

Abstract
Extracellular vesicles (EVs) have shown promise as biological delivery vehicles, but therapeutic applications require efficient cargo loading. Here, we developed new methods for CRISPR/Cas9 loading into EVs through reversible heterodimerization of Cas9‐fusions with EV sorting partners. Cas9‐loaded EVs were collected from engineered Expi293F cells using standard methodology, characterized using nanoparticle tracking analysis, western blotting, and transmission electron microscopy and analysed for CRISPR/Cas9‐mediated functional gene editing in a Cre‐reporter cellular assay. Light‐induced dimerization using Cryptochrome 2 combined with CD9 or a Myristoylation‐Palmitoylation‐Palmitoylation lipid modification resulted in efficient loading with approximately 25 Cas9 molecules per EV and high functional delivery with 51% gene editing of the Cre reporter cassette in HEK293 and 25% in HepG2 cells, respectively. This approach was also effective for targeting knock‐down of the therapeutically relevant PCSK9 gene with 6% indel efficiency in HEK293. Cas9 transfer was detergent‐sensitive and associated with the EV fractions after size exclusion chromatography, indicative of EV‐mediated transfer. Considering the advantages of EVs over other delivery vectors we envision that this study will prove useful for a range of therapeutic applications, including CRISPR/Cas9 mediated genome editing.
Keywords: CRISPR/Cas9 delivery, exosomes, extracellular vesicles, gene editing, optogenetics
1. INTRODUCTION
Clustered regularly interspaced short palindromic repeats (CRISPR)‐associated‐endonuclease‐9 (Cas9) allows for precise gene editing and holds great promise for therapeutic treatment of genetic disorders. One of the main challenges for the therapeutic application of CRISPR/Cas9 is the safe, efficacious and targeted delivery of Cas9 protein and its associated machinery to diseased cells (Martinez‐Lage et al., 2018). In particular, the large size of Streptococcus pyogenes Cas9 (4113 nucleotides/ 1371 amino acids) poses a significant challenge as this exceeds the passenger capacity of regular AAV viral vectors for DNA‐based delivery of Cas9, and it remains challenging to encapsulate such a large protein in nanoparticles (Rehman et al., 2016). Research efforts into CRISPR/Cas9 therapeutic delivery using viral and lipid nanoparticle‐based delivery vehicles are rapidly expanding, but there is a clear need for the development of safe and more efficient delivery approaches (Luther et al., 2018).
Extracellular vesicles (EVs), including exosomes and microvesicles among other subtypes, are nanometre sized particles released by virtually every cell type and are involved in the transfer of functionally active proteins, lipids, and nucleic acids between cells (Yáñez‐Mó et al., 2015). EVs play important roles in many physiological and pathological processes related to cancer (Steinbichler et al., 2017), cardiovascular (Osteikoetxea et al., 2016), and neurodegenerative diseases (Soria et al., 2017). Several groups have started to explore the use of EVs for the delivery of various therapeutic cargoes including drugs (Jang et al., 2013; Saari et al., 2015; Schindler et al., 2019), nucleic acids (Kamerkar et al., 2017) and proteins (Fuhrmann et al., 2015; Wang et al., 2018). Therapeutic cargo can be loaded into the EVs by passive loading into the particle during co‐incubation of the purified vesicles with drug, or by applying transient physical disruption of the lipid bilayer using techniques such as electroporation, freeze‐thaw, sonication, extrusion, to allow the cargo to redistribute into the particles (Liu & Su, 2019). Alternatively, producer cells can be genetically engineered to release EVs which already carry the therapeutic cargo. This approach eliminates all the manipulation steps required to load cargo after EV isolation, and could facilitate large scale production for therapeutic application. Without modifications for active sorting into EVs the size of the cargo to be loaded into EVs is typically limited to smaller proteins and short stretches of RNA. Loading of multiple copies of large protein cargoes into EVs remains challenging (Fuhrmann et al., 2015; Liu & Su, 2019).
To overcome the challenge of loading large proteins into EVs we explored various protein heterodimerization techniques as a way of reversibly securing the cargo into EVs. One of the partners of the heterodimer was fused to Cas9 as cargo. The other partner was fused to a protein that accumulates in EVs (e.g., one of the tetraspanins that are highly abundant in EVs), or modified with a fatty acid moiety, both of which are referred to as the EV‐sorting motif. This results in membrane localization and preferential incorporation of these modified proteins into EVs (Fang et al., 2007; Lai et al., 2015). Having control over protein heterodimerization allows the EV loading system to switch between active loading during EV production (ON conditions to drive heterodimerization), and release of the cargo in the recipient cell to allow the Cas9 cargo to freely traffic to the nucleus, which is the site of action of the gene editing event (OFF condition to stop heterodimerization). The dimerization systems explored in this work are summarized in Table 1. These comprise three light‐inducible systems and a small molecule‐inducible system. In addition to the different heterodimerization systems, we also screened several EV‐sorting motifs for their ability to drive accumulation of cargo into the EVs.
TABLE 1.
Summary of protein dimerization systems used in this study for Cas9 EV loading
| Partner System | Size | Dimerization trigger | References | |
|---|---|---|---|---|
|
A B |
Cryptochrome 2 (CRY2) Cryptochrome‐interacting basic helix‐loop‐helix 1 (CIBN) |
57 kDa 19 kDa |
450–488 nm light | (Choi et al., 2020; Kennedy et al., 2010; Lin & Todo, 2005; Yim et al., 2016) |
|
A B |
Truncated Phytochrome B (PHYB) Phytochrome‐interacting factor 6 (PIF6) |
49 kDa 41 kDa |
650 nm light | (Levskaya et al., 2009; Müller et al., 2013; Uda et al., 2017) |
|
A B |
Vivid‐based Magnet positively‐charged (pMags) Vivid‐based Magnet negatively‐charged (nMags) |
17 kDa 17 kDa |
470 nm light | (Kawano et al., 2015) |
|
A B |
FK506‐binding protein (FKBP) FKBP‐rapamycin‐binding domain (FRB) |
12 kDa 12 kDa |
Rapalog ligand | (Heath et al., 2019; Putyrski & Schultz, 2012) |
Different binding partners of each dimerization system were fused to either Cas9 or to a range of EV‐sorting motifs to allow for Cas9 loading into EVs.
One of the most widely used systems for light‐inducible protein dimerization is that of Cryptochrome 2 (CRY2) and its interacting protein Cryptochrome‐interacting basic helix‐loop‐helix 1 (CIB1) or CIB1 truncated version (CIBN) (Kennedy et al., 2010). CIBN and CRY2 require exposure to blue light (450–488 nm) for dimerization in the presence of flavin adenine dinucleotide (FAD) chromophore as a photo‐absorbing chromophore (Lin & Todo, 2005). Interestingly, the CIBN‐CRY2 system has been successfully used for loading of Cre recombinase (Cre) and the NF‐κB pathway suppressor srIκB into EVs (Choi et al., 2020; Yim et al., 2016).
Phytochrome B (PHYB) and its interacting protein Phytochrome‐interacting factor 6 (PIF6) requires light (630 nm) and the presence of phycocyanobilin (PCB) for heterodimerization. One drawback of this approach is that the photoabsorbing chromophore PCB is not present in mammals and needs to be added exogenously for the system to be functional (Levskaya et al., 2009). Alternatively, the biosynthetic pathway can be introduced in the mammalian cells by introduction of the bacterial genes involved in synthesis of PCB from heme (Müller et al., 2013), which was the approach applied in this study.
