cellular and humoral immunity. J. Med. Virol. 2002;67:187–199. doi: 10.1002/jmv.2207. [DOI] [PubMed] [Google Scholar]
- Mulamba G.B., Hu A., Azad R.F., Anderson K.P., Coen D.M. Human cytomegalovirus mutant with sequence-dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922) Antimicrob. Agents Chemother. 1998;42:971–973. doi: 10.1128/aac.42.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevozhay D., Adams R.M., Murphy K.F., Josic K., Balázsi G. Negative autoregulation linearizes the dose-response and suppresses the heterogeneity of gene expression. Proc. Natl. Acad. Sci. USA. 2009;106:5123–5128. doi: 10.1073/pnas.0809901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M., Wang J., Wakata A., Sakamoto K., Mori Y. Crystal structure of the DNA-binding domain of human herpesvirus 6A immediate early Protein 2. J. Virol. 2017;91 doi: 10.1128/JVI.01121-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak B., Sullivan C., Sarnow P., Thomas R., Bricout F., Nicolas J.C., Fleckenstein B., Levine A.J. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology. 1984;132:325–338. doi: 10.1016/0042-6822(84)90039-4. [DOI] [PubMed] [Google Scholar]
- Nyquist H. Regeneration theory. Bell Syst. Tech. J. 1932;11:126–147. [Google Scholar]
- Paterson T., Everett R.D. The regions of the herpes simplex virus type 1 immediate early protein Vmw175 required for site specific DNA binding closely correspond to those involved in transcriptional regulation. Nucleic Acids Res. 1988;16:11005–11025. doi: 10.1093/nar/16.23.11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelson A.S. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2002;2:28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- Perez J.C., Groisman E.A. Evolution of transcriptional regulatory circuits in bacteria. Cell. 2009;138:233–244. doi: 10.1016/j.cell.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret J., Boivin G. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev. Med. Virol. 2014;24:186–218. doi: 10.1002/rmv.1787. [DOI] [PubMed] [Google Scholar]
- Pollard K.S., van der Laan M.J. Statistical inference for simultaneous clustering of gene expression data. Math. Biosci. 2002;176:99–121. doi: 10.1016/s0025-5564(01)00116-x. [DOI] [PubMed] [Google Scholar]
- Ramratnam B., Bonhoeffer S., Binley J., Hurley A., Zhang L., Mittler J.E., Markowitz M., Moore J.P., Perelson A.S., Ho D.D. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- Reed, L.J.M., and H. (1938). A simple method of estimating fifty percent endpoints. J. Epidemiol. 27, 493-497.
- Renzette N., Pokalyuk C., Gibson L., Bhattacharjee B., Schleiss M.R., Hamprecht K., Yamamoto A.Y., Mussi-Pinhata M.M., Britt W.J., Jensen J.D., Kowalik T.F. Limits and patterns of cytomegalovirus genomic diversity in humans. Proc. Natl. Acad. Sci. USA. 2015;112:E4120–E4128. doi: 10.1073/pnas.1501880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro R.M., Bonhoeffer S. Production of resistant HIV mutants during antiretroviral therapy. Proc. Natl. Acad. Sci. USA. 2000;97:7681–7686. doi: 10.1073/pnas.97.14.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G.P., Schrier R.D., Oldstone M.B. Cytomegalovirus infects human lymphocytes and monocytes: Virus expression is restricted to immediate-early gene products. Proc. Natl. Acad. Sci. USA. 1984;81:6134–6138. doi: 10.1073/pnas.81.19.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J., Rummel C., Barth S.W., Gerstberger R., Hübschle T. Molecular aspects of fever and hyperthermia. Neurol. Clin. 2006;24:421–439. doi: 10.1016/j.ncl.2006.03.004. v, v. [DOI] [PubMed] [Google Scholar]
- Savageau M.A. Comparison of classical and autogenous systems of regulation in inducible operons. Nature. 1974;252:546–549. doi: 10.1038/252546a0. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnipper L.E., Crumpacker C.S. Resistance of herpes simplex virus to acycloguanosine: Role of viral thymidine kinase and DNA polymerase loci. Proc. Natl. Acad. Sci. USA. 1980;77:2270–2273. doi: 10.1073/pnas.77.4.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen M., Thomas S.M., Kim S., Yeh J.I., Ferris R.L., Johnson J.T., Duvvuri U., Lee J., Sahu N., Joyce S., et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen M., Tosca P.J., Zwayer C., Ryan M.J., Johnson J.D., Knostman K.A., Giclas P.C., Peggins J.O., Tomaszewski J.E., McMurray T.P., et al. Lack of toxicity of a STAT3 decoy oligonucleotide. Cancer Chemother. Pharmacol. 2009;63:983–995. doi: 10.1007/s00280-008-0823-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P.P., Tanowitz M., Bennett C.F. Selective tissue targeting of synthetic nucleic acid drugs. J. Clin. Invest. 2019;129:915–925. doi: 10.1172/JCI125228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton G.D., Calcutt N.A., Garrett R.S., Gu D., Sarvetnick N., Campana W.M., Powell H.C. Necrotizing myopathy induced by overexpression of interferon-gamma in transgenic mice. Muscle Nerve. 1999;22:156–165. doi: 10.1002/(sici)1097-4598(199902)22:2<156::aid-mus3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Shenk T.E., Stinski M.F. Human cytomegalovirus. Preface. Preface. Curr. Top. Microbiol. Immunol. 2008;325:v. [PubMed] [Google Scholar]
- Sinzger C., Digel M., Jahn G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008;325:63–83. doi: 10.1007/978-3-540-77349-8_4. [DOI] [PubMed] [Google Scholar]
