
Figure 1.
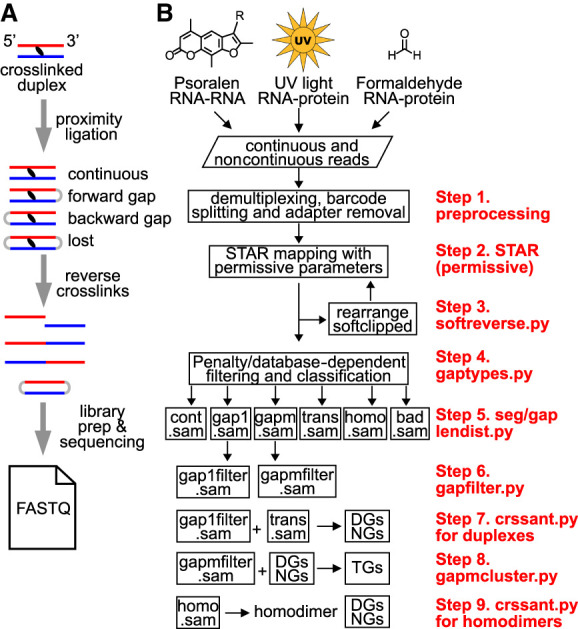
Overview of RNA crosslink-ligation experiments and analysis pipeline. (A) Outline of a typical crosslink-ligation experiment leading to FASTQ output files. The proximity ligation of crosslinked duplexes can produce both forward and backward arrangements. Circularized RNAs are rare and lost during library preparation because they cannot be ligated to adaptors. Similarly, concurrent crosslinking at multiple locations and subsequent ligation of them produce multigapped reads (gapm in panel B). (B) Several different types of crosslinking methods, such as psoralen, UV, and formaldehyde, together with proximity ligation produces noncontinuous reads that can be used to determine RNA structures. Newly developed computational tools and optimized parameters are listed on the right in nine steps (steps 1–9). Sequencing data that include both continuous and noncontinuous reads are demultiplexed, and the adapter/primer sequences are removed using published tools, for example, FASTX and Trimmomatic (step 1). The processed reads are mapped to genome references using optimized STAR parameters (permissive parameters, step 2). After the first round of STAR mapping, continuous alignments with softclips (indicating unmapped segments) are rearranged for a second round of STAR mapping (step 3). All alignments from the two rounds of STAR mapping are combined and filtered based on the gap penalty and a database of gapped alignments with longer segments, and then classified into six alignment types, including continuous (cont.sam in SAM format), one-gap (gap1), multigap (gapm), trans interactions (trans), homotypic interactions (homodimers, or homo), and miscellaneous bad alignments (bad) (step 4, using the gaptypes.py script) (see details in Fig. 3A–D; Supplemental Fig. S4). Data quality is checked using seglendist.py and gaplendist.py scripts, which calculate segment and gap length distributions (step 5). After removal of splicing events and reverse transcription artifacts, for example, short 1- to 2-nt gaps (step 6, using gapfilter.py), each of these alignment types is further processed to extract information for duplexes (step 7) (see Fig. 4 for details), high-level structures (step 8) (see Fig. 5 for details), and RNA homodimers (step 9, homo.sam) (see Fig. 6 for details). In step 7, two types of alignments, gap1filter.sam and trans.sam, are used to generate duplex groups and non-overlapping groups (DGs and NGs). In step 8, gapmfilter.sam alignments and the precomputed DGs and NGs are used to build trisegment groups (TGs). In step 9, overlapping chimeras are used to build potential homodimers. Detailed descriptions of these steps are in the Methods section and Supplemental Material.