Vivid‐based magnet heterodimerization is based on charge‐matched engineered versions of Vivid (VVD) proteins in which one heterodimer partner is engineered to display positive charges at its surface and the other carries negative charges, favouring heterodimerization over the homodimer, which is the normal oligomer for VVD when exposed to blue light (470 nm) (Kawano et al., 2015).
Lastly, we explored a small molecule‐induced dimerization system that we have shown previously to be suitable for Cre recombinase loading into EVs (Heath et al., 2019). This system consists of FK506‐binding protein (FKBP) and its interacting protein FKBP‐rapamycin‐binding domain (FRB). The two protein domains heterodimerize in the presence of a rapalog dimerizing ligand (Heath et al., 2019; Putyrski & Schultz, 2012).
Here, we developed and compared the efficiency of the above protein heterodimerization systems for loading of a large protein cargo, Cas9, into EVs and assessed Cas9 transfer to target cells.
To test for efficient functional delivery of Cas9 we repurposed an existing a Cre‐Lox fluorescent reporter by designing a CRISPR single guide RNA (sgRNA) targeting both LoxP sites. This enabled us to determine the percentage of cells where Cas9 was functionally delivered by their change in fluorescence. In the absence of editing the reporter cells show green fluorescent protein (GFP) expression, whereas following successful editing they switch colour due to red fluorescent protein (RFP) expression.
This modified Cre‐Lox reporter enabled us to determine the functionality of the Cas9 fusion proteins, as well as the efficacy for EV delivery of Cas9 with the different heterodimerization systems and EV sorting motifs. The response of the reporter cells to Cas9 loaded EVs was measured in two ways: following treatment with isolated EVs, and in a transwell model where the reporter cells were separated from the Cas9 EV secreting cells by a 0.4 μm membrane.
After comparing Cas9 delivery to reporter cells by EVs loaded using different strategies we next proceeded with the most efficient combination to test whether a therapeutically relevant gene could be edited with EVs choosing the Proprotein convertase subtilisin/kexin type 9 (PCSK9) gene as a target. PCSK9 regulates the life cycle of low‐density lipoprotein (LDL) receptor, binding to its extracellular domain and targeting it for endosomal degradation. Ultimately, this leads to a decrease in the levels of circulating LDL cholesterol with extensive evidence that PCSK9 knockout/down‐regulation reduces circulating LDL cholesterol levels, resulting in lower risk of atherosclerotic cardiovascular disease (Gennemark et al., 2021; Musunuru et al., 2021; Rothgangl et al., 2021; Wang et al., 2021). Gene editing of PCSK9 was measured using Tracking of Indels by DEcomposition (TIDE) (Brinkman et al., 2014) and amplicon next‐generation sequencing.
2. MATERIALS AND METHODS
2.1. Cell culture
Human embryonic kidney Expi293F (Thermo Fisher Scientific) cells were cultured in a shaking incubator with Expi293 media (Thermo Fisher Scientific) at 37°C in 8% CO2 with 125 rpm. HEK293‐loxP‐GFP‐RFP (puromycin) Cre reporter cells (Amsbio), referred to as reporter cells throughout the text, were cultured in high glucose DMEM, supplemented with 1% GlutaMAX, 10% FCS (all Gibco), and 5 μg/ml Puromycin (Sigma Aldrich) at 37°C in 5% CO2. Hek293T (ECACC Cat# 12022001, Lot: 18K217) and HepG2 (ECACC CCat# 85011430, Lot: 19B009) cells were cultured in high glucose DMEM, supplemented with 1% GlutaMAX, 10% FCS (all Gibco).
2.2. Cell transfection
Transfection of Expi293F for Cas9 EV production: 40 kDa PEI max (Polysciences) transfection reagent was prepared at 1 mg/ml, sterile filtered (0.2 μm) and stored at ‐20°C. 24 h after seeding Expi293F cells at 2 × 106 cells/ml, PEI max‐DNA complexes were prepared at a ratio of 25 μg DNA with 100 μl PEI Max and incubated at room temperature for 15 min before addition to 25 ml of cells. In conditions where Cas9 and the EV recruiting protein without sgRNA were co‐transfected the plasmid DNA ratio was 1:1. In conditions where Cas9, the EV recruiting protein, and sgRNA were co‐transfected the plasmid DNA ratio was 2:2:1, respectively. 24 h after transfection, cells were pelleted by centrifugation at 300 × g for 10 min to wash away residual PEI‐max‐DNA complexes. The cell pellet was washed again in PBS and re‐suspended in 50 ml of fresh media. Cells conditioned the media for 48 h before EVs were harvested. For rapalog induced loading of EVs, transfected cells were incubated with AP21967 (Takara A/C heterodimerizer) during the 48 h of EV media conditioning period. For light induced loading of EVs, transfected cells were placed under a blue or red LED light during the 48 h of EV media conditioning period.
For plasmid control conditions and LoxP sgRNA expression in reporter cells FuGENE HD (Promega) was used according to manufacturer instructions. For 5000 reporter cells per well seeded in 384‐well plates (Greiner Bio‐One), 1.75 μl OptiMEM Media with 0.035 μg DNA and 0.12 μl of FuGENE HD for each well were first mixed and incubated at room temperature for 15 min and added to cells 16 h before starting experiments with EV treatments.
2.3. DNA constructs design
In order to use reporter cells for Cas9 detection a LoxP sgRNA was designed with the following sequence, ATTATACGAAGTTATCACCA, and cloned in a sgRNA expression plasmid under human U6 promoter. For gene editing of PCSK9 a sgRNA was designed with the following sequence, TCATCCGCCCGGTACCGTGG, and cloned in a sgRNA expression plasmid under human U6 promoter.
Constructs for Cas9 and EV sorting protein expression were generated by restriction enzyme, site directed mutagenesis, or Gibson assembly cloning of the relevant DNA sequences into mammalian expression plasmid under CMV promoter. All amino acid sequences of the protein domains used are shown in Table S2. Throughout the manuscript all fusion proteins names are given accounting for their architecture such that Cas9CRY2 represents N terminus‐Cas9‐CRY2‐C‐terminus and CIBN‐CD9 represents N‐terminus‐CIBN‐CD9‐C‐terminus. Fusion protein schematics are available in Figure S1. Notably, Cas9 fusion proteins contain an N‐Terminal SV40 nuclear localization signal (NLS) with amino acid sequence PKKKRKV and protein dimerization domains were fused to the C‐terminus.
2.4. EV collection
For each cargo loading condition 45 ml of cell conditioned media was processed with an ultracentrifugation protocol adapted from (Osteikoetxea et al., 2015). Cell viability at time of EV isolation was 95.4%±2.1% (mean ± SD). Cell‐conditioned media was first centrifuged at 300 × g for 10 min to pellet cells and then at 2500 × g for 15 min to pellet dead cells and larger EVs. Remaining supernatant was transferred to 38 ml polyallomer centrifuge tubes (Beckman Coulter) and centrifuged in an Optima L‐90K ultracentrifuge (Beckman Coulter) using a SW32 Ti rotor (k‐Factor 204), at 16,000 × g for 30 min to deplete for remaining large and intermediate size EVs. Then a 100,000 × g spin for 70 min was used to pellet EVs for this study. The supernatant was discarded and pellets were collected in 1.5 ml PBS and re‐pelleted at 100,000 × g using a SW32 Ti rotor. The resulting pellet was re‐suspended in a final volume of 200 μl of PBS followed by aliquoting for downstream experiments and storage at ‐80°C until use. For control experiments with additional purification by size exclusion chromatography (SEC), 150μl of the final EV isolates were applied to qEVsingle / 70 nm columns (IZON Cat# SP2) and manufacturer's instructions were followed to separate an initial 1 ml void fraction, and subsequently collect 10 consecutive 0.5 ml fractions.
