

Abstract
Plasmodium parasites that cause malaria produce plasmepsins (PMs), pepsin-like aspartic proteases that are important antimalarial drug targets due to their role in host hemoglobin degradation. The enzymes are synthesized as inactive zymogens (pro-PMs), and the mechanism of their conversion to the active, mature forms has not been clearly elucidated. Our structural investigations of vacuolar pro-PMs with truncated prosegment (pro-tPMs) reveal that the formation of the S-shaped dimer is their innate property. Further structural studies, biochemical analysis, and molecular dynamics simulations indicate that disruption of the Tyr-Asp loop (121p-4), coordinated with the movement of the loop L1 (237–247) and helix H2 (101p–113p), is responsible for the extension of the pro-mature region (harboring the cleavage site). Consequently, under acidic pH conditions, these structural changes result in the dissociation of the dimers to monomers and the protonation of the residues in the prosegment prompts its unfolding. Subsequently, we demonstrated that the active site of the monomeric pro-tPMs with the unfolded prosegment is accessible for peptide substrate binding; in contrast, the active site is blocked in folded prosegment form of pro-tPMs. Thus, we propose a novel mechanism of auto-activation of vacuolar pro-tPMs that under acidic conditions can form a catalytically competent active site. One monomer cleaves the prosegment of the other one through a trans-activation process, resulting in formation of mature enzyme. As a result, once a mature enzyme is generated, it leads to the complete conversion of all the inactive pro-tPMs to their mature form.
Keywords: enzyme activation, histo-aspartic protease, plasmepsin I, plasmepsin II, plasmepsin IV
Introduction
Malaria is an infectious disease responsible for causing globally half a million cases of death per year [1]. Five unicellular eukaryotic protozoans of genus Plasmodium (P. falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi) are causative agents of human malaria [2]. Among them, P. falciparum and P. vivax are the deadliest and result in nearly all human malarial deaths [3]. Several drugs, including chloroquine, sulfadoxine, and artemisinin, are used to treat malaria patients [4]. However, due to the development of drug-resistant malaria parasites, there is an urgent need to design new antimalarial compounds with novel targets. In the human erythrocytes, the parasite hydrolyzes host hemoglobin in its food vacuole [5] in order to obtain amino acids for protein synthesis [6,7]. Four pepsin-like aspartic proteases which share 50–79% amino acid sequence identity, known as plasmepsins (PMs), are present in the food vacuole of P. falciparum [8,9]. These vacuolar PMs, plasmepsin I (PMI), plasmepsin II (PMII), histo-aspartic protease (HAP), and plasmepsin IV (PMIV), have been shown to be directly involved in the process of hemoglobin degradation [8,10,11], making them potential targets for developing novel drugs against malaria [12].
Similar to other pepsin-like aspartic proteases, vacuolar PMs are synthesized as inactive zymogens, in order to prevent unwarranted degradation of proteins [13]. These zymogens containing a 124-residue N-terminal prosegment are referred to here as proplasmepsins (pro-PMs). The prosegments of the vacuolar pro-PMs are ~ 70 residues longer than those of other pepsin-like aspartic proteases. A transmembrane region (~ 50 residues) is located at the N terminus of the prosegments of pro-PMs and is nonessential for the soluble expression of the recombinant truncated proplasmepsins (pro-tPMs) [14,15]. Here, we will be referring to the truncated form of the prosegment (77p-123p, ‘p’ refers to the residues of the prosegment) as just prosegment. The prosegment consists of an initial β-strand followed by two α-helices and a pro-mature region (segment connecting the second α-helix with the mature part of the protein, Lys119p-Leu8). The mature enzyme is formed through the removal of the prosegment during activation. However, despite extensive biochemical and structural studies of vacuolar PMs, the activation mechanism of their zymogens is not well understood.
Recently, we have shown that the truncated prosegment assists in correct folding of pro-tPMII, with a mechanism similar to that of pepsin [16]. Crystal structures of pro-tPMs have been determined for P. falciparum PMII (pro-tPMII) [17,18], HAP (pro-tHAP) [19], PMIV (pro-tPMIV) [18], and P. vivax PM (Pv-pro-tPM) [20]. These structures revealed that the conformation of the prosegment of pro-tPMs is unique and different from its counterpart in other pepsin-like aspartic proteases (Fig. 1A). The prosegment comprises of an initial β-strand followed by two α-helices and a loop region that harbors the Tyr-Asp loop (121p-4) [17] (Fig. 1B). The inactive form of pro-tPMs is stabilized by the interactions of the prosegment with the C-terminal domain of the enzyme. Such interactions keep the N- and C-terminal domains apart, thereby separating the two catalytic aspartates (Asp and His in pro-tHAP) in the active site of pro-tPMs [21]. In contrast, a lysine residue in the prosegment of pepsinogen physically blocks the active site, thus keeping the zymogen in an inactive form. It is assumed that the salt bridge interactions stabilizing the position of the prosegment across the active site of pepsin are disrupted at low pH. This process releases the prosegment and opens the substrate-binding cleft, leading to auto-activation catalyzed by the active site of the same pepsin molecule [21]. Although based on the crystal structure of pro-tPMII, a similar auto-activation process has been proposed for pro-tPMs at low pH, this mechanism is still in question. Since the pro-mature region is located distant to the active site [17], a large structural change would have to occur in order to bring it near the active site for auto-activation. The mechanism facilitating such a process remains unclear. It has also been reported that in vitro auto-activation of recombinant pro-tPMII takes place at a different site (+2 and + 12 upstream) compared to the native cleavage site (G123p-S1) (Fig. 1C) [14,22], and that the location of the cleavage site varies depending on the reaction conditions [7,23,24]. Therefore, delineation of the auto-activation mechanism requires further investigations of the structural features of pro-tPMs. Since vacuolar PMs are involved in hemoglobin degradation, deciphering their mechanism of auto-activation could guide the development of inhibitors preventing maturation that could act as novel antimalarial drugs.
Fig. 1.
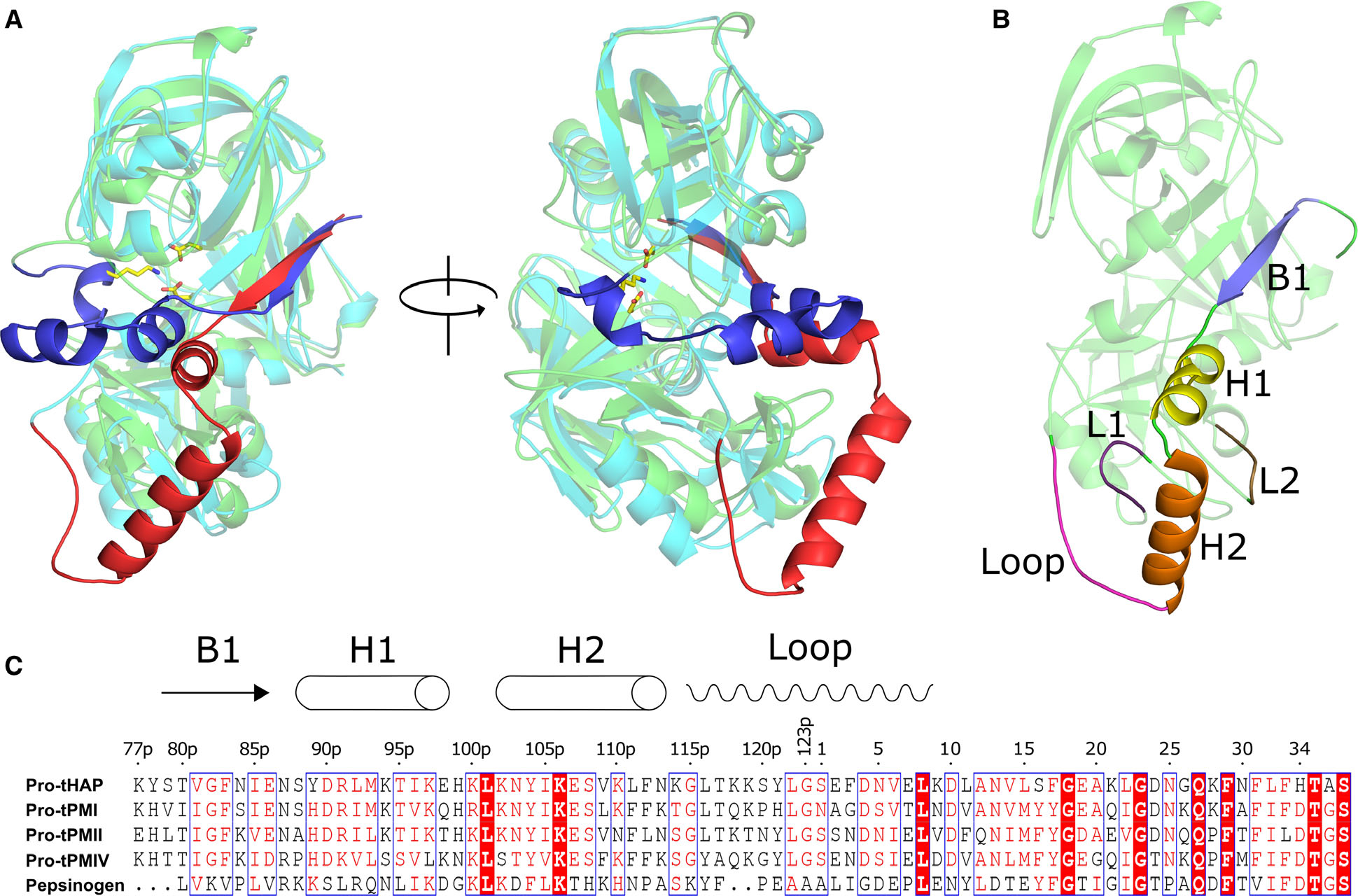
Structural and sequence alignment of the pro-tPMs and pepsinogen. (A) Structural superposition of Pv-pro-tPM (1MIQ) and porcine pepsinogen (3PSG), with their mature parts in cyan and green; truncated prosegments in red and blue, respectively. (B) Domain architecture of pro-tPMs; the mature polypeptide is colored green and the secondary structural elements of the truncated prosegment are distinctly colored. The protein structural representations have been generated with the PyMOL molecular graphics system (Schrödinger LLC, New York, NY, USA; version 2.3.2). (C) Multiple sequence alignment of pro-tPMs (uniprot accession number: pro-tHAP-Q9Y006; pro-tPMI-P39898; pro-tPMII-P46925; pro-tPMIV-W7FF86; porcine pepsinogen-P00791) and pepsinogen was performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), with the secondary structural elements of pro-tHAP shown on the top of the alignment. The highly conserved residues are shown as white on red background as generated by ESPript 3 [60]. The numbers on the top of the alignment represent the residue position in the pro-tHAP structure. The letter ‘p’ in the sequence numbering implies the residues in the prosegment.
In this study, we propose a unique mechanism of the activation of pro-tPMs. Our data show that pro-tPMs form a stable S-shaped dimer and that their promature region is intrinsically flexible. The disruption of the Tyr-Asp loop interactions in the prosegment under acidic conditions is essential for pro-mature region flexibility and facilitates dissociation of the dimer. Based on the detailed in vitro biochemical assays and structural analysis on pro-tPMs, we introduce for the first time a novel trans-activation mechanism for the conversion of inactive pro-tPMs to the mature plasmepsins (mPMs).
Results
Pro-mature region in the structure of pro-tHAP
Pro-tHAP has been crystallized in three different conditions (HAP-zymol (6KUB): 200 mM ammonium citrate tribasic pH 7.0, 100 mM imidazole pH 7.0, 20% w/v polyethylene glycol monomethyl ether 2000; HAP-zymo2 (6KUC): 200 mM lithium citrate tribasic tetrahydrate, 20% w/v polyethylene glycol 3350; HAP-zymo3 (6KUD): 200 mM ammonium phosphate dibasic, 20% w/v polyethylene glycol 3350) and the medium-to-high-resolution structures have been well refined (Table 1). The previously solved crystal structure of pro-tHAP (3QVC) was crystallized in 0.2M Tri-potassium citrate, pH 8.1, 20% (w/v) PEG 3350.
Table 1.
Data processing and refinement statistics.
| HAP-zymo1 | HAP-zymo2 | HAP-zymo3 | |
|---|---|---|---|
|
| |||
| Data processinga | |||
| Space group | C2 | C2 | C2 |
| Unit cell parameters | |||
| a, b, c (Å) α, β, γ (°) | 122.9, 69.3, 73.3 | 122.6, 69.7, 73.0 | 122.2, 69.9, 73.3 |
| 90.0, 126.1, 90.0 | 90.0, 126.1, 90.0 | 90.0, 125.7, 90.0 | |
| Temperature (K) | 100 | 100 | 100 |
| Wavelength (Å) | 1.54182 | 1.54182 | 1.54182 |
| Resolution range (Å) | 35.0–2.0 (2.1–2.0) | 36.0–2.5 (2.6–2.9) | 29.0–2.9 (3.0–2.9) |
| Rmergeb (%) | 8.0 (110.2) | 10.3 (160.3) | 11.8 (96.0) |
| Completeness (%) | 99.2 (99.7) | 98.4 (97.9) | 97.4 (96.8) |
| Mean I/σ(I) | 10.9 (1.2) | 13.12 (1.2) | 12.15 (1.7) |
| Total reflections | 123585 (16568) | 65010 (7129) | 41682 (3932) |
| Unique reflections | 33708 (4599) | 17267 (1890) | 11027 (1033) |
| Redundancy | 3.7 (3.6) | 3.8 (3.7) | 3.8 (3.8) |
| Refinement statistics | |||
| Resolution range (Å) | 35.0 – 2.0 | 36.0 – 2.5 | 29.0 – 2.90 |
| Working set: no. of reflections | 32021 | 16407 | 10474 |
| Rfactor (%) | 18.31 | 19.18 | 18.43 |
| Test set: no. of reflections | 1686 | 864 | 552 |
| Rfree (%) | 23.64 | 24.83 | 25.05 |
| Protein atoms | 3088 | 3021 | 2988 |
| Water molecules | 349 | 118 | 27 |
| Geometry statistics r.m.s.d. | |||
| Bond distance (Å) | 0.012 | 0.012 | 0.014 |
| Bond angle (°) | 1.6 | 1.9 | 1.7 |
| Ramachandran plot (%) | |||
| Favored region | 96.7 | 95.2 | 95.3 |
| Allowed regions | 99.7 | 99.7 | 99.7 |
| Outlier | 0.26 | 0.27 | 0.27 |
| PDB code | 6KUB | 6KUC | 6KUD |
Values in parentheses correspond to highest resolution shell.
