
Figure 1:
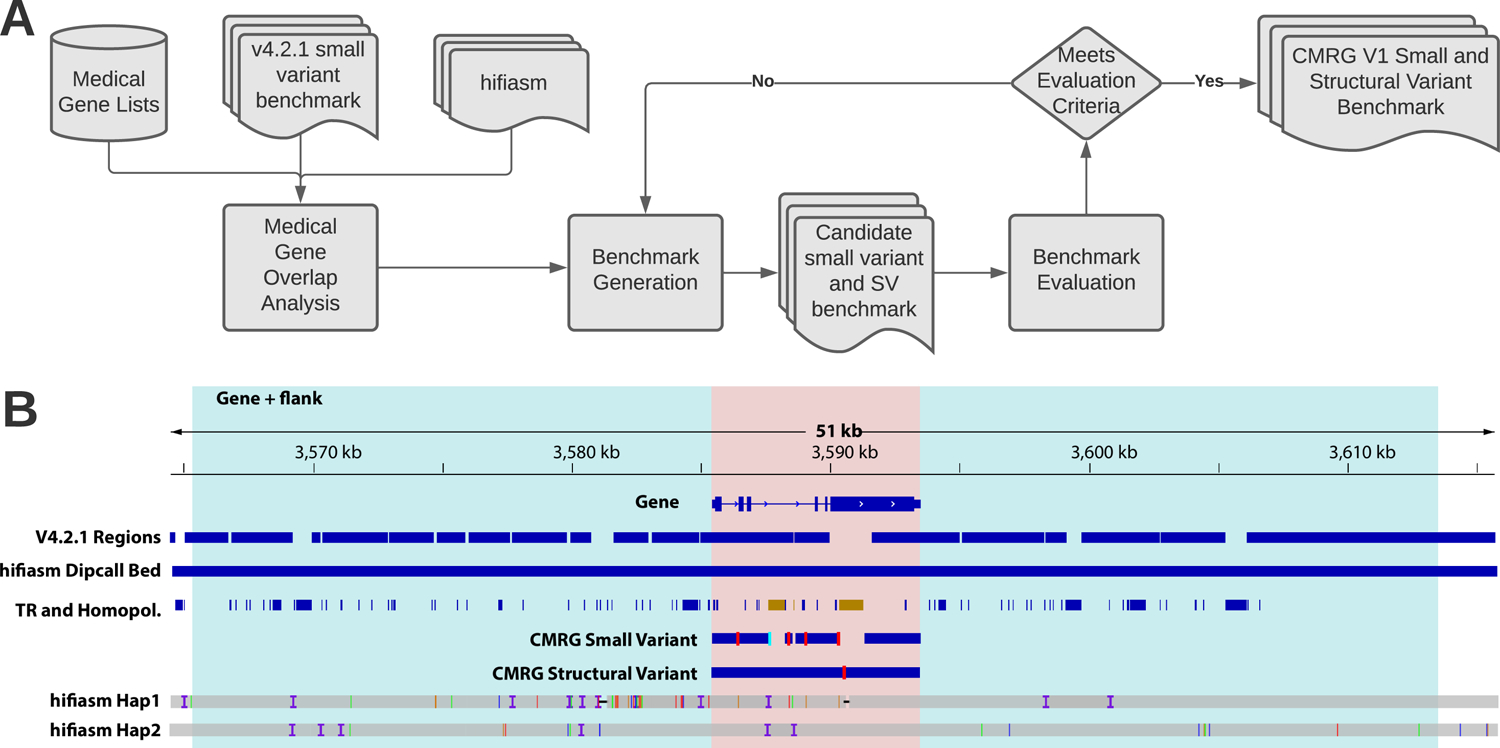
GIAB developed a process to create new phased small variant and structural variant benchmarks for 273 challenging, medically relevant genes. (A) We developed a list of 4,701 autosomal potentially medically relevant genes. We generated a new benchmark for 273 of the 4,701 genes that were completely resolved by our hifiasm haplotype-resolveddiploid assembly and <=90% included in the v4.2.1 GIAB small variant benchmark for HG002 (V4.2.1 Regions). (B) We required that the entire gene region (pink) and 20 kb flanking sequence on each side (blue) were completely resolved by both haplotypes in the assembly (hifiasm Hap1 and hifiasm Hap2), indicated with the hifiasm Dipcall Bed track. In addition, we required that any segmental duplications overlapping the gene were completely resolved. From the small variant benchmark regions (CMRG Small Variant blue bars), we excluded SVs and any tandem repeats or homopolymers overlapping SVs (right TR and Homopol. region in brown). The left TR and Homopol. region in brown is excluded from the small variant benchmark regions because the larger tandem repeat contains an imperfect homopolymer longer than 20 bp, which we exclude because long homopolymers have a higher error rate in the assembly. All regions of this gene were included in the SV benchmark regions (CMRG Structural Variant blue bar). The vertical red lines in CMRG Small Variant and CMRG Structural Variant indicate locations of benchmark small variants and SVs, respectively. Finally, we evaluated the small variant and structural variant benchmarks with manual curation and long range PCR, and also ensured they accurately identify false positives and false negatives after excluding errors found during curation.