2.5. Transmission electron microscopy
EV samples were fixed for 30 min in paraformaldehyde 2% (Sigma‐Aldrich) in PBS, and incubated for 15 min, at 4°C, on top of carbon‐coated 100‐mesh copper grids, previously glow discharged at 7.2 V for 60 s, using a Bal‐Tec MED 020 Coating System. Grids were washed in PBS, fixed in glutaraldehyde 1% (Sigma‐Aldrich) for 5 min at 4°C, washed again and blotted dry with filter paper. EVs on the grids were then negatively stained with aqueous uranyl acetate 2% (Sigma‐Aldrich) for 2 min, followed by washing, drying and analysis using a FEI Tecnai G2 Spirit transmission electron microscope (ThermoFisher Scientific), equipped with a Morada digital camera (Olympus Soft Image Solutions GmbH). All the procedures were performed at the Electron Microscopy facility of the University of Valencia.
2.6. Western blotting
EV and cell samples were measured by NanoDrop spectrophotometer (Thermo Fisher Scientific) to determine protein content. Cell lysates and EV samples were each mixed with 4× LDS sample buffer (NuPAGE Thermo Fisher) plus 10 % β‐mercaptoethanol for reducing conditions (Sigma Aldrich), and heated at 70°C for 10 min. For non‐reducing conditions, samples were mixed with 4× LDS sample buffer (NuPAGE Thermo Fisher) and heated at 70°C for 15 min.
For each sample equal protein amounts (6 μg) were resolved on 4–12% gradient Bis/Tris polyacrylamide gels (Life Technologies). Chameleon Duo Pre‐stained Protein Ladder (LI‐COR, Lincoln, NE) protein standards were used. Proteins were transferred using Trans‐Blot Turbo Mini or Midi polyvinylidene fluoride transfer packs (Bio‐Rad Laboratories, Hercules, CA, USA). Subsequently the membranes were blocked using Odyssey TBS Blocking Buffer (LI‐COR) for 1 h at room temperature. Next, membranes were incubated with primary antibodies over‐night with agitation at 4°C in Odyssey TBS Blocking Buffer (LI‐COR). After three washes with 0.1% TBS‐Tween (TBS‐T), the secondary antibodies were added to membranes for 1 h at room temperature with agitation. After three washes for 5 min in TBS‐T the membranes were visualized using the Odyssey CLx imaging system (LI‐COR) and data was analysed with Image Studio v.4.0 software. See Table S1 for detailed information on antibodies.
2.7. EV size distribution and concentration measurement
EV size distribution and concentration were determined using Nanoparticle Tracking Analysis with a NS300 instrument (Malvern) with a blue 405 nm laser and sCMOS camera. All samples were measured with constant equipment settings for five repeats of 60 s each acquired under flow with a syringe pump speed of 30 using camera level 15, screen gain 2, and detection threshold of 7. Data were collected and analysed with software version NTA 3.2 Dev Build 3.2.16.
2.8. EV treatments
5000 reporter cells were plated per well in a 384 well plate (Greiner Bio‐One). EVs were added at 5.5 × 104 EVs/reporter cell and incubated for 24 h. After 24 h media was changed for fresh media without EVs and plates were analysed for gene editing at 72 h after EV addition.
2.9. Transwell assays
Cas9 cargo transfer by EV was tested in 0.4 μm membrane transwell co‐culture setups. 96,000 reporter cells were seeded per well in a 24 well plate (Greiner Bio‐One). For plasmid control conditions and LoxP sgRNA expression in reporter cells FuGENE HD (Promega) was used to transfect the necessary plasmids as described above in the Cell Transfection section. For Cas9CRY2 EV secreting conditions 40 kDa PEI max (Polysciences) was used to transfect Expi293F cells with the necessary plasmids as described above in the Cell Transfection section. Thincert (Greiner Bio‐One) transparent PET cell culture inserts for 24 well plates with a pore diameter of 0.4 μm were placed into each well and 96,000 cells secreting Cas9CRY2 EV from various experimental condition were seeded onto the inserts.
2.10. Cell fluorescence measurements
Plates were imaged on an IncuCyte S3 every 2 h and the 72 h time point after EV addition was chosen for analysis. Images were captured using a 20× objective and image analysis was performed with the IncuCyte S3 software S3 2019. Given the long half‐life of GFP it could still be detected in cells at the point of analysis even in cells where Cas9 cleavage occurred. Thus, a mask was created to identify all cells based on GFP positivity meanwhile another mask was created for RFP positive cells indicative of Cas9 mediated cleavage at both LoxP sites. Percentage of RFP‐positive cells was defined as the mask area of RFP positive cells divided by the mask area of all cells based on GFP positivity and multiplied by 100. In addition to IncuCyte measurements, cell fluorescence was also assessed CytoFLEX S N2‐V3‐B5‐R3 Flow Cytometer (Beckman Coulter Cat# B78557). In this case cells were also assessed at the 72 h time point after EV addition and percentage positivity of GFP and RFP or tdTomato was measured collecting at least 10,000 events in the cell gate per measurement with a medium flow rate.
2.11. DNA preparation and next‐generation amplicon sequencing
Treated HEK 293T cells were washed with PBS and lysed in 100 μl QuickExtract DNA extraction solution (Lucigen). For DNA extraction, samples were then vortex, heated at 65°C for 6 min, vortexed again and heat‐inactivated at 98°C for 2 min. A two‐step PCR protocol was then performed to generate ready‐to‐sequence PCSK9 amplicons. PCSK9 amplicons were generated in the first‐step PCR (25 μl reaction) using KAPA HiFi HotStart ReadyMix (Roche), 300 nM of PCSK9‐specific primers with adapter extensions (Forward: 5′‐TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGTTGCCCCATGTCGACTAC‐3′; Reverse: 5′‐GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCGAGAAGTGGAAACCACCA‐3′; Sigma‐Aldrich) and 11 μl of the extracted DNA, incubated on a C1000 Touch (Bio‐Rad, CA) with the following conditions: 95°C for 3 min; five cycles at 98°C for 20 s, 75°C for 15 s, 72°C for 15 s; 30 cycles at 98°C for 20 s, 65°C for 15 s, 72°C for 15 s; 72°C for 2 min. Generated amplicons were then purified using the HighPrep PCR Clean‐up system (MAGBIO). Briefly, PCR reaction was mixed with beads at 1:1.8 ratio for 10 min for DNA binding, DNA was washed twice with 80% ethanol for 30 s, and eluted in elution buffer (Qiagen). Samples were then indexed in the second‐step PCR (25 μl reaction) using KAPA HiFi HotStart ReadyMix (Roche), 500 nM of indexing primers (Forward: 5′‐AATGATACGGCGACCACCGAGATCTACACNNNNNNNNTCGTCGGCAGCG*T*C‐3′; Reverse: 5′‐CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTCTCGTGGGCTC*G*G‐3′; IDT) and 0.5 ng of purified PCSK9 amplicons, incubated on a C1000 Touch (Bio‐Rad) as follows: 72°C for 3 min, 98°C for 30 s; 10 cycles at 98°C for 10 s, 63°C for 30 s, 72°C for 3 min; 72°C for 5 min. Indexed amplicons were then purified as above using a PCR reaction‐to‐beads reaction of 1:1. Amplicon size and concentration, and absence of non‐specific products was verified on a by capillary electrophoresis on a Fragment Analyzer (Agilent) using a standard sensitivity dsDNA method. Samples were pooled to a concentration of 25 nM. The concentration was confirmed by measurement with high sensitivity dsDNA Qubit assay (ThermoFisher Scientific) prior to further dilution and denaturation of the pool, according to guidelines provided by Illumina. Final loading concentration used as input to the sequencing was 6 pM. The samples were sequenced on a MiSeq instrument (Illumina) with paired‐end 150 bp read length setting. Samples depth of sequencing was between 0.24 and 0.32 million read pairs. Read pairs were merged using FLASH (Magoc & Salzberg, 2011) were mapped to the predicted PCSK9 amplicon sequence using bwa (Li & Durbin, 2009). The generated alignment files were processed to determine the insertion/deletion (indels) of bases at the relevant sgRNA target site, and percentage of indels relative to the total reads amount was calculated for each sample.