Rmerge = ∑|I – 〈I〉 |∑ I where I is the observed integrated intensity, 〈I〉 is the average integrated intensity obtained from multiple measurements, and the summation is over all observed reflections.
The residues in the pro-tPMs are numbered according to the scheme shown above the alignment presented in Fig. 1C. The numbering with ‘p’ corresponds to the residues of the prosegment while without it refers to the mature part of the pro-tPMs. The overall structural fold of these new pro-tHAP structures is similar to the one described before [19]. The prosegment (Lys77p-Gly123p) in pro-tHAP comprises a β-strand (B1, 79p–86p) followed by the first α-helix (H1, 88p-98p), a short helical turn, a second α-helix (H2, 101p–113p), and a short coil connection to the mature enzyme (Fig. 2A,B). The pro-mature region (Lys119p-Leu8) has been unambiguously modeled within the electron density map of acceptable quality (Fig. 2B) in our newly determined structures of pro-tHAP (HAP-zymo1: 6KUB and HAP-zymo2: 6KUC). The third, lower resolution structure (HAP-zymo3: 6KUD) that we have determined and reported in this study has fewer missing residues (Tyr121p-Ser1) in the pro-mature region as compared to the one reported previously (3QVC, missing residues: Ser120p-Phe3) [19]. The numbering of the residues in the current structures of pro-tHAP is different from the previously reported structure of pro-tHAP (3QVC), where the latter follows pepsin numbering. The interactions between the prosegment and the mature fragment of pro-tHAP are mainly polar. However, H2 of the prosegment forms several hydrophobic contacts with two loops of the mature part of pro-tHAP, L1 (237–247) and L2 (275–286). Structural superpositions of the previously reported pro-tHAP structure (3QVC) with the newly solved structures in this study HAP-zymo1, HAP-zymo2, and HAP-zymo3 have produced root-mean-square deviation (r.m.s.d.) values of 0.42 Å, 0.44 Å, and 0.42 Å, respectively, indicating minimal structural rearrangements among them. Surprisingly, the only substantial differences are found in the pro-mature regions (Fig. 2C). The main chain conformations of a region of the prosegment comprising residues 77p-117p, and the mature part of the protein that includes residues 11–328 are almost identical in all four structures. The conformation of the N terminus of the mature peptide (7–10) is different in the structures with and without the ordered pro-mature region. The peptides connecting Lys119p and Leu8 have coil secondary structure and adopt different conformations in HAP-zymo1 and HAP-zymo2. It is also important to note that conformational variability of the incomplete pro-mature region is observed in HAP-zymo3, as well as in previously reported structure (3QVC). It has been shown that in the previously published crystals structures of pro-tPMII (1PFZ, 5BWY), pro-tPMIV (5JOD), and Pv-pro-tPM (1MIQ), the pro-mature region is held by a Tyr-Asp loop through hydrogen bonds between Tyr121p-Asp4 and a salt bridge interaction between Asp4-Lys238 [8]. Although these Tyr, Asp, and Lys residues are conserved in pro-tHAP, they do not form similar loop structures in HAP-zymo1 and HAP-zymo2. Our analysis also reveals that the Tyr-Asp loop is missing in the C-subunit of the pro-tPMII (1PFZ) structure; no justification was provided to rationalize such lack of this structural feature in the previous structural report [17]. Moreover, in the present study it was noted that the C-terminal part (116p-120p) of the prosegment of pro-tPMIV is not well defined in a fragmented electron density [18]. It is likely that the C-terminal part of the prosegment and the pro-mature region of pro-tHAP, as well as of other pro-tPMs, is more flexible compared to the other parts of the structure. Therefore, upon acidification, this segment of the polypeptide may unfold and become prone to hydrolytic cleavage. The importance of the flexibility of the promature region for the activation of pro-tPMs is discussed below.
Fig. 2.
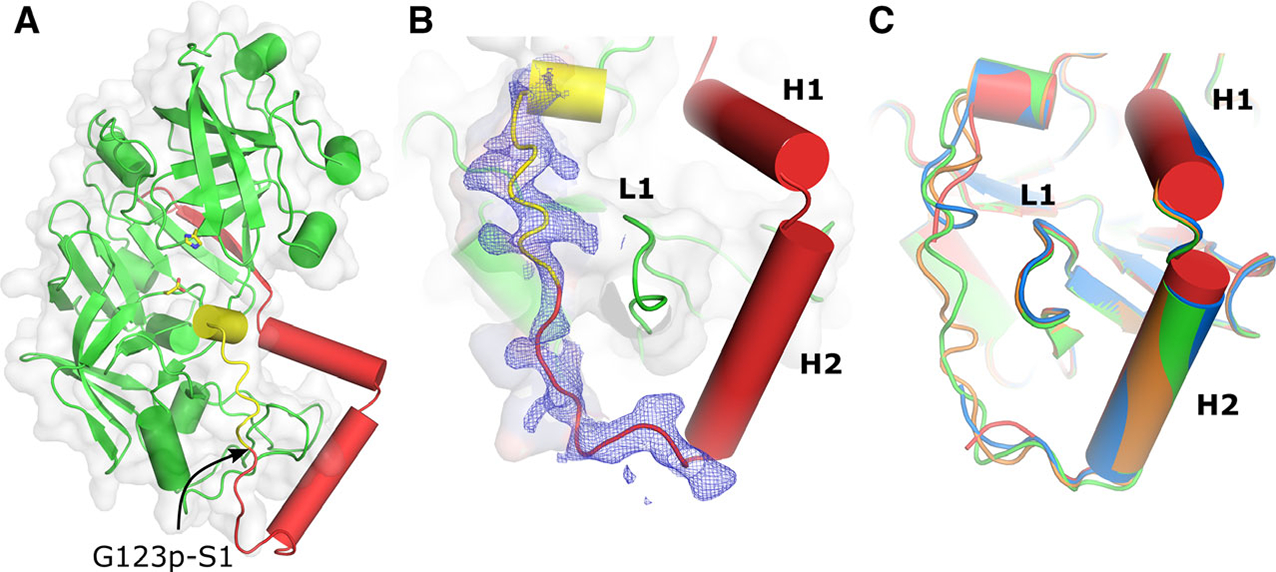
Structural fold and the secondary structure of the prosegment of pro-tPMs. (A) Overall structural fold of pro-tHAP. The prosegment is colored red and the polypeptide that would form the first β-strand of the mature enzyme is colored yellow. The active site residues are shown as sticks and an arrow indicates the native cleavage site. (B) 2Fo–Fc electron density (contoured at 0.8 σ) is shown around the promature region and the N terminus of the mature enzyme. The important secondary structure elements of the prosegment H1, H2, and L1 are also marked. (C) Conformational variabilities of the pro-mature regions of different pro-tHAP structures, HAP-zymo1 (green; 6KUB), HAP-zymo2 (orange; 6KUC), HAP-zymo3 (blue; 6KUD) from the current study and from the previously reported structure (red; 3QVC). The protein structural representations have been generated with the PyMOL molecular graphics system (Schrödinger LLC, New York, NY, USA; version 2.3.2).
S-shaped dimers of pro-tHAP and other pro-tPMs
Pro-tHAP has been crystallized in three different conditions with same space group and almost identical cell dimensions (Table 1). An analysis of contacts in the available crystal structures reveals several important features of pro-tPMs. The pro-tHAP forms an S-shaped dimer in the crystals (Fig. 3A). Formation of this dimer is mediated by extensive stacking interactions between hydrophobic residues of helix H2 and loop L1 of the two monomers (Fig. 3A), adjacent to the pro-mature region. A similar interface has been reported for pro-tPMIV [18]. Although pro-tPMII, pro-tPMIV, and Pv-pro-tPM have been crystallized in different space groups, analysis of the crystal contacts indicates that these proteins also create S-shaped dimers in their crystals (Fig. 3B–D). Sequence analysis of the dimer interface reveals conservation of the residues in both H2 and L1 across all the vacuolar pro-tPMs, suggesting the importance of these residues in maintaining a stable dimer. Interestingly, in the dimers of pro-tPMII and pro-tPMIV, the Tyr-Asp loops of the two monomers are quite close to each other. The tyrosine residues (Tyr121p) of the monomers are in proximity to each other and interact via a water molecule in Pv-pro-tPM structure (1MIQ). A corresponding water molecule is also most likely present between the Tyr121p residues in the pro-tPMII structure (1PFZ) [17] due to the clear presence of a positive Fo–Fc density; however, it has not been previously modeled. Importantly, the same water molecule forms hydrogen bonds with Ser1 from each monomer.
Fig. 3.
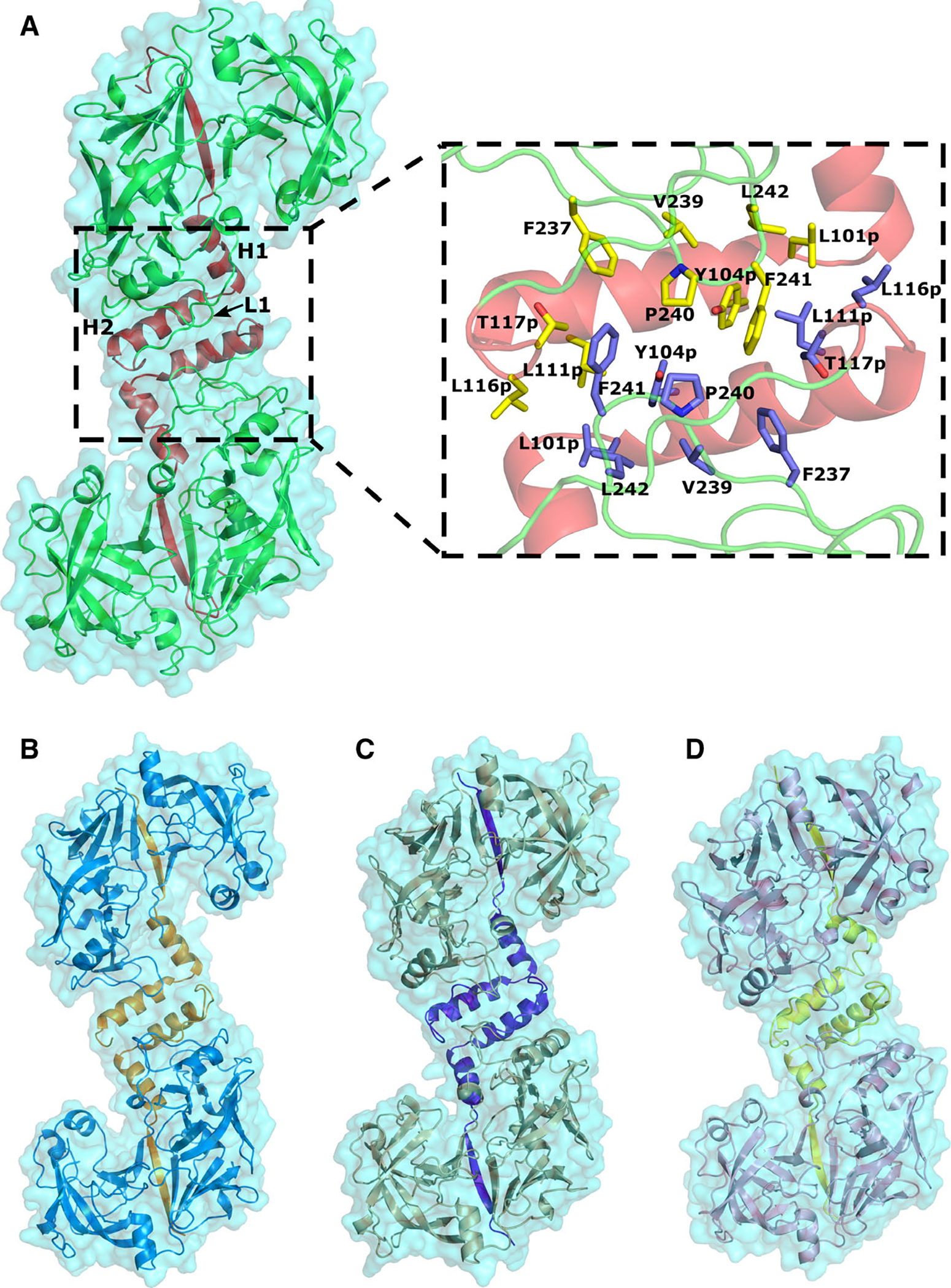
The crystallographic S-shaped dimers of pro-tPMs. (A) Pro-tHAP dimer (HAP-zymo1; 6KUB) with prosegment colored red. The inset shows the residues involved in formation of the dimer interface. (B) The pro-tPMII dimer (1PFZ) with orange color prosegment. (C) The pro-tPMIV dimer (5JOD) with blue color prosegment. (D) The Pv-pro-tPM dimer (1MIQ) with yellow color prosegment. The protein structural representations have been generated with the PyMOL molecular graphics system (Schrödinger LLC, New York, NY, USA; version 2.3.2).