- Slabaugh M., Roseman N., Davis R., Mathews C. Vaccinia virus-encoded ribonucleotide reductase: sequence conservation of the gene for the small subunit and its amplification in hydroxyurea-resistant mutants. J. Virol. 1988;62:519–527. doi: 10.1128/jvi.62.2.519-527.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C.A., Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 2017;25:1069–1075. doi: 10.1016/j.ymthe.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi R., Choi M., Stoddard B.L. Redesign of extensive protein-DNA interfaces of meganucleases using iterative cycles of in vitro compartmentalization. Proc. Natl. Acad. Sci. USA. 2014;111:4061–4066. doi: 10.1073/pnas.1321030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M.W., Bolovan-Fritts C., Dar R.D., Womack A., Simpson M.L., Shenk T., Weinberger L.S. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell. 2012;151:1569–1580. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhune S., Torigoi E., Moorman N., Silva M., Qian Z., Shenk T., Yu D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007;81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiry E., Meurens F., Muylkens B., McVoy M., Gogev S., Thiry J., Vanderplasschen A., Epstein A., Keil G., Schynts F. Recombination in alphaherpesviruses. Rev. Med. Virol. 2005;15:89–103. doi: 10.1002/rmv.451. [DOI] [PubMed] [Google Scholar]
- Tigano M., Vargas D.C., Tremblay-Belzile S., Fu Y., Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature. 2021;591:477–481. doi: 10.1038/s41586-021-03269-w. [DOI] [PubMed] [Google Scholar]
- Van Antwerp D.J., Martin S.J., Kafri T., Green D.R., Verma I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Vanarsdall A.L., Johnson D.C. Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2012;2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi N., Chaturvedi S., Weinberger L.S. Feedback-mediated signal conversion promotes viral fitness. Proc. Natl. Acad. Sci. USA. 2018;115:E8803–E8810. doi: 10.1073/pnas.1802905115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger L.S., Dar R.D., Simpson M.L. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat. Genet. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- Weller S.K., Coen D.M. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 2012;4:a013011. doi: 10.1101/cshperspect.a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werther R., Hallinan J.P., Lambert A.R., Havens K., Pogson M., Jarjour J., Galizi R., Windbichler N., Crisanti A., Nolan T., Stoddard B.L. Crystallographic analyses illustrate significant plasticity and efficient recoding of meganuclease target specificity. Nucleic Acids Res. 2017;45:8621–8634. doi: 10.1093/nar/gkx544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildy P. Recombination with herpes simplex virus. J. Gen. Microbiol. 1955;13:346–360. doi: 10.1099/00221287-13-2-346. [DOI] [PubMed] [Google Scholar]
- Xi S., Gooding W.E., Grandis J.R. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: Implications for cancer therapy. Oncogene. 2005;24:970–979. doi: 10.1038/sj.onc.1208316. [DOI] [PubMed] [Google Scholar]
- Yang Y., Wang C., Cheng P., Zhang X., Li X., Hu Y., Xu F., Hong F., Dong G., Xiong H. CD180 ligation inhibits TLR7- and TLR9-mediated activation of macrophages and dendritic cells through the lyn-SHP-1/2 axis in murine lupus. Front. Immunol. 2018;9:2643. doi: 10.3389/fimmu.2018.02643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Guo P., Xie X., Wang Y., Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017;21:648–657. doi: 10.1111/jcmm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.C., Pesce C.G., Colman-Lerner A., Lok L., Pincus D., Serra E., Holl M., Benjamin K., Gordon A., Brent R. Negative feedback that improves information transmission in yeast signalling. Nature. 2008;456:755–761. doi: 10.1038/nature07513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D., García-Sastre A., Xiao X., Wang R., Palese P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLOS Pathog. 2005;1:e4. doi: 10.1371/journal.ppat.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Servos M.R., Liu J. Instantaneous and quantitative functionalization of gold nanoparticles with thiolated DNA using a pH-assisted and surfactant-free route. J. Am. Chem. Soc. 2012;134:7266–7269. doi: 10.1021/ja3014055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended FD86 treatment over 60 days does not appear to generate dominant mutations. Sanger sequencing of IE86 (Exon 5) from virus in the continuous culture experiment in the presence of FD86 (Figure 5B). Viral DNA was extracted, IE86 exon 5 was PCR amplified (see Table S1 for primer sequences), Sanger sequenced, and aligned with the IE86 sequence for homology analysis. Despite apparent transient SNPs (e.g., at nucleotide position 640), no mutations “sweep” the population or are maintained in the consensus sequence over time.
Sequencing analysis of the CMV promoter region from the continuous culture experiment in the presence of FD86 (Figure 5B). Viral DNA was extracted and 250bp upstream and downstream of the crs was amplified by PCR (see Table S1 for primer sequences), Sanger sequenced, and then aligned with the viral promoter sequence for homology analysis. No acquired consensus mutations are observed over 14 replication cycles.
Sequencing analysis of IE86 (Exon 5) using virus obtained by culturing FD86-treated continuous culture supernatants (days 16, 32, and 48) in the presence or absence of FD86 (Figure S5C). Viral DNA was extracted, IE86 exon 5 was PCR amplified (see Table S1 for primer sequences), Sanger sequenced, and aligned with the IE86 sequence for homology analysis. No acquired consensus mutations are observed.
Sequencing analysis of the CMV promoter region using virus obtained by culturing FD86-treated continuous culture supernatants (days 16, 32, and 48) in the presence or absence of FD86 (Figure S5C). Viral DNA was extracted and 250-bp upstream and downstream of the crs was amplified by PCR (see Table S1 for primer sequences), Sanger sequenced and aligned with the viral promoter sequence for homology analysis. No acquired consensus mutations are observed.
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. Complete model details for simulations are provided in this paper, and this paper does not report original code.