2.12. Triton X‐100 detergent controls
To control that Cas9 is delivered by membrane enclosed EVs and not by other non‐lipidic particles, EVs were treated with 0.1% Triton X‐100 for 5 min as described earlier (Osteikoetxea et al., 2015). Next equal nominal amounts of EVs from non‐treated or detergent‐lysed condition were administered to reporter cells resulting in 5.5 × 104 EVs/reporter cell. Due to the high dilution of treated stock EVs the final concentrations of Triton X‐100 in cell media were below 0.003% which showed no evidence of cell death or different morphology compared to Triton X‐100 free conditions.
2.13. RNA isolation and droplet digital PCR analysis
Prior to RNA isolation, EV samples were incubated with RNase A (DNase and protease‐free) 2 mg/ml (ThermoFisher Scientific), at 37°C for 15 min, for degradation of extravesicular RNA. RNAse activity was then inhibited with RNase inhibitor 1 U/μl (Applied Biosystems) and EVs lysed with QIAzol (EVs: Qiazol ratio (vol/vol) = 1:3). Cell pellets were washed with PBS and lysed with QIAzol as per manufacturer's instructions. EVs and cells RNA was isolated with miRNeasy micro kit, following manufacturer's instructions. Concentration of isolated RNA was measured with Qubit RNA HS assay kit, according to the manufacturer's protocol. Equal amounts of RNA (20 ng) for all samples were then reverse transcribed using the first‐strand cDNA synthesis kit SuperScript IV VILO with ezDNase enzyme (ThermoFisher Scientific), following manufacturer's protocol. Droplet digital PCR reactions were set up with equal amounts of cDNA (200 ng) for all samples, 2 × ddPCR Supermix for EvaGreen, sgRNA specific primers (Forward: 5′‐GGCGACGTAAAGTTTTAGAGCT‐3′; Reverse: 5′‐CGACTCGGTGCCACTTTT‐3′; Sigma‐Aldrich). Droplets were then generated using a Bio‐Rad's QX200 AutoDG instrument using droplet generation oil for EvaGreen, followed by PCR amplification in a C1000 Touch thermal cycler (BioRad) according to the manufacturer's instructions. All samples were analysed in triplicates. Droplet analysis was performed with the QX200 Droplet reader (Bio‐Rad) using ddPCR Droplet Reader Oil (Bio‐Rad). Results were analysed using the QuantaSoft software, version 1.7.4.0917 (Bio‐Rad) with the absolute quantification algorithm.
2.14. Data analysis
Data was handled in Excel and analysed and plotted using GraphPad 8.0. Data was tested for normal distribution and statistical significance was calculated using one‐way ANOVA. Where data was not normally distributed, Kruskal‐Wallis test was used instead. Asterisks show level of statistical significance: * P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. We have also submitted all relevant data of our experiments to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV210345).
3. RESULTS
3.1. Inducible protein dimerization does not significantly alter EV properties
We first characterized EV size and morphology obtained from the Expi293F cell line to be used for EV production. Transmission electron microscopy of the EV preparations confirmed the presence of membrane‐enclosed vesicles with a cup‐like shape in isolates from control non‐transfected cells compared to those transfected with constructs for inducible protein cargo loading (Figure 1A). The four inducible protein dimerization loading systems, CIBN‐CRY2, PHYB‐PIF6, p/nMags, and FKBP‐FRB, are schematically depicted along with dimerization signal in Figure 1B. The size distribution of EVs was measured by nanoparticle tracking analysis and this revealed that expression of heterodimerization proteins caused no significant changes in EV size (Figure 1C), the number of EVs released per cell, or cell viability (Figure S2). Western blot showed the presence of either membrane and intraluminal proteins characteristic of EVs, such as markers ALIX, TSG‐101, flotillin‐1, β‐actin, syntenin‐1, CD63 and CD81, which were enriched in non‐transfected or engineered EVs compared to the lysates of secreting cells. When loading equal amounts of proteins, EVs from non‐transfected cells or from cells over‐expressing heterodimerization proteins contained relatively similar amounts of EV markers (Figure 1D). These results indicate that expression of heterodimerization proteins for EV loading had no impact upon EV biogenesis.
FIGURE 1.