In order to ascertain that this type of S-shaped dimer is also present in solution and is not just a crystallographic artifact, small-angle X-ray scattering (SAXS) was performed for pro-tPMI. Although no crystal structure of pro-tPMI is available, SAXS results show that it forms a dimer in solution (Fig. 4). All profiles in the very low angle region are well approximated by Guinier’s formula [25]. The radius of gyration (Rg) of the dimer is 35.4 Å at 298 K after correcting for concentration effects. The pair correlation functions, that is, P(r) functions, have unimodal shapes with maximum dimensions of 116.8 Å (Fig. 4A). The homology model of a dimer of pro-tPMI fits well the envelope obtained from SAXS data (Fig. 4B). These results clearly suggest the presence of an S-shaped dimer of pro-tPMI in solution as observed for other pro-tPMs in their crystals. Hence, we propose that such dimeric structure might be a common structural feature of all pro-tPMs. Further, we also evaluated the conformational variabilities of the structures in pro-tPM dimers.
Fig. 4.
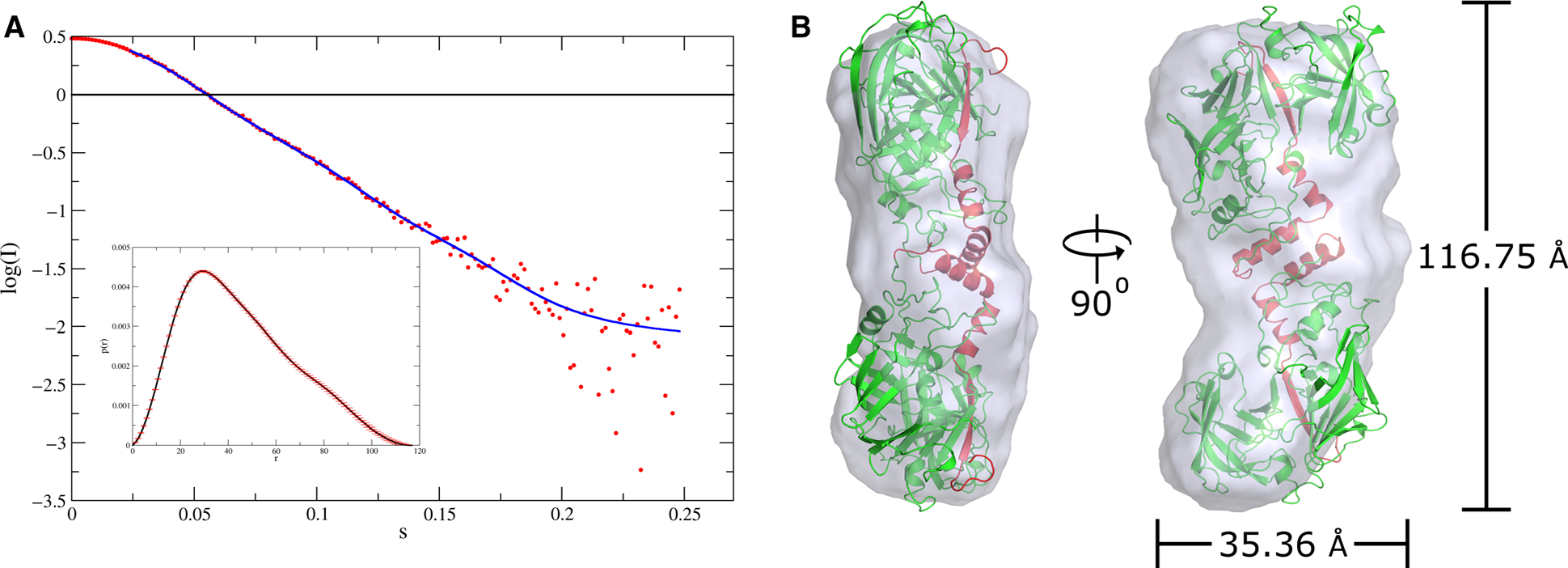
Dimeric nature of pro-tPMs in solution as revealed by SAXS experiment. (A) The scattering curve of pro-tPMI. The inset shows the distance distribution function. (B) Superimposed S-shaped dimeric model of pro-tPMI in the SAXS envelope (gray). The radius of gyration and maximum dimension of the envelope obtained from SAXS data are also shown. The model of dimeric pro-tPMI was fitted in the envelope using DAMMIN [54]. The protein structural representations have been generated with the PyMOL molecular graphics system (Schrödinger LLC, New York, NY, USA; version 2.3.2).
Interestingly, superposition of all pro-tPM dimers with respect to monomer 1 of HAP-zymo1 reveals a very large change in the orientation with respect to monomer 2 of HAP-zymo1 (Fig. 5A). Comparison of monomer 2 of HAP-zymo1 with 1PFZ chain C, 5BWY chain B, 1PFZ chain D, 1MIQ chain B, and 5JOD chain B shows relative angular movement (Amd) of 8.25°, 20.61°, 25.72°, 31.55°, and 34.18°; and a relative distance (Dmd) of 9.45Å, 18.69 Å, 22.78 Å, 20.28 Å, and 24.82 Å, respectively (Fig. 5B). We have observed a direct correlation between the variations of the angles and the distances in the compared structures (Fig. 5C). Notably, the surface area enclosed between the dimers (Sd) of HAP-zymol, 1PFZ (chain A and C), 5BWY (chain A and B), 1PFZ (chain B and D), 1MIQ (chain A and B), and 5JOD (chain A and B) are 804.3 Å2, 610.7 Å2, 826.4 Å2, 828.9 Å2, 938.8 Å2, and 914.7 Å2, and the corresponding interaction energy of the dimers (Ed) is −14.2, −14.2, −17, −15.4, −17.3, and −17.1 kcal·mol−1, as calculated using PISA server (http://www.ebi.ac.uk/pdbe/pisa/) (Fig. 5C), respectively. These results indicate a direct correlation between the enclosed surface area and the energy of interaction among the dimers of the pro-tPMs. Based on the analysis of these four parameters in the pro-tPMs crystal structures, we propose that large conformational changes in the dimers might play an important role in their activation, as discussed later.
Fig. 5.
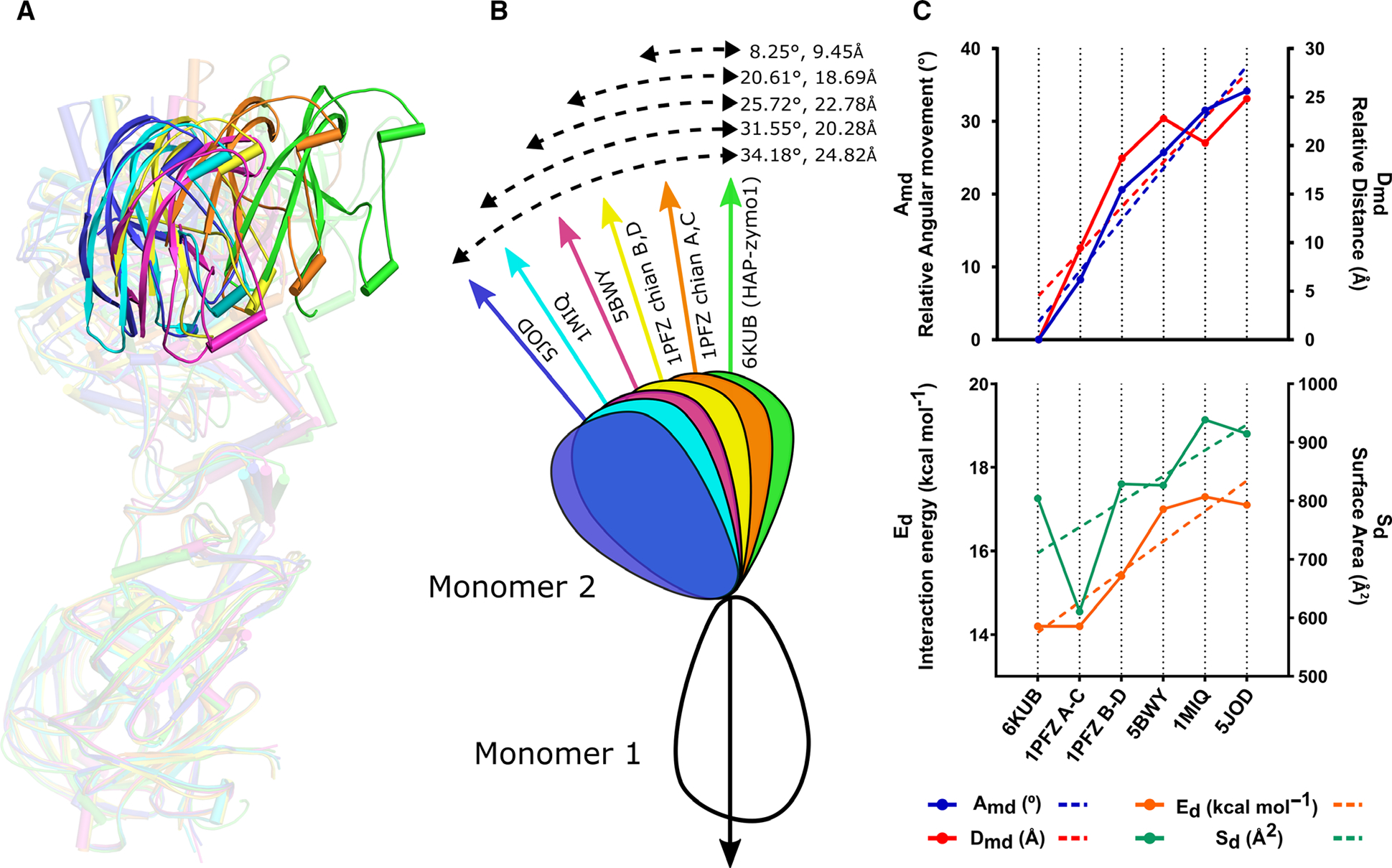
Comparison of the relative orientations of the monomers in the dimeric structures of pro-tPMs. (A) Superposition of the dimeric forms of HAP-zymo1 (green; 6KUB) with pro-tPMII (1PFZ; chain A and C in orange), pro-tPMII (1PFZ; chain B and D in yellow), pro-tPMII (5BWY in magenta), Pv-pro-tPM (1MIQ in cyan), and pro-tPMIV (5JOD in blue) with respect to monomer 1. The transparency of the dimers is increased to highlight the relative movement of the structures. The protein structural representations have been generated with the PyMOL molecular graphics system (Schrodinger LLC, New York, NY, USA; version 2.3.2). (B) Schematic representation of the superposed dimers of pro-tPMs with respect to HAP-zymo1 (6KUB) and the relative movements (angle and distance) observed. (C) Graphs representing the variation of angle (Amd) and distance (Dmd) in the structures of pro-tPMs (upper panel); energy of interaction (Ed) and the area enclosed in the dimers (Sd) of pro-tPMs (lower panel).
Conformational flexibility of the pro-mature region in pro-tPMs
We had previously compared the conformations of the prosegment of pro-tHAP (3QVC) with pepsinogen, pro-tPMII, and Pv-pro-tPM [19]. However, we had not commented on the pro-mature region of pro-tHAP, as this region could not be modeled in that structure. Recently, a high-resolution crystal structure of pro-tPMIV has been determined [18]. These structures, along with our newly determined two pro-tHAP structures (6KUB and 6KUC) with well-defined prosegments, enabled us to undertake a detailed analysis of the conformational flexibility of the pro-mature regions among pro-tPMs. Superposition of pro-tPMII (A chain of 1PFZ), pro-tPMII (A chain of 5BWY), pro-tHAP (HAP-zymol), pro-tPMIV (A chain of 5JOD), and Pv-pro-tPM (A chain of 1MIQ) revealed similarities and variations among these structures. It is evident that helix H2 and the pro-mature region (the peptide connecting the prosegment to the mature enzyme) in pro-tHAP assume different conformations compared to other vacuolar pro-tPMs (Fig. 6A). Although helix H2 starts in almost the same position in these pro-tPMs (except for Pv-pro-tPM), its C-terminal part assumes a substantially different conformation in pro-tHAP, as it protrudes away from the loop L1 of the mature enzyme (Fig. 6A). A conformational change of H2 in pro-tHAP is also coordinated with a downward movement of L1. The pro-mature region in pro-tHAP is in an extended coil conformation, whereas in other pro-tPM structures the corresponding region adopts a constricted Tyr-Asp loop conformation (Fig. 6A). The most important interaction within this loop is the hydrogen bond between Tyr121p and Asp4, which is essential for keeping the prosegment in its strained conformation. In the crystal structure of pro-tHAP, the Tyr-Asp loop is extended and these two residues are separated by a distance of 20.5 Å (Fig. 6B). A unique feature of pro-tPMI is the substitution of Tyr121p by a histidine. The formation of pro-tPMI dimer, as seen in SAXS experiment, suggests that in this loop histidine may play a similar role to tyrosine.
Fig. 6.