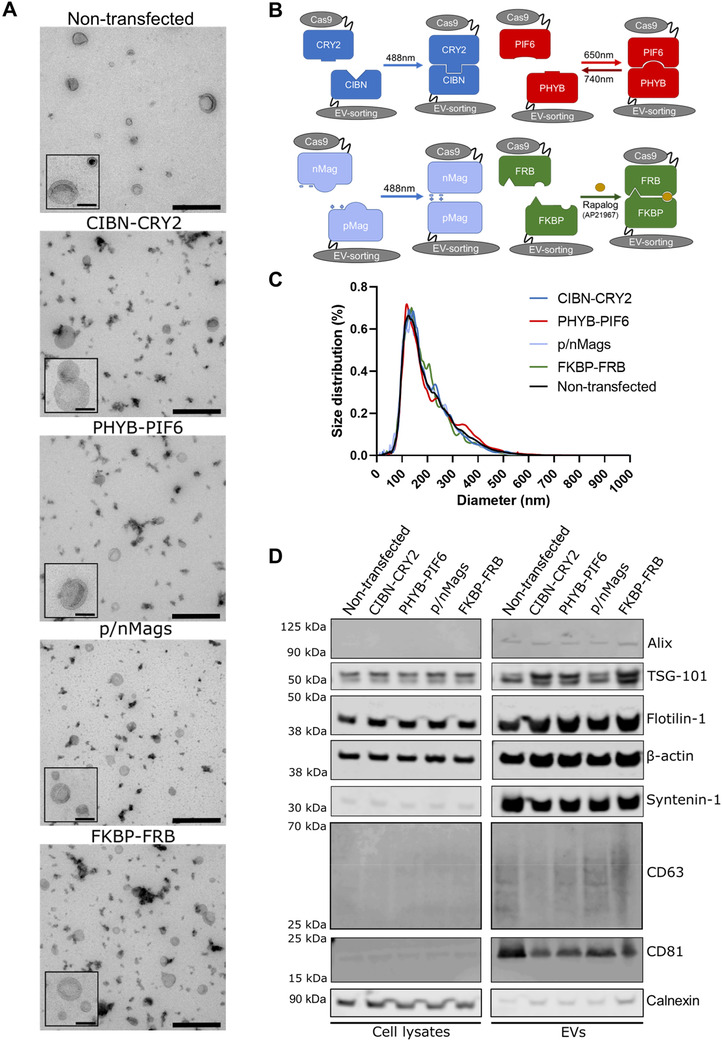
EV characterization. (A) Transmission Electron Microscopy reveals the presence of vesicles with the typical size and lipid‐enclosed morphology observed in EV preparations (insert scale bar 100 nm, whole field scale bar 400 nm). (B) Schematic diagram of the four inducible dimerization systems used in this study. (C) EV size distribution determined by nanoparticle tracking analysis reveals no significant differences in diameters of EV secreted by wild‐type cells and by cells engineered to express each of the inducible dimerization systems with the MysPalm EV sorting motif and Cas9 cargo (n = 4 per condition). (D) Western blot shows that EV isolates from non‐transfected conditions, or from each of the inducible dimerization systems with the MysPalm EV sorting motif and Cas9 cargo, are enriched compared to their secreting cells in proteins commonly found in EVs, such as Alix, TSG‐101, Flotilin‐1, β‐actin, syntenin‐1 and the most accepted EV markers CD63 and CD81 (6 μg total protein loaded per sample)
3.2. Cas9 loading efficiencies for optogenetic and small molecule approaches
We screened a small library of proteins and lipid modifications to identify the best approach to target the heterodimer into EVs. We tested seven EV associated proteins or lipid motifs fused to CIBN for their ability to recruit Cas9CRY2 (NLS‐Cas9‐CRY2) under blue light into EVs. The EV sorting motifs tested were CD9, Rab5c, CD81, Myristoylation (Mys), Palmitoylation (Palm), Myristoylation‐Palmitoylation‐Palmitoylation (MysPalm), and Prenylation (Prenyl). The protein motifs were introduced as C‐terminal fusions to CIBN connected by a short unstructured glycine serine linker. The lipid modifications were introduced as a short amino sequence motif added to the N‐ (Mys, Pam, MysPalm) or C‐ terminus (Prenyl) of CIBN, and these motifs were recognized by the cellular machinery for lipid modification resulting in addition of lipid anchor. First, the EV sorting motifs were screened by Western blotting loading the same protein amount per well, and using the EV protein marker ALIX as a control for EV loading at similar levels, as shown in Figure S3A. Relative quantification of western blot band intensities revealed that CD9, CD81, Mys, and MysPalm recruited high levels of Cas9 into EVs (Figure S3B). We selected CD9 and MysPalm as the most efficient representatives of Cas9 protein loading into EVs by two distinct mechanisms, the enrichment of characteristic EV proteins during their biogenesis and the improved association of lipidated proteins with biological membranes, respectively. Next, we explored the ability of CD9 and MysPalm to load Cas9 into EVs using the other inducible dimerization proteins. We used Western blotting with a standard of recombinant purified Cas9 to quantify the amount of Cas9 protein loaded per EV (Figure 2A) and shows representative Western blots for each system (Figure 2B). All four approaches tested revealed a detectable band of Cas9 cargo at the expected position in the EV fraction, albeit there was a dispersed pattern for PIF6/PHYB suggestive of instability of the fusions leading to some degradation. The Western blot results clearly show that among the EV sorting motifs and Cas9 constructs assessed CRY2‐CIBN proved superior to other dimerization systems for loading of Cas9 into EVs. Using densitometry analysis, we estimated the number of Cas9 molecules present per EV (Figure 2C). This quantification revealed that the CIBN‐CRY2 system contained the highest amount of Cas9 per EV reaching more than 20 molecules per EV (Figure 2C).
FIGURE 2.
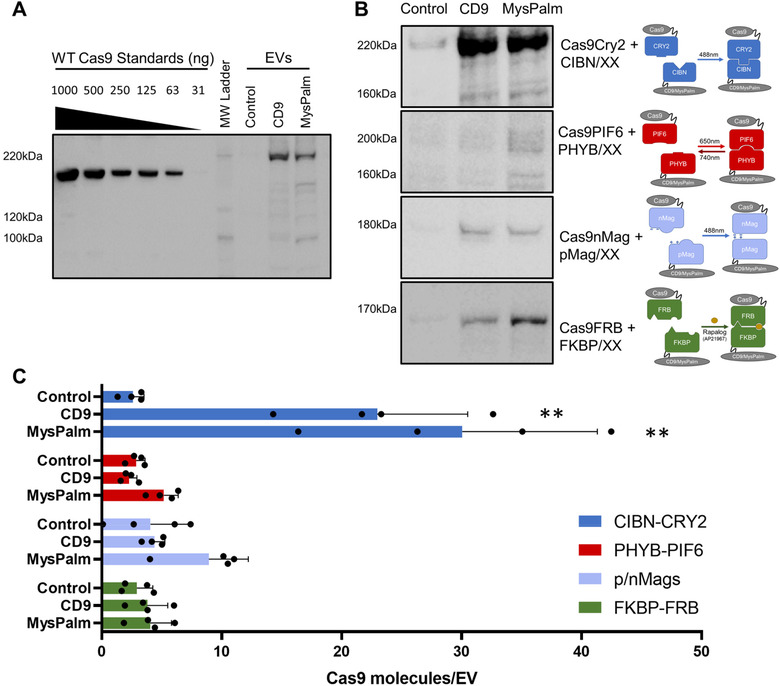
Cas9 loading into EVs. (A) Cas9 quantification using calibration curve of wild type (WT) recombinant Cas9 protein alongside tested EV isolates (n = 4 per condition, representative blot for CIBN‐CRY2 loading shown). (B) Comparison of Cas9 loading by the different heterodimerization systems using CD9 or MysPalm for recruitment into EV, compared to the control condition without heterodimerization partner (n = 4, representative blot shown), (C) Cas9 molecules/EV calculated from quantitative blots as shown in C and NTA particle quantification for each isolate (Mean ± SD, n = 4 per condition, statistical significance determined with Kruskal‐Wallis test)
3.3. Cas9 fusion proteins remain functionally active
Having established that Cas9 can be loaded into EVs through inducible protein dimerization, we next assessed whether the protein engineering impacted Cas9 functionality. We used a Cre fluorescent reporter system to measure gene editing by Cas9. We adapted a commercially available HEK293‐loxP‐GFP‐STOP‐loxP‐RFP Cre reporter cell line (AMSbio, described in Heath et al., 2019) by designing a sgRNA capable of targeting the LoxP sites. This design allows for Cas9 mediated cleavage at both LoxP sites resulting in GFP‐STOP excising after repair by the cells’ machinery for DNA double‐strand break repair, primarily through the non‐homologous end joining (NHEJ) pathway (Chang et al., 2017) (Figure 3A). Untreated cells express GFP and display green fluorescence. LoxP targeted gene editing will result in removal of the GFP‐STOP cassette, and cells will start to express RFP resulting in red fluorescence. Using this system, we evaluated Cas9 mediated gene editing in vitro by transfecting the reporter cells with pcDNA3.1+ plasmids encoding each of the Cas9 fusion constructs and WT Cas9 as a comparator. We quantified the extent of gene editing by imaging the red fluorescence with an IncuCyte S3 as shown in representative images (expressed as percentage of RFP positive cells, Figure 3B). Expression of WT Cas9 together with transcription of the LoxP sgRNA encoded on the same plasmid resulted in a significant percentage of red cells in the reporter cells. All four fusion proteins were active although the editing efficiency was 30–50% of that of WT Cas9 (Figure 3C). Having confirmed that the Cas9 fusion protein remains functionally active, we proceeded with functional delivery analysis of Cas9‐loaded EVs.
FIGURE 3.
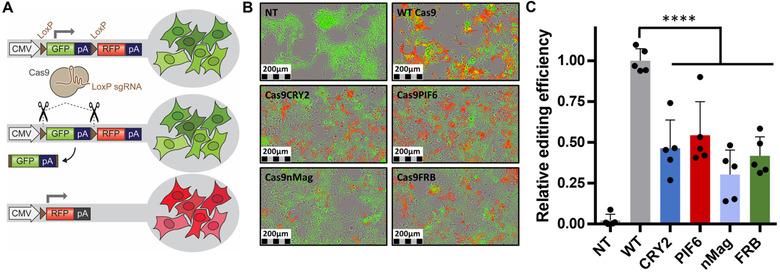
Impact of fusion proteins on Cas9 functionality. (A) Schematic of Cre‐Lox reporter cassette adapted for Cas9 cleavage detection with a sgRNA design targeting both LoxP sites. Cas9 is targeted by LoxP sgRNA to cleave DNA at both LoxP sites resulting in excision of GFP‐STOP cassette and expression of RFP. (B) Visualization of gene editing in HEK293‐loxP‐GFP‐STOP‐loxP‐RFP cells after transfection with plasmids encoding the indicated Cas9 fusion proteins, with appearance of RFP upon successful editing of loxP‐GFP (n = 5 per condition, representative images). (C) Relative gene editing efficiency of Cas9 fusion proteins compared to Wild Type (WT) Cas9 and Non‐Transfected (NT) Cells (Mean ± SD, n = 5 per condition, statistical significance determined with one‐way ANOVA)
3.4. Functional delivery of Cas9 by EVs
We next investigated whether EVs loaded with Cas9 were capable of delivering functional cargo in vitro to reporter cells. EVs were isolated from cells transfected to express Cas9‐loading constructs alone (apo‐Cas9) or co‐transfected also with a plasmid encoding LoxP sgRNA, which resulted in co‐recruitment of the sgRNA as passenger in complex with Cas9 into EVs. Transmission electron microscopy confirmed the presence of EVs in all conditions (Figure S4A) and droplet digital PCR (ddPCR) was used to quantify the amount of sgRNA molecules present in the isolated EVs (Figure S4B). Results showed the number of sgRNA molecules loaded per ng of RNA in EVs increased in the conditions of active loading where dimerization of CRY2 with CIBN fused with CD9 or MysPalm drive Cas9 loading compared to control passive loading conditions of Cas9CRY2 without dimerization with EV sorting motifs. In contrast to the increased levels of sgRNA in EVs in actively loaded conditions, cellular sgRNA levels decreased in CD9 or MysPalm actively loaded conditions compared to the passive loading condition suggesting that the cellular sgRNA is recruited and secreted by EVs.
Functional delivery of EVs loaded with Cas9 alone was tested in reporter cells that were transfected with the plasmid encoding LoxP sgRNA prior to addition of EVs (Figure 4A). EVs loaded with Cas9 and LoxP sgRNA were tested directly in the non‐transfected (NT) reporter cells (Figure 4B). For all conditions tested, EVs were administered to reporter cells for 24 h at a concentration corresponding to 5.5 × 104 EVs/reporter cell followed by a media change and final fluorescent imaging at 72 h after EV addition. Gene editing of reporter cells treated with Cas9 EVs loaded with control condition without heterodimerization, CD9, or MysPalm was compared to non‐EV‐treated reporter cells for which the percentage of RFP positive cells was set to 0%. The plasmid control was the percentage of RFP positive cells for reporter cells transfected with WT Cas9 and LoxP sgRNA encoding plasmids. Efficient gene editing for apo‐Cas9 delivery was detected in the LoxP sgRNA expressing reporter cells treated with Cas9CRY2 EVs loaded through MysPalm reaching 51% editing (Figure 4A). Interestingly, we observed significant gene editing for delivery of Cas9‐sgRNA complex using EVs at around 42% (Figure 4B). Intriguingly, the same CRY2‐CIBN loading approach coupled to CD9 as the EV‐sorting partner did not lead to any significant gene editing (Figures 4A and B). In addition, the other optogenetic and ligand induced heterodimerization approaches used for EV loading (all with lower loading levels of Cas9) did not deliver any statistically significant levels of gene editing (Figure S5).
FIGURE 4.
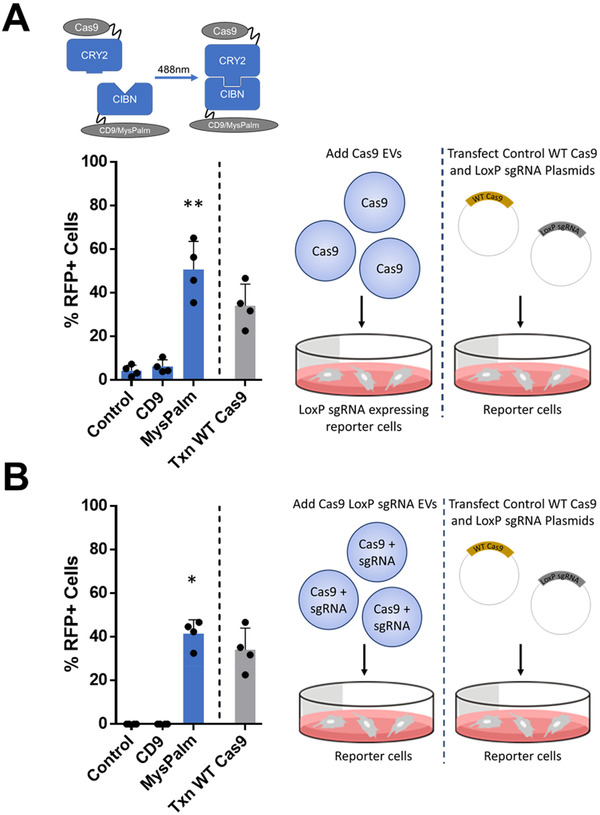
In vitro Cas9 delivery by EVs. (A) Cas9 was delivered by EVs loaded with and complemented by sgRNA from plasmid transfection of reporter cells. Gene editing occurs in transfected cells with the wild type Cas9 and sgRNA and with Cas9CRY2 EVs loaded through MysPalm achieving 50.7% while CD9 loaded, or control passively loaded EVs without CIBN resulted in 4.4‐6.3% gene editing. (Mean ± SD, n = 4, statistical significance determined with Kruskal‐Wallis test). (B) Cas9 and LoxP sgRNA was delivered by EVs. Gene editing occurs in transfected cells with the wild type Cas9 and sgRNA and with Cas9CRY2 and LoxP sgRNA EVs loaded through MysPalm achieving 41.6% while while CD9 loaded, or control passively loaded EVs without CIBN resulted in no gene editing (Mean ± SD, n = 4, statistical significance determined with Kruskal‐Wallis test). Cells were stimulated with an equal amount of EVs (5.5 × 104 EVs/cell)
To further test the EV mediated transfer of functional Cas9 to recipient cells we performed experiments in a transwell co‐culture setting in which the EV expressing cells and reporter cells are separated by a 0.4 μm membrane. In the transwell co‐culture setting we, demonstrated similar levels of gene editing activity to that seen with EV isolates, 28% and 31% in absence or presence of sgRNA in EVs (Figure 5A). We also decided to investigate if another cell type could be edited and chose HepG2 cells transfected to express a different loxP‐tdTomato‐STOP‐loxP‐GFP Cre reporter construct depicted in Figure S6. For this experiment we decided to test the most efficient combination of Cas9CRY2 EVs loaded through MysPalm co‐loaded with or without LoxP sgRNA dosing 5.5 × 104 EVs/cell followed by a media change and further 72 h incubation and flow cytometric analysis. Here, Cas9 loaded EVs as well as Cas9 and LoxP sgRNA loaded EVs resulted in 25.4% and 19.2% gene editing, respectively, in HepG2 cells (Figure 5B).