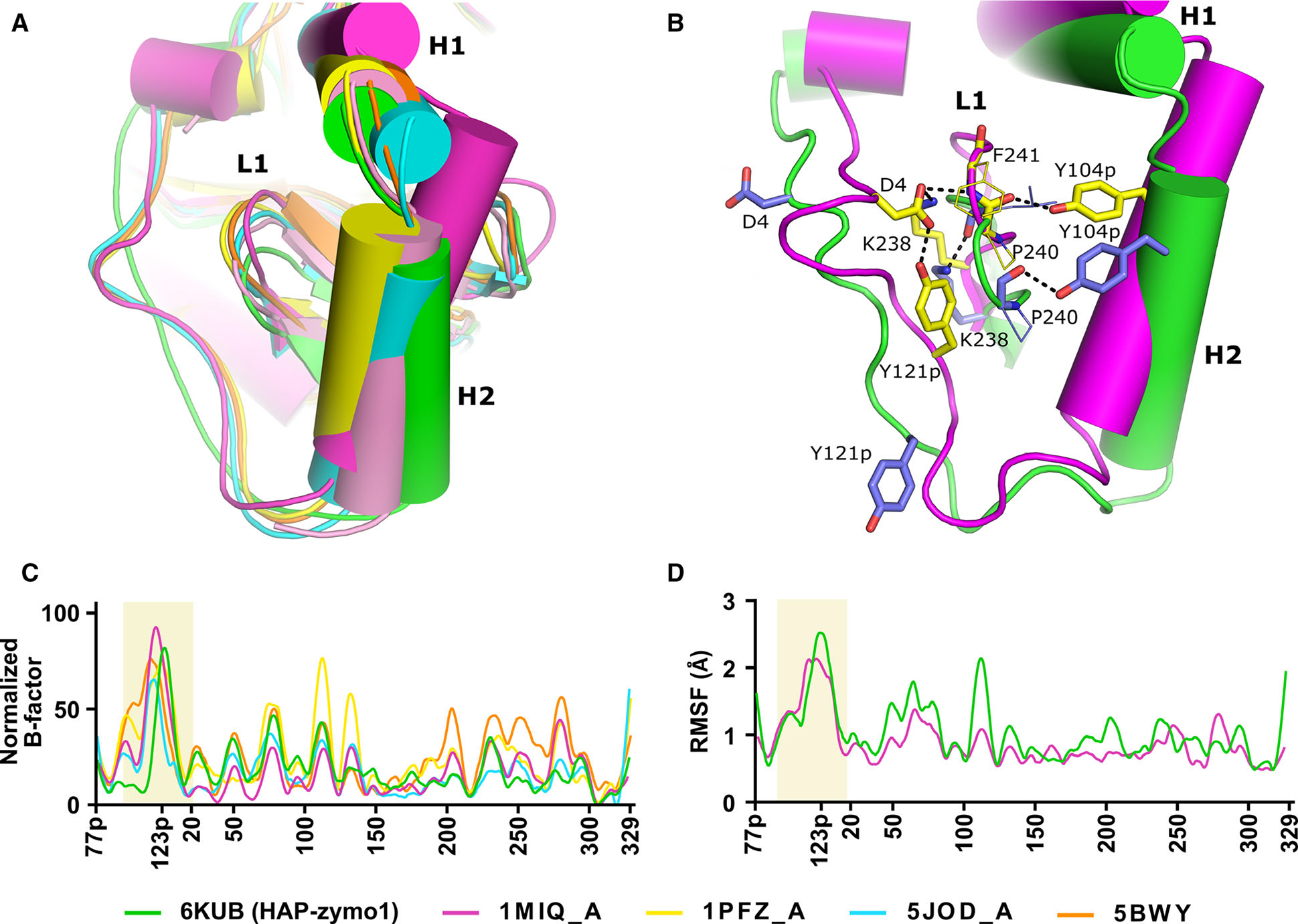
Comparison of the regions of flexibility in the structure of pro-tPMs. (A) Superposition of pro-tHAP (HAP-zymo1; 6KUB) in green, Pv-pro-tPM (1MIQ, chain A) in magenta, pro-tPMII (1PFZ, chain A) in yellow, pro-tPMII (1PFZ, chain C) in salmon pro-tPMIV (5JOD, chain A) in cyan, pro-tPMII (5BWY) in orange. (B) Interactions essential for stabilization of the Tyr-Asp loop of pro-mature region. Secondary structural elements of Pv-pro-tPM (1MIQ) and pro-tHAP (6KUB are shown in magenta and green, respectively. The side chains of residues of Pv-pro-tPM (1MIQ) and pro-tHAP (6KUB) are shown in yellow and blue, respectively. Important polar interactions are shown as black dotted lines. The protein structural representation has been generated with the PyMOL molecular graphics system (Schrödinger LLC, New York, NY, USA; version 2.3.2). (C) Normalized B-factors of the Cα atoms derived from the crystal structures of pro-tPMs; pro-tHAP (6KUB) in green, Pv-pro-tPM (1MIQ) in magenta, pro-tPMII (1PFZ) in yellow, pro-tPMIV (5JOD) in cyan, pro-tPMII (5BWY) in orange. (D) The R.M.S.F. of the Cα atoms obtained from the simulation of pro-tHAP (6KUB) in green and Pv-pro-tPM (1MIQ) in magenta.
Notably, the amino acids in the H2 and L1 regions are highly conserved among the vacuolar pro-tPMs, highlighting the importance of the residues located in these structural elements. Analysis of the crystal structure of Pv-pro-tPM indicates that an intricate hydrogen-bonding network between the pro-mature region, H2 and L1, leads to stability and rigidity of the prosegment (Fig. 6B). Two additional interactions, a salt bridge between the side chains of Lys238 and Asp4, and a hydrogen bond between Asp4 side chain of Tyr-Asp loop and the main chain amide of Phe241 of L1, restrict the movement of L1. Tyr104p from H2 interacts with the main chain carbonyl group of Pro240 in L1, stabilizing the motion of H2. These interactions reduce the overall dynamics of the pro-mature region, as well as L1. Similar interactions are also observed in pro-tPMII and pro-tPMIV. In contrast, in pro-tHAP (HAP-zymo1, 6KUB), hydrogen bonds between the pro-mature region and L1 are absent, resulting in flexibility of the region. An interaction between Tyr104p of H2 and the main carboxyl of Pro240 of L1 is still present; however, it does not confer enough stability, leading to the downward movement of L1 along with H2.
We have analyzed the individual subunits of the available structure of pro-tPMII (1PFZ). Interestingly, one of the subunits (C chain) assumes a different conformation (Fig. 6A) compared to the other subunits and the pro-mature region is not visible in that monomer, similar to that observed previously for pro-tHAP (3QVC) (Fig. 2C). Superposition of pro-tHAP (6KUB) and the C-subunit of pro-tPMII (1PFZ) reveal that L1 and H2 also have downward conformational movement in pro-tPMII, again similar to that observed in pro-tHAP Fig. 6A. However, similar movement of L1 and H2 was not observed in the other three chains of 1PFZ. The temperature factors (B-factors) (Fig. 6C) of the pro-mature region are also higher compared to the other regions of the proteins in pro-tPMII (5BWY), pro-tPMIV (5JOD), and Pv-pro-tPM (1MIQ), despite the well-defined positions of this region and the Tyr-Asp loop in the electron density maps. This result indicates the flexible nature of this segment of the polypeptide irrespective of its constricted or extended conformation of the Tyr-Asp loop.
As pro-tPMs show extensive conformational variability in their pro-mature regions, we further studied the dynamics of the pro-mature region by molecular dynamics (MD) simulations. The root-mean-square fluctuation (r.m.s.f.) plots (Fig. 6D) of the simulation trajectories of pro-tHAP and Pv-pro-tPM demonstrate the intrinsically flexible nature of the pro-mature regions. An analysis of hydrogen-bonding interactions of the Tyr-Asp loop regions in these two structures also reveals that the carboxylate group of the aspartate side chain can form alternate polar interactions, contributing toward the flexibility of the pro-mature regions. Hence, it is evident from structural analysis and MD simulations that the pro-mature regions of pro-tPMs are intrinsically flexible.
Dimer-to-monomer transition in the activation pathway
As shown by our SAXS experiment, pro-tPMs exist as dimers in solution. In order to probe the various oligomeric states of pro-tPMs in solution, we have performed all our experiments with pro-tPMII only, due to low yield of pro-tPMI, pro-tHAP, and pro-tPMIV. Size-exclusion chromatography (SEC) of pro-tPMII (at pH 7.5) shows elution at ~ 9.0 ml, corresponding to a molecular weight of ~84 kDa (Fig. 7A), thus indicating its dimeric population. Our structural analysis highlights the importance of the Tyr-Asp loop in maintaining the stability of the pro-mature region, L1, H2, as well as the integrity of the dimer of pro-tPMs. Therefore, it is expected that mutation of Tyr121p or Asp4 of the Tyr-Asp loop to alanine should disrupt the formation of this loop and other stabilizing interactions, eventually preventing formation of a dimer. The size-exclusion chromatography (SEC) of the pro-tPMII Y121pA mutant elutes at 10 mL, indicating a molecular weight of ~ 55 kDa (Fig. 7A), corresponding to a monomer. The apparent higher molecular weight than expected (~ 43 kDa) is likely due to an increase in hydrodynamic radius because of the extended pro-mature region and the exposure of the previously buried surface area between the dimer. These findings are further corroborated by the dynamic light scattering (DLS) data (Fig. 7C), which shows that pro-tPMII has a larger hydrodynamic radius than that of the pro-tPMII Y121pA mutant. Thus, the stability of the Tyr-Asp loop is important in maintaining the dimeric state of pro-tPMs.
Fig. 7.
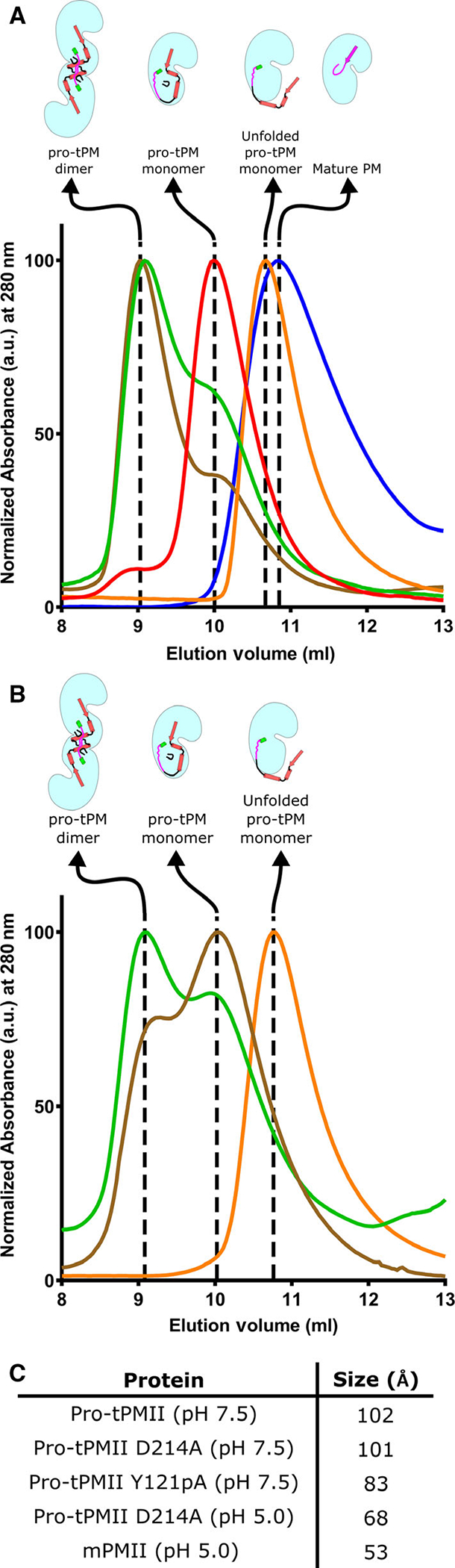
Size-exclusion chromatography analysis of the WT and the mutants of pro-tPMII, mature PMII and the refolding of prosegment of pro-tPMII. (A) The size-exclusion chromatography elution profiles for the dimeric form of pro-tPMII WT and D214A mutant are shown in brown and green, respectively; the peak of the monomeric pro-tPMII Y121pA mutant is shown in red. The orange and blue peaks denote elution of the monomeric prosegment unfolded pro-tPMII D214A mutant and the mature PMII, respectively. (B) Gel filtration elution profiles of the prosegment folded, unfolded, and refolded pro-tPMII D214A mutants are represented in green, orange, and brown, respectively. A cartoon representation of conformational states of the protein is shown above each curve. (C) Hydrodynamic radius (in Å) of different forms of PMII as calculated from DLS. The experiments were performed in triplicate (n = 3) and representative data are depicted.
Unfolding of the prosegment in acidic conditions
Previous studies on the activation of pro-tPMs have proposed that the activation of the inactive form of the enzyme to their mature form can occur through a sequential or direct mechanism [26]. To understand the activation process, we have slowed the maturation of pro-tPMI by employing 10-fold molar excess of pep-statin A (PepA, a potent inhibitor of pepsin-like aspartic proteases). The processing of PepA-treated pro-tPMI in acidic conditions was analyzed by SDS/PAGE. In the absence of any inhibitor, we have observed complete activation of pro-tPMI to mature PMI (mPMI) in 1 h by the band shift from ~ 43 kDa to ~ 37 kDa on SDS/PAGE. However, in the presence of PepA we observed formation of several polypeptide intermediates with molecular weight in the range between the pro-tPMI and the mPMI (Fig. 8). This result indicates that under acidic condition the prosegment unfolds, thus allowing the sequential cleavage along its length during the activation process. This observation also justifies the presence of various cleavage intermediates and products formed under acidic conditions, as previously reported for pro-tPMs [7,27].
Fig. 8.
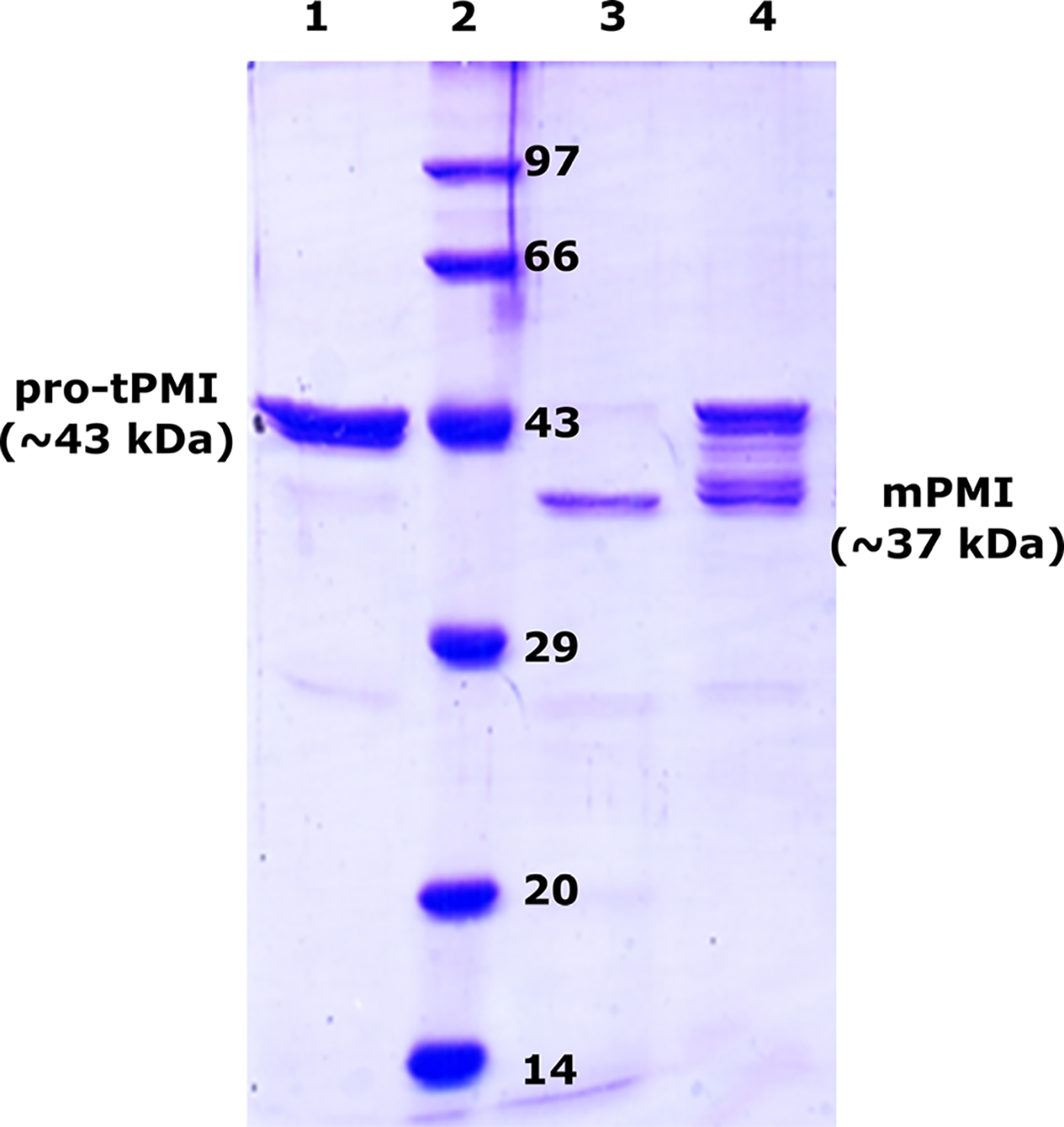
Effect of pepstatin A (PepA) on pro-tPMI processing to mPMI. Pro-tPMI controlat ~ 43 kDa (lane 1); protein molecular weight marker in kDa (lane 2); pro-tPMI acidified with 0.1 m sodium acetate, pH 4.5 for 1 h without PepA (lane 3); PepA-treated pro-tPMI in acidic condition incubated for 1 h (lane 4). The concentration of pro-tPMI used was 0.01 mM and inhibitor in 10-fold molar excess (0.1 mM). The experiments were performed in triplicate (n = 3) and representative SDS/PAGE shown.
To capture an intermediate state during the activation process, we created an active site mutant (D214A) of pro-tPMII that could not form the mature enzyme under acidic conditions. As this mutant cannot undergo activation, it prevents generation of multiple species upon acidification and allows capturing of conformational states of pro-tPMII. In acidic buffer (pH 5.0) pro-tPMII D214A mutant elutes in SEC at 10.6 mL, corresponding to molecular weight of ~43 kDa (Fig. 7A), suggesting the protein is in its monomeric form. The pro-tPMII D214A mutant is still dimeric at pH 7.5 and the elution profile is similar to the WT pro-tPMII (Fig. 7A). Interestingly, the hydrodynamic radius of pro-tPMII D214A mutant is slightly larger in pH 5.0 than that of mPMII, which elutes at 10.8 mL (corresponding to ~37 kDa), and smaller than of the monomeric pro-tPMII Y121pA mutant (Fig. 7A). It has been proposed that the prosegment is displaced away from the mature part of the pro-tPMs under acidic conditions. Thus, the form that is observed between the monomeric pro-tPMII and the mature PMII (mPMII) is likely an intermediate state with unfolded prosegment. Our SEC data for all the forms of pro-tPMII and mPMII correlate very well with the size of these species observed in DLS measurements (Fig. 7C).
Further, in order to confirm the existence of an intermediate with unfolded prosegment during activation, we investigated the unfolding and refolding of the prosegment at acidic as well as alkaline pH, respectively. At pH 7.5, the SEC of pro-tPMII D214A mutant shows both dimeric and monomeric forms (Fig. 7B, green peak). Acidification of this protein sample (with both forms) in pH 5.0 buffer resulted in a single peak (Fig. 7B, orange peak) in close proximity to the previously described elution volume of mPMII, indicating formation of monomers with unfolded prosegment. It is interesting to observe that the transfer of pro-tPMII D214A mutant from an acidic (pH 5.0) to an alkaline buffer (pH 7.5) leads to complete refolding of the prosegment, as indicated by dimer and monomer peaks in SEC (Fig. 7B, brown peak), similar to the natively folded pro-tPMII. These observations suggest that the prosegment of pro-tPMII in acidic conditions undergoes reversible unfolding and that sequential cleavage occurs along the prosegment.
Prosegment dynamics and accessibility of the active site
Based on our structural analysis and size distribution of pro-tPMII at different pH values, we speculate that unfolding of the prosegment in acidic conditions should lead to formation of an accessible active site and closure of the flap. Trp41 present near the active site undergoes conformational changes upon flap closure triggered by unfolding of the prosegment and by substrate or inhibitor binding. Therefore, fluorescence of Trp41 was used as a probe to monitor the flap dynamics and the conformational changes that accompany the unfolding of the prosegment of pro-tPMII during its conversion to mPMII. In order to capture these changes without the prosegment being cleaved, catalytically inactive pro-tPMII D214A mutant was utilized. The fluorescence spectra of pro-tPMII D214A mutant measured at pH 7.5 and 5.0 show distinct differences. A significant decrease in fluorescence intensity and a 4 nm λmax blue shift (342 → 338 nm) is observed at pH 5.0, as compared to pH 7.5 (Fig. 9A). Here, mPMII and mPMII D214A mutant are taken as controls to monitor Trp fluorescence upon removal of the prosegment. Further decrease in fluorescence intensity and a 2 nm λmax blue shift (338 → 336 nm) was also observed when the protein was incubated at pH 5.0 with PepA (Fig. 9A). Titration of the pro-tPMII D214A mutant in pH 5.0 with increasing concentrations of PepA showed a concentration-dependent decrease in the fluorescence intensity up to equimolar protein-PepA ratio (Fig. 9B). Therefore, these results indicate that, in acidic pH, unfolding of the prosegment leads to opening of the substrate-binding site area in the active site of mature enzyme and, hence, allows PepA binding.
Fig. 9.
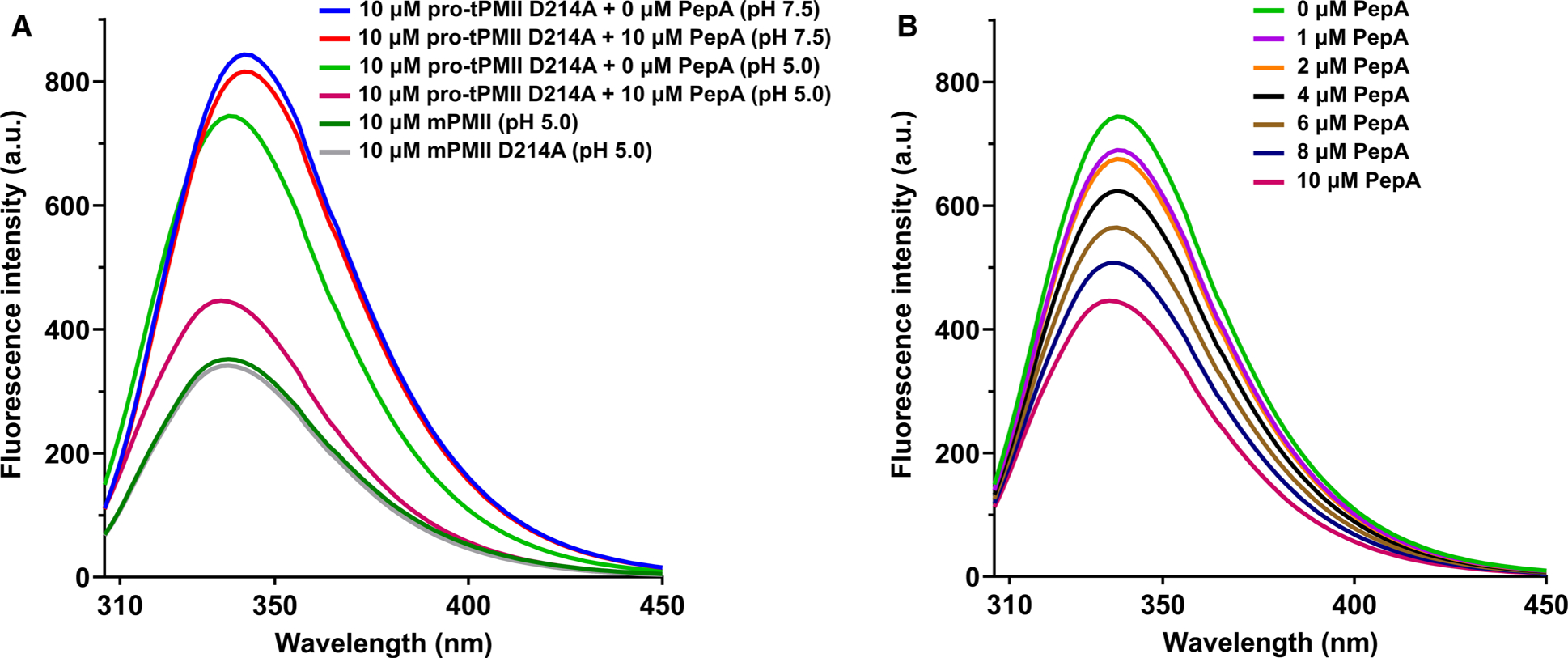
Tryptophan fluorescence quenching of pro-tPMII D214A mutant. (A) Quenching of fluorescence of pro-tPMII D214A in pH 7.5 and 5.0, with and without inhibitor; mature PMII and mature PMII D214A mutant are taken as positive controls. (B) Fluorescence quenching of pro-tPMII D214A mutant at pH 5.0 in the presence of PepA (0–10 μm). The experiments were performed in quintuplicate (n = 5), and the data are represented as a mean of all the measurements.
Discussion
The activation mechanism of vacuolar proplasmepsins (pro-PMs) is not yet fully understood, although these enzymes belong to the family of extensively studied pepsin-like aspartic proteases and the first structure of pro-tPMII was determined more than two decades ago. It was assumed (without any substantial evidence) that vacuolar pro-tPMs follow a similar activation mechanism as the one proposed for pepsinogen [21,28]. Moreover, despite the fact that several crystal structures of pro-tPMs have been reported, the lack of structural analysis and detailed biochemical investigations has limited the understanding of their activation mechanism. Our study reports for the first time the flexible nature of the pro-mature region, its influence on the dimer-to-monomer transition, the pH-dependent unfolding of the prosegment of the pro-tPMs, and implications of these phenomena in the activation process. These results indicate a novel mechanism for activation of pro-tPMs, not described previously for any other pepsin-like-aspartic proteases.