FIGURE 5.
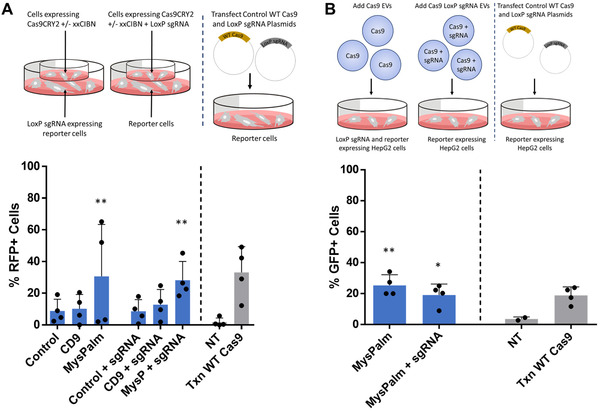
Cas9 transfer by EVs is also observed in transwell co‐cultures or other cell backgrounds. (A) Co‐cultures separated by 0.4 μm transwells show statistically significant gene editing with cells secreting Cas9CRY2 loaded EVs with or without LoxP sgRNA through MysPalm achieving 28.2% and 30.6%, respectively, while cells secreting Cas9CRY2 loaded EVs with all other conditions resulted in 8.6–12.8% gene editing (Mean ± SD, n = 4, statistical significance determined with Kruskal‐Wallis test). (B) Cas9 or Cas9 and LoxP sgRNA was delivered by EVs to HepG2 cells transfected to express a loxP‐tdTomato‐STOP‐loxP‐GFP Cre reporter construct. Gene editing efficiency was determined by flow cytometry to be 25.4% for with Cas9CRY2 EVs loaded through MysPalm while EVs loaded with both Cas9Cry2 and LoxP sgRNA resulted in 19.2% gene editing (Mean ± SD, n = 4, statistical significance determined with one‐way ANOVA)
So far, assessing Cas9 delivery to fluorescent reporter cells allowed for easily and rapidly testing all the multiple conditions for Cas9 EV loading investigated. Despite these advantages, we decided to also investigate whether a therapeutically relevant gene could be edited and chose PCSK9 as a target due to extensive research on this gene as a potential candidate for circulating cholesterol level reduction. For this we decided to test the most efficient combination of Cas9CRY2 EVs loaded through MysPalm co‐loaded with or without PCSK9 sgRNA (Figure 6). After addition of EVs to Hek293T cells for 24 h at a concentration corresponding to 5.5 × 104 EVs/cell followed by a media change and further 72 h incubation, the cells were harvest for genomic DNA isolation and samples were sent for next‐generation and sanger sequencing. Analysis of next‐generation sequences revealed that Cas9 loaded EVs as well as Cas9 and PCSK9 sgRNA loaded EVs resulted in 5.8% and 4.4% gene editing, respectively (Figure 6). Gene editing was also detected using the TIDE analysis of Sanger sequences (Figure S7).
FIGURE 6.
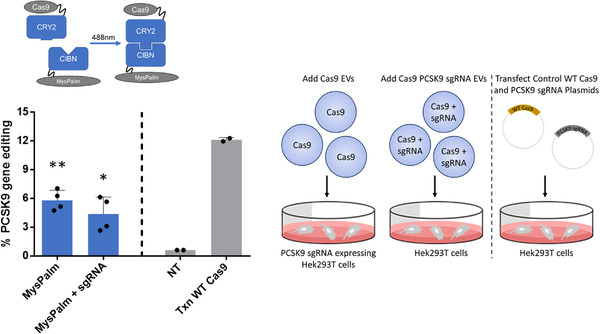
In vitro Cas9 delivery by EVs for PCSK9 gene editing. Cas9 or Cas9 and PCSK9 sgRNA was delivered by EVs. In the case Cas9 alone delivery by EVs, PCSK9 sgRNA was delivered by plasmid transfection of Hek293T cells. Gene editing efficiency was determined by next‐generation sequencing to be 5.8% for with Cas9CRY2 EVs loaded through MysPalm while EVs loaded with both Cas9Cry2 and PCSK9 sgRNA resulted in 4.4% gene editing (Mean ± SD, n = 4, statistical significance determined with one‐way ANOVA). Cells were stimulated with an equal amount of EVs (5.5 × 104 EVs/cell)
To confirm Cas9 delivery occurred through transfer by lipid membrane enclosed EVs we performed similar experiments as detailed in Figure 4, but first EV isolates were pretreated for 5 min with 0.1% Triton X‐100, to disrupt all vesicular structures as described previously (Osteikoetxea et al., 2015; Théry et al., 2018). If Cas9 transfer was occurring independently of EVs, potentially through contaminating soluble Cas9 (protein, mRNA, or DNA), then treatment with low concentration of detergent would disrupt the EV dependent transfer but still allow for transfer of the non‐vesicular components. Indeed, our findings showed that upon Triton X‐100 pretreatment our EV isolates lost the ability to transfer Cas9 to target cells, as detected by the absence of red fluorescence (Figure S8). Additionally, we further purified Cas9 EVs loaded with MysPalm and Cas9CRY2 using SEC and tested the gene editing activity of each recovered fraction. Similarly, to the Triton X‐100 control, we expected that any gene editing activity due to transfer of contaminating soluble Cas9 (protein, mRNA, DNA) would be observed in the non‐vesicular fractions recovered by SEC purification due to the significant differences in size and biophysical parameters of EVs compared to these contaminants. Here we also observed that gene editing activity was retained in EV fractions (Figure S9).