In this study, we demonstrated that pro-tPMs form S-shaped dimers both in solution (Fig. 4) and in crystals (Fig. 3). Implications of this characteristic feature of the pro-tPMs in their activation mechanism were not discussed in previous reports. The dimers reveal two important conformations of the pro-mature region, namely constricted and extended forms. We examined the constricted pro-mature region with intact Tyr-Asp loop in the previously determined structures of pro-tPMIV (5JOD), pro-tPMII (5BWY), Pv-pro-tPM (1MIQ), and pro-tPMII (1PFZ chain A, B, and D) (Fig. 6B). We also could capture for the first time the unique conformation of the pro-mature region of pro-tHAP that is present in an extended conformation in the three crystal structures. The interactions in the Tyr-Asp loop in these structures are disrupted and Tyr121p moves ~ 20 Å away from Asp4, from their usual hydrogen-bonding distance in the intact form. The subsequent breaking of the inter-residue hydrogen bond is coupled with the downward movement of loop L1 and helix H2 (Movie S1). These observations suggest that any perturbations in the interactions in the Tyr-Asp loop and the dimer interface could play a significant role in the dynamics of the pro-tPM dimer. Based on extended pro-mature region and the disrupted Tyr-Asp loop in the crystal structures of pro-tHAP, it is expected that it should be in the monomeric form (as observed from our SEC profile of the pro-tPMII Y121pA mutant, with disrupted Tyr121p and Asp4 interaction Fig. 7A); however, its existence as a dimer in the crystal form is unique. Similar conformational variability of the pro-mature region can also be seen in one subunit of the pro-tPMII structure (1PFZ, chain C), although its significance has not been previously discussed [17].
A crucial step in the activation of the pro-tPMs is the dissociation of the dimer to monomers. Disruption of the dimerization interface is highlighted by the deviations in the angle between the monomers of the pro-tPMs (Movie S2). Analysis of four different parameters in the five crystal structures of pro-tPMs provides significant insights into the stability of the dimer, as well as to the dynamics of the pro-mature region. We have observed correlation between Amd (relative angular movement between the monomers of the superposed ‘S’-shaped dimers of pro-tPMs) and Dmd (relative distance between the monomers of the superposed ‘S’-shaped dimers of pro-tPMs), as well as Sd (surface area enclosed between the dimers) and Ed (interaction energy of the dimers) in different dimers of pro-tPMs. The structures of pro-tPMs with constricted pro-mature region and intact Tyr-Asp loop show a significant change in Amd of ~ 34.0° and Dmd of ~ 25 Å with respect to HAP-zymo1, whereas the C chain of 1PFZ, with extended pro-mature region, shows much less difference in Amd and Dmd (Fig. 5). The pro-tHAP structures (6KUB, 6KUC, 6KUD) reported in this study have the most extended conformation of pro-mature region that disrupts the formation of the Tyr-Asp loop. As the pro-mature region undergoes extension, this leads to the reduction in Amd and Dmd. A similar trend is observed in Sd and Ed and the extension of the pro-mature region caused a decrease in Sd and the Ed due to reduction in the interactions at the dimer interface. These structural analyses provide an evidence that the intact Tyr-Asp loop is essential for the stability of the dimer. Therefore, extension of the pro-mature region causes movement of the monomers that weakens their interactions at the dimer interface and leads to the dissociation of the dimer.
The importance of the Tyr-Asp loop toward the stability of the dimer is further supported by the size distribution of the pro-tPMII Y121pA mutant, which could not form the Tyr-Asp loop. The Y121pA mutation causes disruption of multiple interactions and destabilizes the dimer interface. That explains why the mutant protein exists only in the monomeric form, while the wild-type protein is dimeric. It also correlates with the significantly reduced values of Sd and the Ed in pro-tHAP as compared to other pro-tPMs, where the Tyr-Asp loop is absent, due to an extended conformation of its pro-mature region. We conclude that, due to its dynamic nature, the pro-mature region is able to adopt both the constricted and extended conformations. Thus, the pro-tHAP dimer that was observed in our crystal structure with the most extended conformation of pro-mature region might be an intermediate state just prior to its dissociation to form monomers. Therefore, it could be inferred that the breakage of the Tyr-Asp loop and the extension of the pro-mature region are the initial triggers in the conversion from a dimer to monomers.
The next crucial event in the activation process is the unfolding of the prosegment, preceding the conversion of pro-tPM to the mature enzyme. We were able to capture a pH-dependent change in the hydrodynamic radius of pro-tPMII. In acidic conditions, there is a reduction in the hydrodynamic radius of pro-tPMII D214A mutant due to the reversible unfolding of the prosegment. While only the prosegment gets unfolded, the mature domain of the enzyme remains compact. Transferring the pro-tPMII with an unfolded prosegment to alkaline pH results in refolding of the prosegment and complete conversion to a mixture of both dimeric and monomeric fractions (Fig. 7B). The existence of various activation intermediates of pro-tPMI in the presence of PepA (Fig. 8) suggests sequential cleavage at multiple cleavage sites along the unfolded prosegment. Thus, we present experimental evidences for the unfolding of the prosegment, which is an important conformational change that leads to the loss of its secondary structure, followed by the sequential cleavage of the prosegment.
The prosegment of pro-tPMs acts as a harness keeping the active site inaccessible and the two domains apart, rendering the protein inactive [28]. The unfolding of the prosegment, leading to a large conformational change, is captured by tryptophan fluorescence. Three Trp residues are present in pro-tPMII. Two of them (Trp128 and Trp193) are located at the interface of the two domains and form NH-π interactions through their side chains. An analysis of the crystal structures of the pro-tPMs and the mature PMII reveals that the conformation and environment of both these tryptophan residues remain unperturbed, thus should not contribute to the change in fluorescence intensity and the shift in λmax. However, Trp41 is located in the vicinity of the active site that includes the flexible ‘flap’. That residue undergoes significant changes in its conformation and its surrounding environment during the process of unfolding and cleavage of the prosegment. In pro-tPMII, the flap is open and Trp41 is surrounded by hydrophobic residues, whereas in the mature enzyme it flips almost 90° and forms a hydrogen bond with the Tyr77 in the flap. Thus, fluorescence of Trp41 serves as a probe for monitoring the open and closed conformations of the flap and the unfolding of the prosegment. In the inactive state of pro-tPMs, the flap is in an open conformation due to steric constrains from the folded prosegment. However, under acidic conditions the prosegment of pro-tPMs unfolds and the flap can close, forming a hydrogen bond between Tyr77 and Trp41. At pH 7.5, the prosegment of pro-tPMII D214A mutant is intact and the flap is in an open state, where maximum fluorescence is observed. No significant change in fluorescence is observed after addition of equimolar concentration of PepA at pH 7.5, suggesting that the active site is not accessible as the prosegment is folded. However, at pH 5.0 the prosegment unfolds and the flap can close, leading to formation of a hydrogen bond between Tyr77 and Trp41 and thus changing the electronic environment near Trp41. This change results in the reduction in the fluorescence intensity and a λmax blue shift of the emission spectra. At the same time, the addition of PepA to the pro-tPMII D214A mutant protein at pH 5.0 shows further decrease in fluorescence and λmax blue shift. As the concentration of PepA is increased, a concentration-dependent decrease in fluorescence is observed, indicating that indeed the inhibitor binds in the active site of the pro-tPMII D214A mutant with unfolded prosegment. The lowest fluorescence is observed for the mPMII and its mPMII D214A mutant where the flap is in closed conformation. It is thus evident that, under acidic conditions, the prosegment unfolds and the two domains harboring the catalytic aspartates (Asp34 and Asp214) come close to each other, forming a competent active site. Such an active site is accessible to the substrate, indicating a possibility that the enzyme can cleave its substrate peptide. Thus, these evidences clearly suggest that the pro-tPMs can be transiently active and involve in trans-activation leading to production of mature enzyme.
Our observations on pro-tPM activation are consistent with previous studies, which have shown that the zymogens of some eukaryotic aspartic proteases are catalytically active. In prorenin, the binding of its receptor or antibodies specific to the prosegment, or the binding of renin inhibitors, or exposure to lower pH and temperature facilitates reversible nonproteolytic conversion from a closed to an open intermediate state [29–35]. Such open and closed states have also been proposed for β-secretase (BACE), wherein in its open conformation the BACE zymogen binds an inhibitor in a manner similar to the mature form [36]. It has also been shown that the prosegment does not suppress the activity of the mature part, which then can cleave a substrate peptide [37]. It was observed that pro-tPM from P. berghei could bind a substrate in the active site cleft even in the presence of the prosegment [38]. The pro-tPMV zymogen is catalytically active [39,40] and no release of prosegment is observed under acidic conditions, both during auto-activation and in vivo [39,41]. These earlier studies on zymogens of pepsin-like aspartic proteases also provide sufficient evidence to strengthen our proposition that the two domains of pro-tPMs with unfolded prosegment could come close to each other. Such a conformation of the pro-tPMs enables the two catalytic aspartates to form a catalytically competent active site capable of peptide hydrolysis.
Retroviral aspartic proteases are synthesized as monomers and function as dimers, with each monomer contributing a catalytic aspartate. These proteases are synthesized as part of the polyprotein with the N terminus flanked by a trans-frame region (TFR) and reverse transcriptase at the C terminus [42,43]. TFR acts as a prosegment for HIV-1 protease and inhibits its activity by preventing dimer formation. While HIV-1 protease is monomeric in the polyprotein, transient dimers are sometimes formed which can bind the N-terminal cleavage site, leading to autoprocessing and formation of the mature enzyme [43]. The existence of such transient species suggests the possibility of similar transient interactions between the competent active site of one pro-tPM and the unfolded prosegment of the other protein, resulting in cleavage of the prosegment and leading to formation of mature enzyme by transactivation.
In summary, we propose a unique mechanism of activation of pro-tPMs to its mature form, which has not been previously reported for any other pepsin-like aspartic protease (Fig. 10). Pro-tPMs are inactive and form S-shaped dimers via the Tyr-Asp loop, L1, and H2, as depicted in Fig. 10A. Under acidic conditions, the compact pro-mature region is extended by disruption of the interaction in the Tyr-Asp loop, leading to the downward movement of L1 and H2. This extension causes the dimer to undergo an angular movement and reduces interactions at the dimer interface (Fig. 10B), ensuing formation of monomers (Fig. 10C). A decrease in pH causes protonation of the residues of the prosegment that leads to its unfolding (Fig. 10D). With our current understanding of the dynamics of pro-tPMs, we propose that the unfolded prosegment imparts flexibility and allows the two domains of the enzyme to come closer, forming the catalytically competent active site. In contrast to pepsinogen, the cleavage site of pro-tPMs is far away from the active site. It is therefore unlikely that the pro-tPMs can self-cleave its own prosegment. Hence, subsequently, this catalytically competent pro-tPM which is formed is able to cleave the prosegment of another unfolded pro-tPM (Fig. 10E), leading to formation of an initial mature enzyme (Fig. 10F). Once the first molecule of mature enzyme is formed, it rapidly converts pro-tPMs to their mature forms and the cycle continues until all protein is activated (Fig. 10G). Such maturation mechanism of pro-tPMs has not been postulated before for any other pepsin-like aspartic proteases. Our data might also explain the molecular basis for the catalytic activity of a few zymogens (e.g., PMV and BACE) belonging to pepsin-like aspartic proteases. Our findings could also contribute toward development of novel interventions to prevent activation of these pepsin-like aspartic proteases to their active mature form, which are involved in life-threatening diseases.
Fig. 10.
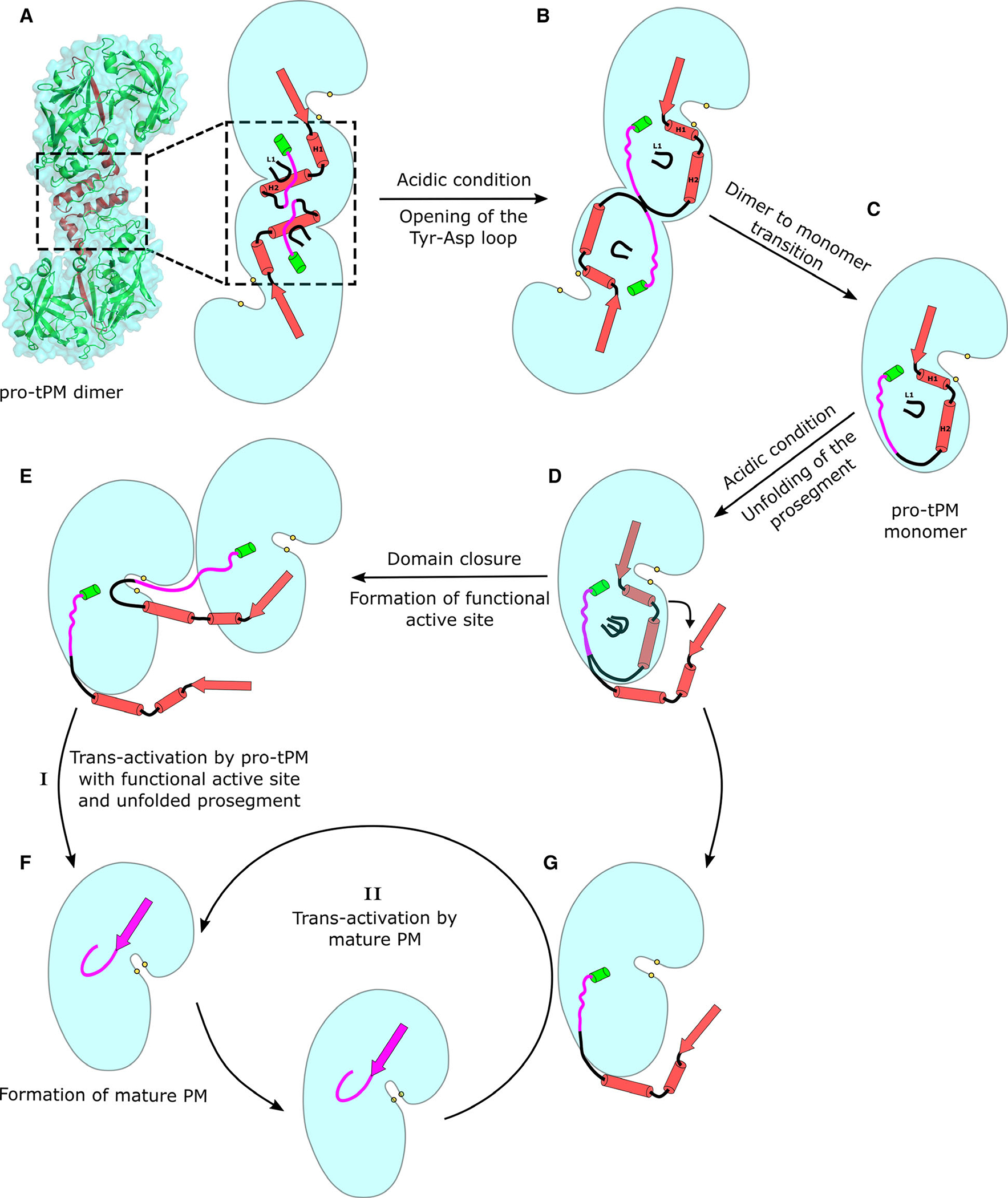
Schematic representation of the overall mechanism of trans-activation of pro-tPMs. The secondary structural elements of the truncated prosegment are represented as cartoon in red, and the pro-mature region is colored in magenta. The cyan shaded region represents the surface of the different forms of PMs (pro-tPMs and mPMs). The panels represent various stages of trans-activation of pro-tPMs to mPMs. The protein structural representation in panel(a) has been generated with the PyMOL molecular graphics system (Schrödinger LLC, New York, NY, USA; version 2.3.2).
Materials and methods
Expression and purification of pro-tPMs
The recombinant expression constructs in pET-32b of pro-tPMI, pro-tPMII, and pro-tHAP used in this study have the truncated prosegment lacking the transmembrane regions [44]. They contain thioredoxin fused at the N terminus followed by a thrombin cleavage site, an enterokinase cleavage site, and 6x His-tag, as described previously [15,44,45]. The pET-32b construct of each pro-tPM was transformed in Rosetta-gami B(DE3) pLysS, with ampicillin, chloramphenicol, kanamycin, and tetracycline utilized as selection markers. The glycerol stock of E. coli Rosetta-gami B(DE3) pLysS cells containing recombinant expression plasmid was inoculated into fresh 50 mL Luria–Bertani (LB) broth with ampicillin, chloramphenicol, kanamycin, and tetracycline (50 μg-mL−1, 34.1 μg-mL−1, 12.5 μg-mL−1, and 12.5 μg-mL−1, respectively). The culture was grown overnight under shaking conditions at 37 °C. The overnight culture was used to inoculate 6 L of LB medium with the same antibiotics at their respective concentration mentioned above and grown under shaking at 37 °C till O.D. at 600 nm reached to 1.0. Overexpression of the protein was performed by inducing the cells with a final concentration of 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG). The cells were grown for 4 h at 30 °C and were later harvested by centrifugation at 6000 g for 10 min at 4 °C.
Purification of trx-pro-tPMII were performed as previously described [19,44], with a few modifications. The cell pellet was resuspended in sodium phosphate buffer (50 mM sodium phosphate, 300 mM NaCl, 0.2% CHAPS at pH 7.5). Lysis was performed by addition of 1.0 mg-mL−1 lysozyme, 10 μg-mL−1 DNase, and 10 mM MgCl2. The contents were mixed continuously for 4 h at room temperature. The cells lysate was centrifuged at 16 000 g for 40 min, the pellet was discarded, and the supernatant was filtered through a 0.2-μm polyvinylidene difluoride (PVDF) membrane and loaded to HisTrap (GE Healthcare, Chicago, IL, USA) column equilibrated with 50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole at pH 7.5. Washing was done with sodium phosphate buffer containing 25 mM imidazole. Elution was carried out with a step gradient of sodium phosphate buffer containing 75 mM, 125 mM, and 250 mM imidazole and the fractions were collected separately. The eluted fractions were checked on SDS/PAGE. The protein-containing fractions were pooled and concentrated in 10 kDa molecular weight cutoff centrifugal concentrator. The concentrated sample was loaded onto a Superdex-75 16/600 (GE Healthcare) pre-equilibrated with 50 mM sodium phosphate, 150 mM NaCl, 0.2% CHAPS at pH 7.5. The samples were again analyzed on SDS/PAGE, and the protein-containing fractions were pooled and concentrated. Purification of the recombinant trx-pro-tPMI and trx-pro-tHAP was also performed with the same procedure as mentioned for trx-pro-tPMII.
Site-directed mutagenesis
Site-directed mutagenesis was performed using pET-32b-PMII to generate alanine-substituted trx-pro-tPMII mutants at residues Tyr121p and Asp214. Mutations were confirmed by DNA sequencing. The mutant proteins were overexpressed in Rosetta-gami B(DE3) pLysS and the same purification protocol was followed as for the trx-pro-tPMII.
Size-exclusion chromatography of wild-type and mutant forms of pro-tPMII
The thioredoxin tags of the WT and mutants of pro-tPMII were cleaved by treatment with enterokinase and the tags were removed by size-exclusion chromatography. The mature form of the enzyme was obtained as described previously [44]. The purified pro-tPMII (both WT and mutants) and mPMII were then concentrated and loaded onto Superdex-75 10/300 (GE Healthcare) pre-equilibrated with 50 mM sodium phosphate, 150 mM NaCl pH 7.5 or 100 mM sodium acetate pH 5.0. The elution profiles for each protein were monitored with absorbance at 280 nm.
Effect of pepstatin A on pro-tPMI processing
The 5 mM stock solution of PepA was prepared in 100% DMSO and stored at −20 °C. PepA was used in 10x molar excess (0.1 mM) with respect to the concentration of the protein (0.01 mM). The reaction mixture was acidified by fourfold dilution with 100 mM sodium acetate, pH 4.5 and incubated at 37 °C for 1 h. The reaction was stopped and subsequently analyzed on SDS/PAGE. The acidified pro-tPMI without the inhibitor was used as a positive control.
Tryptophan fluorescence quenching of pro-tPMII D214A
Fluorescence measurements were obtained by monitoring the changes in intrinsic tryptophan fluorescence emission of the protein samples. The measurements were carried out in a 10-mm path length quartz cell using a Jasco FP-8500 spectrofluorometer equipped with thermostatically controlled cell holder maintained at 25 °C. The pro-tPMII D214A (concentration ~ 10 μM) in the buffers containing 100 mM sodium phosphate pH 7.5 or 100 mM sodium acetate pH 5.0 was titrated with increasing concentration of PepA (0–10 μM) and the quenching of tryptophan fluorescence emission spectra was recorded between 305 and 450 nm, following excitation at 295 nm. Each spectrum is an average of five replicates and was corrected for the contribution of the buffer without the protein. The data from the experiment were analyzed using nonlinear regression in graphpad prism software (San Diego, CA, USA).
Dynamic light scattering
The dynamic light scattering (DLS) measurements of samples were performed at 25 °C using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK). The backscattered light from a 4 mW He-Ne laser (λ = 633 nm) was measured at an angle of 173°. Particle size was obtained as an average of three independent experiments. Samples of WT and mutants of pro-tPMII at the final concentration of 10 μM were prepared in buffer containing 100 mM sodium phosphate pH 7.5. Mature PMII was prepared in 100 mM sodium acetate pH 5.0 at a concentration of 10 μM. Samples were equilibrated for 60 s prior to measurement. The solvent and protein refractive indexes chosen for calculations were 1.331 and 1.450, respectively.
Crystallization of pro-tHAPs
The purified pro-tHAP sample was concentrated to 10 mg-mL−1 and used for crystallization. Crystallization screens were set up with a Phoenix crystallization robot (Protein Crystallography Facility, IIT Bombay) using commercial screens (JCSG CORE I and PEG-Suite (Qiagen, Hilden, Germany); INDEX, PEG-Ion and PEG-Rx (Hampton Research)), by the sitting drop vapor diffusion method at 295 K. Each well contained 0.3 μL protein and 0.3 μL of reservoir solution, equilibrated against 50 μL of reservoir solution. Crystals of pro-tHAP were obtained in three different crystallization conditions: (a) 200 mM ammonium citrate tribasic pH 7.0, 100 mM imidazole pH 7.0, 20% w/v polyethylene glycol monomethyl ether 2000; (b) 200 mM lithium citrate tribasic tetrahydrate, 20% w/v polyethylene glycol 3350; and (c) 200 mM ammonium phosphate dibasic, 20% w/v polyethylene glycol 3350. The crystals grew to their maximum size within 4–5 days. The pro-tHAP structures solved with the crystals obtained in crystallization conditions a, b, and c are referred to here as HAP-zymo1, HAP-zymo2, and HAP-zymo3, respectively.
Diffraction data collection
For diffraction data collection, crystals of pro-tHAP were cryoprotected with a mother liquor containing 30% glycerol and then flash-frozen in a liquid nitrogen stream at 100 K. Diffraction data were collected by the rotation method with a Rigaku MicroMax 007HF generator equipped with R-Axis IV++ detector using CuKα X-ray radiation (1.5418 Å) at the Protein Crystallography Facility, IIT Bombay, Mumbai. Diffraction data for HAP-zymo1, HAP-zymo2, and HAP-zymo3 were collected with an exposure of 10 min, at a detector distance of 140, 180, and 200 mm, respectively. All data sets were indexed, integrated, and scaled with XDS [46]. The intensities were converted to structure factors with the programs F2MTZ and CAD of CCP4 [47].
Structure solution and refinement
The initial phases of pro-tHAP were obtained by molecular replacement using the structure of pro-tHAP (3QVC). The calculated Mathews coefficients for HAP-zymo1, HAP-zymo2, and HAP-zymo3 are 2.97, 2.98, and 2.99 Å3 Da−1, respectively, indicating the presence of one molecule in the asymmetric unit. The correct orientation of the molecule was found by PHASER [48]. Further rounds of manual model building and electron density interpretation were performed in coot [49], refmac5 [50], and phenix.refine of the PHENIX package [51] which were used for refinement. Crystal structures were modeled with TLS and were divided into four TLS groups: −4–117p, 118p-9, 10–186, 187–328; −1–122p, 123p-4, 5–196, 197–328; and 77p-119p, 120p-10, 11–218, 219–328 for HAP-zymo1, HAP-zymo2, and HAP-zymo3, respectively [52]. The electron density around the pro-mature region was poor but still sufficient to build the main chain and fit some of the side chains, except in the case of HAP-zymo3.
Small-Angle X-ray Scattering of pro-tPMI
Small-Angle X-ray Scattering (SAXS) data were collected using Rigaku BioSAXS-2000 at InSTEM, National Centre for Biological Sciences, Bangalore. The camera is based on a two-dimensional Kratky design mounted on the open port of a Rigaku rotating anode X-ray generator. Pro-tPMI was purified in 50 mM sodium phosphate, 150 mM sodium chloride, 0.2% CHAPS, pH 7.5, and three different concentrations (3, 4, and 6 mg-mL−1) were used for data collection. Data were collected for 30 min for each protein concentration, and a reading of the buffer was used for background subtraction. The measurements were performed at 25 ± 1 °C using a custom-made thermostat cell with a sample volume of 30 μL. For each data point, a total of five measurements of 0.1 s integration time were recorded. Data were image-corrected and circularly averaged; the five profiles for each condition were averaged to improve signal quality. Data were automatically processed by the AAP module of ATSAS [53] and its quality was evaluated by Kratky, Guinier, and P(r) distribution plots. After averaging in DAMAVER, the ‘damstart’ model was used for a round of refinement in DAMMIN [54], yielding the final SAXS envelope.
Molecular dynamics simulation
Two structures, HAP-zymo1 (6KUB) and Pv-pro-tPM (1MIQ), were used for molecular dynamics simulations. Topology and force field parameters for all atoms were assigned from CHARMM22 with CMAP correction parameter set [55–57]. The proteins 6KUB and 1MIQ were first solvated using 12301 and 13875 TIP3P water molecules within a rectangular box with dimensions 98 × 64 × 76 and 98 × 74 × 74 Å3, respectively. 35 and 49 Na+ and 36 and 39 Cl− ions were added to HAP-zymo1 and 1MIQ, respectively, to mimic the physiological ion concentration of 0.15 m. Long range electrostatic interactions were calculated using particle mesh Ewald (PME) method with a cutoff of 12 Å [58]. A cutoff for both electrostatic interactions and van der Waals interactions was set to 12 Å. Bonds to hydrogen atoms were constrained using the SHAKE algorithm. The equation of motion was integrated every 2 fs, and snapshots were saved every 0.2 ps. The temperature was controlled using Langevin dynamics with friction coefficient of 1 ps−1. Water molecules and ions present in the system were first subjected to energy minimization for 10 000 steps to remove any bad contacts with strong restraints on the protein (500 kcal·mol−1·Å−2). This was followed by minimization of the entire system without any restraints for another 10 000 steps. The system was then gradually heated to 298 K in 14 steps of 2 ps each at constant volume. Weak restraints of 10 kcal·mol−1·Å−2 were applied to the protein. The density of the system was then relaxed for 40 ps by switching to NPT ensemble with equilibrium pressure of 1 atm and temperature of 298 K. Constant pressure was maintained using Nosé–Hoover Langevin piston pressure control. The restraints on the protein were then removed and the system was further equilibrated for 100 ps. Finally, production simulations of ~10 ns were carried out for each system in the NPT ensemble at 298 K and 1 atm pressure. All simulations were performed using namd v2.9 [59].
Supplementary Material
Acknowledgements
We acknowledge the Protein Crystallography Facility at IIT Bombay for providing crystallization and diffraction facilities. We thank Prof. P. Sunthar, Department of Chemical Engineering, IIT Bombay, and Prof. Kamendra Sharma, Department of Chemistry, IIT Bombay, for providing access to the DLS instrument. The authors would also like to thank Nicholas Grant for critical reading of the manuscript and Suman Pandey for collecting SAXS data at National Centre for Biological Sciences, NCBS, Bangalore. IR acknowledges the Ph.D. fellowship from the University Grant Commission (UGC). VM thanks Department of Biotechnology (DBT), Ministry of Science and Technology, India, for Ph.D. fellowship. The work was supported by the Ramalingaswami Reentry Fellowship (DBT), research seed grant from IRCC, IIT Bombay, to Prasenjit Bhaumik, and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The financial support of the Natural Sciences and Engineering Research Council of Canada (HX and RYY) is also gratefully acknowledged.
Abbreviations
- HAP
histo-aspartic protease
- mPM
mature plasmepsin
- PMI
plasmepsin
- PMII
plasmepsin II
- PMIV
plasmepsin IV
- PMs
plasmepsins
- pro-PMs
proplasmepsins
- pro-tPMs
truncated proplasmepsins
- Pv
Plasmodium vivax
- trx-pro-tPM
thioredoxin tagged truncated proplasmepsin
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Database
Atomic coordinates and structure factors have been submitted in the Protein Data Bank (PDB) under the PDB IDs 6KUB, 6KUC, and 6KUD.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Movie S1. Conformational changes occurring in the promature region of pro-tPMs with the disruption of the interactions in the Tyr-Asp loop.Movie S2. Structural changes occurring in the S-shaped dimeric form of pro-tPMs due to the extension of promature region and breakage of contacts in the Tyr-Asp loop.
References
- 1.WHO (2016) World Malaria Report 2016.
- 2.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA & Dondorp AM (2014) Malaria. Lancet 383, 723–735. [DOI] [PubMed] [Google Scholar]
- 3.Mackintosh CL, Beeson JG & Marsh K (2004) Clinical features and pathogenesis of severe malaria. Trends Parasitol 20, 597–603. [DOI] [PubMed] [Google Scholar]
- 4.Aguiar ACC, da Rocha EMM, De Souza NB, Franca TCC & Krettli AU (2012) New approaches in antimalarial drug discovery and development: a review. Mem Inst Oswaldo Cruz 107, 831–845. [DOI] [PubMed] [Google Scholar]
- 5.Francis SE, Banerjee R & Goldberg DE (1997) Biosynthesis and maturation of the malaria aspartic hemoglobinases plasmepsins I and II. J Biol Chem 272, 14961–14968. [DOI] [PubMed] [Google Scholar]
- 6.Kolakovich KA, Gluzman IY, Duffin KL & Goldberg DE (1997) Generation of hemoglobin peptides in the acidic digestive vacuole of Plasmodium falciparum implicates peptide transport in amino acid production. Mol Biochem Parasitol 87, 123–135. [DOI] [PubMed] [Google Scholar]
- 7.Xiao H, Tanaka T, Ogawa M & Yada RY (2007) Expression and enzymatic characterization of the soluble recombinant plasmepsin I from Plasmodium falciparum. Protein Eng Des Sel 20, 625–633. [DOI] [PubMed] [Google Scholar]
- 8.Bhaumik P, Gustchina A & Wlodawer A (2012) Structural studies of vacuolar plasmepsins. Biochim Biophys Acta 1824, 207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedi RK, Patel C, Mishra V, Xiao H, Yada RY & Bhaumik P (2016) Understanding the structural basis of substrate recognition by Plasmodium falciparum plasmepsin V to aid in the design of potent inhibitors. Sci Rep 6, 31420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M & Goldberg DE (2002) Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci USA 99, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J & Mottram JC (2001) Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol 17, 532–537. [DOI] [PubMed] [Google Scholar]
- 12.Berry C, Humphreys MJ, Matharu P, Granger R, Horrocks P, Moon RP, Certa U, Ridley RG, Bur D & Kay J (1999) A distinct member of the aspartic proteinase gene family from the human malaria parasite Plasmodium falciparum. FEBS Lett 447, 149–154. [DOI] [PubMed] [Google Scholar]
- 13.Koelsch G, Mares M, Metcalf P & Fusek M (1994) Multiple functions of pro-parts of aspartic proteinase zymogens. FEBS Lett 343, 6–10. [DOI] [PubMed] [Google Scholar]
- 14.Hill J, Tyas L, Phylip LH, Kay J, Dunn BM & Berry C (1994) High level expression and characterisation of Plasmepsin II, an aspartic proteinase from Plasmodium falciparum. FEBS Lett 352, 155–158. [DOI] [PubMed] [Google Scholar]
- 15.Xiao H, Sinkovits AF, Bryksa BC, Ogawa M & Yada RY (2006) Recombinant expression and partial characterization of an active soluble histo-aspartic protease from Plasmodium falciparum. Protein Expr Purif 49, 88–94. [DOI] [PubMed] [Google Scholar]
- 16.Jaafar AH, Xiao H, Dee DR, Bryksa BC, Bhaumik P & Yada RY (2016) The prosegment catalyzes native folding of Plasmodium falciparum plasmepsin II. Biochim Biophys Acta – Proteins Proteomics 1864, 1356–1362. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein NK, Cherney MM, Loetscher H, Ridley RG & James MN (1999) Crystal structure of the novel aspartic proteinase zymogen proplasmepsin II from Plasmodium falciparum. Nat Struct Biol 6, 32–37. [DOI] [PubMed] [Google Scholar]
- 18.Recacha R, Jaudzems K, Akopjana I, Jirgensons A & Tars K (2016) Crystal structure of Plasmodium falciparum proplasmepsin IV: the plasticity of proplasmepsins. Acta Cryst F 72, 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhaumik P, Xiao H, Hidaka K, Gustchina A, Kiso Y, Yada RY & Wlodawer A (2011) Structural insights into the activation and inhibition of histo-aspartic protease from Plasmodium falciparum. Biochemistry 50, 8862–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein NK, Cherney MM, Yowell CA, Dame JB & James MNG (2003) Structural insights into the activation of P vivax plasmepsin. J Mol Biol 329, 505–524. [DOI] [PubMed] [Google Scholar]
- 21.Khan AR, Khazanovich-Bernstein N, Bergmann EM & James MN (1999) Structural aspects of activation pathways of aspartic protease zymogens and viral 3C protease precursors. Proc Natl Acad Sci U S A 96, 10968–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyas L, Gluzman IY, Moon RP, Rupp K, Westling J, Ridley RG, Kay J, Goldberg DE & Berry C (1999) Naturally-occurring and recombinant forms of the aspartic proteinases plasmepsins I and II from the human malaria parasite Plasmodium falciparum. FEBS Lett 454, 210–214. [DOI] [PubMed] [Google Scholar]
- 23.Parr CL, Tanaka T, Xiao H & Yada RY (2008) The catalytic significance of the proposed active site residues in Plasmodium falciparum histoaspartic protease. FEBS J 275, 1698–1707. [DOI] [PubMed] [Google Scholar]
- 24.Moon RP, Tyas L, Certa U, Rupp K, Bur D, Jacquet C, Matile H, Loetscher H, Grueninger-Leitch F, Kay J et al. (1997) Expression and characterisation of plasmepsin I from Plasmodium falciparum. Eur J Biochem 244, 552–560. [DOI] [PubMed] [Google Scholar]
- 25.Glatter O & Kratky O (1982) Small Angle X-ray Scattering. London, UK: Academic Press. [Google Scholar]
- 26.Richter C, Tanaka T & Yada RY (1998) Mechanism of activation of the gastric aspartic proteinases: pepsinogen, progastricsin and prochymosin. Biochem J 335, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YM, Lee MH, Piao TG, Lee JW, Kim JH, Lee S, Choi KM, Jiang JH, Kim TU & Park H (2006) Prodomain processing of recombinant Plasmepsin II and IV, the aspartic proteases of Plasmodium falciparum, is auto- and trans-catalytic. J Biochem 139, 189–195. [DOI] [PubMed] [Google Scholar]
- 28.Bernstein NK & James MN (1999) Novel ways to prevent proteolysis – prophytepsin and proplasmepsin II. Curr Opin Struct Biol 9, 684–689. [DOI] [PubMed] [Google Scholar]
- 29.Morales R, Watier Y & Böcskei Z (2012) Human prorenin structure sheds light on a novel mechanism of its autoinhibition and on its non-proteolytic activation by the (Pro)renin receptor. J Mol Biol 421, 100–111. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki F, Hayakawa M, Nakagawa T, Nasir UM, Ebihara A, Iwasawa A, Ishida Y, Nakamura Y & Murakami K (2003) Human prorenin has “Gate and Handle” regions for its non-proteolytic activation. J Biol Chem 278, 22217–22222. [DOI] [PubMed] [Google Scholar]
- 31.Edalji R, Holzman TF & Gubbins EJ (1991) Active prorenin: Evidence for the formation of a conformational variant of recombinant human prorenin. J Protein Chem 10, 403–406. [DOI] [PubMed] [Google Scholar]
- 32.Derkx FH, Deinum J, Lipovski M, Verhaar M, Fischli W & Schalekamp MA (1992) Nonproteolytic “activation” of prorenin by active site-directed renin inhibitors as demonstrated by renin-specific monoclonal antibody. J Biol Chem 267, 22837–22842. [PubMed] [Google Scholar]
- 33.Derkx FH, Schalekamp MP & Schalekamp MA (1987) Two-step prorenin-renin conversion Isolation of an intermediary form of activated prorenin. J Biol Chem 262, 2472–2477. [PubMed] [Google Scholar]
- 34.Pitarresi TM, Rubattu S, Heinrikson R & Sealey JE (1992) Reversible cryoactivation of recombinant human prorenin. J Biol Chem 267, 11753–11759. [PubMed] [Google Scholar]
- 35.Leckie BJ & McGhee NK (1980) Reversible activation-inactivation of renin in human plasma. Nature 288, 702–705. [DOI] [PubMed] [Google Scholar]
- 36.Ermolieff J, Loy JA, Koelsch G & Tang J (2000) Proteolytic activation of recombinant pro-memapsin 2 (Pro-β-secretase) studied with new fluorogenic substrates. Biochemistry 39, 12450–12456. [DOI] [PubMed] [Google Scholar]
- 37.Shi X-P, Chen E, Yin K-C, Na S, Garsky VM, Lai M-T, Li Y-M, Platchek M, Register RB, Sardana MK et al. (2001) The pro domain of β-secretase does not confer strict zymogen-like properties but does assist proper folding of the protease domain. J Biol Chem 276, 10366–10373. [DOI] [PubMed] [Google Scholar]
- 38.Liu P, Robbins AH, Marzahn MR, McClung SH, Yowell CA, Stevens SM, Dame JB & Dunn BM (2015) Enzymatic characterization of recombinant food vacuole plasmepsin 4 from the rodent malaria parasite Plasmodium berghei. PLoS One 10, e0141758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao H, Bryksa BC, Bhaumik P, Gustchina A, Kiso Y, Yao SQ, Wlodawer A & Yada RY (2014) The zymogen of plasmepsin V from Plasmodium falciparum is enzymatically active. Mol Biochem Parasitol 197, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boonyalai N, Sittikul P & Yuvaniyama J (2015) Plasmodium falciparum Plasmepsin V (Pf PMV): Insights into recombinant expression, substrate specificity and active site structure. Mol Biochem Parasitol 201, 5–15. [DOI] [PubMed] [Google Scholar]
- 41.Klemba M & Goldberg DE (2005) Characterization of plasmepsin V, a membrane-bound aspartic protease homolog in the endoplasmic reticulum of Plasmodium falciparum. Mol Biochem Parasitol 143, 183–191. [DOI] [PubMed] [Google Scholar]
- 42.Louis JM, Clore GM & Gronenborn AM (1999) Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol 6, 868–875. [DOI] [PubMed] [Google Scholar]
- 43.Tang C, Louis JM, Aniana A, Suh J-Y & Clore GM (2008) Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature 455, 693–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mishra V, Rathore I, Arekar A, Sthanam LK, Xiao H, Kiso Y, Sen S, Patankar S, Gustchina A, Hidaka K et al. (2018) Deciphering the mechanism of potent peptidomimetic inhibitors targeting plasmepsins – biochemical and structural insights. FEBS J 285, 3077–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhaumik P, Xiao H, Parr CL, Kiso Y, Gustchina A, Yada RY & Wlodawer A (2009) Crystal structures of the histo-aspartic protease (HAP) from Plasmodium falciparum. J Mol Biol 388, 520–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch W (2010) XDS. Acta Cryst D 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A et al. (2011) Overview of the CCP 4 suite and current developments. Acta Cryst D 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P & Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Cryst D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 50.Murshudov GN, Vagin AA & Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 51.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH & Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst D 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Painter J & Merritt EA (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Cryst D 62, 439–450. [DOI] [PubMed] [Google Scholar]
- 53.Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM & et al. (2017) ATSAS 28: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svergun DI (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 76, 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKerell AD, Feig M & Brooks CL (2004) Improved treatment of the protein backbone in empirical force fields. J Am Chem Soc 126, 698–699. [DOI] [PubMed] [Google Scholar]
- 56.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. JPhys Chem B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 57.Mackerell AD, Feig M & Brooks CL (2004) Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- 58.Darden T, York D & Pedersen L (1993) Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J Chem Phys 98, 10089–10092. [Google Scholar]
- 59.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L & Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robert X & Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.