4. DISCUSSION
Therapeutic genome editing, including editing with CRISPR/Cas9, represent one of the fastest‐growing class of FDA‐approved biologics and genetic medicines and are beginning to make their way to patients (You et al., 2019). The potential of CRISPR/Cas9 therapeutics is undermined by the lack of effective and safe methods for delivery of the CRISPR/Cas9 machinery to diseased cells. Currently, EVs show promise as a vehicle for safe and efficient delivery of CRISPR/Cas9‐based protein systems such as those required for genome, epigenome, base, or primer editing (Adli, 2018; Anzalone et al., 2019). Here we characterize four different systems to load Cas9 into EVs by inducible protein heterodimerization. We observed no significant difference in EV morphology, size, concentration, and protein markers following transfection with constructs to induce Cas9 loading. While we observed differences in the loading efficiency for each heterodimerization system, they all enabled Cas9 loading into EVs by fusing Cas9 and EV‐associated proteins or lipidation motifs to heterodimerizing protein domains. However, we found that Cas9CRY2 loaded through recruitment by CD9 and MysPalm achieved the highest number of Cas9 molecules per EV suggesting the CIBN‐CRY2 may be best suited for loading of therapeutic peptides into EVs. Due to the high molecular weight of Cas9 and gel conditions some band smearing occurred but our data also show that CD9 and MysPalm were important EV recruiting partners to improve loading even though minimal loading was also seen in the absence of EV‐sorting motifs or in the absence of light stimulation. This is consistent with earlier findings from our group where low levels of Cre loading into EVs was observed using the FKBP‐FRB dimerization system in the absence of the ligand or without the corresponding EV sorting motif. However, heterodimerization with CD81 as an EV sorting motif vastly increased loading (Heath et al., 2019). Perhaps the loading observed in the absence of stimulus or EV sorting motif can be explained in the context of high protein overexpression if in addition to stimulus there is also a concentration‐dependency for heterodimerization, as well as concentration‐dependent distribution of cargo into the cell's EVs sorting mechanism. Previously, it has been reported that simple protein overexpression can lead to loading of the overexpressed protein into EVs (Mizrak et al., 2013). While minimal Cas9 loading occurs without EV‐sorting, the improvement in loading through heterodimerization and the ability to deliver functional cargo only in inducibly loaded conditions suggest that heterodimerization is critical for therapeutic Cas9 protein delivery.
We also demonstrate that existing Cre‐Lox reporter systems may be repurposed for detection and quantification of Cas9 gene editing detection by designing sgRNA targeting the LoxP sites. Since the LoxP sequence is not present in mammalian genomes there is little chance of off‐target DNA cleavage when targeting this sequence making it a safer alternative for research than editing of mammalian genes (Yarmolinsky & Hoess, 2015). Because the LoxP does not contain any “NGG” Protospacer adjacent motif (PAM) sequence required for WT Streptococcus pyogenes Cas9, there is the constraint that close to the LoxP sites there must be an NGG sequence that is not within the reporter protein open reading frame and it must be present on both LoxP sites. If these requirements are met, then any existing Cre‐Lox reporter system can be repurposed for Cas9 editing detection. Additionally, we have found that our LoxP sgRNA design has only seven potential off‐target sites with three nucleotide mismatches, and 104 off‐targets with four nucleotide mismatches. No closely matched sites with less than three mismatches to the target site were observed, minimizing the probability of off‐target editing for this guide. For our PCSK9 sgRNA design, in silico off‐target analysis showed no potential off‐target sites with less than four mismatched nucleotides and only 21 potential off‐target sites with four mismatches, highlighting the high degree of specificity of our design. On the other hand, we did not detect any large structural variations at the edited PCSK9 locus in our work, contrarily to previous observations for direct ribonucleoprotein (RNP) delivery (Höijer et al., 2022). This might be due to the lower amount of RNPs delivered by the engineered EVs, suggesting that EV‐based delivery could have a potential advantage compared to other vehicles in circumventing the unwanted effects often observed with CRISPR/Cas9.
One interesting observation is that Cas9 loading through MysPalm is higher than measured for CD9. A previous report investigating lipidation tags and fusion of cargo to plasma membrane proteins also found MysPalm to be more efficient at recruiting into EVs perhaps due to the greater challenge in the transport and proper folding of an integral plasma membrane protein through the endoplasmic reticulum when fused to a larger protein domain (Shen et al., 2011). The increased efficiency of Cas9 loading with MysPalm correlated with the finding that it was the only condition in which significant Cas9 delivery and gene editing was observed. Thus, EVs can only deliver Cas9 if loaded by heterodimerization with the CIBN‐CRY2 system and MysPalm, but not when loaded less efficiently by other heterodimerization conditions or controls lacking heterodimerization. Since the most commonly reported mechanism for EV delivery is that endocytosis occurs and is followed by escape from endosomes resulting in cargo delivery to the cell cytoplasm (Théry et al., 2018) an important advantage of our approach is that it allows for dissociation of the Cas9 cargo from the anchoring tetraspanin or lipid motif due to the reversibility of the protein dimerization system used. After dissociation, concentration gradients between cargo in EVs and the cell will also favour transfer to the cell cytoplasm. Interestingly, one of the other systems studied here that did not result in Cas9 delivery was FKBP‐FRB. In an earlier study using the FKBP‐FRB system for loading Cre protein into EVs our group identified endosomal escape as an important limiting factor to be overcome for the delivery of Cre using EVs (Heath et al., 2019). Thus, another potential explanation for Cas9 transfer occurring when loaded into EVs via MysPalm but not via CD9, despite similar levels of protein present, is that MysPalm provides an advantage over CD9 in terms of endosomal escape. Until this study there had been no evidence about the effect of protein lipidation in the context of EV cargo transfer but there has been extensive evidence that lipidation increases the endosomal escape and delivery efficiency of peptide vectors (Hou et al., 2015).
Another important observation is that co‐expressing sgRNAs in cells that are loading Cas9 into EVs through inducible dimerization is sufficient to recruit sgRNA into EVs without the need to use additional mechanisms. Here, loading of Cas9 with MysPalm also showed the highest levels of sgRNA loading as compared to CD9 or passive loading conditions. In line with our finding, a recent study showed that simple expression of CRISPR sgRNA by EV secreting cells is sufficient for loading into EVs and functional transfer of the sgRNA (De Jong et al., 2020).
Considering the evidence for gene editing with Cas9 loaded EVs isolated by ultracentrifugation we also explored if this observation could be replicated in another cell line or with another system. To that end we repeated experiments using HepG2 cell expressing a fluorescent reporter showing that these cells can also be edited with Cas9 EVs. Additionally, we used 0.4 μm transwell co‐cultures that allows for exchange of Cas9 by EVs, and potentially other soluble forms, smaller than the 0.4 μm filter size. In this setup we also found functional transfer of Cas9 at similar levels as observed with isolated EVs. This observation also revealed that Cas9 functional transfer is not due to potential artifacts introduced during ultracentrifugation or storage but instead cells can transfer Cas9 via EVs without need for isolation.
Having determined that gene editing was possible by EV transfer in a fluorescent reporter system we next set out to test editing of a therapeutically relevant gene, PCSK9. EVs were loaded with Cas9 alone or together with a PCSK9 sgRNA using the MysPalm and CIBN‐CRY2 dimerization condition that resulted in best Cas9 loading and delivery to reporter cells. While statistically significant gene editing of PCSK9 was observed both by next‐generation and Sanger sequencing, it was markedly lower than that observed with the fluorescent reporter system. There is the possibility that differences in gene editing efficiencies could be due to the sgRNA designs as distinct designs can significantly influence in editing efficiencies (Xu et al., 2015; Wang et al., 2019). Another potential reason is the copy number of edited gene per cell. To generate fluorescent reporter cells a lentiviral approach and puromycin selection was employed which results in multiple copies of the reporter integrated per genome. Thus, targeting the reporter construct allows for more gene copies to be edited per cell as compared to the PCSK9 gene which should be present in 2–3 copies in pseudotriploid cells such as Hek293T (Lin et al., 2014). The observation of lower editing efficiency of PCSK9 compared to the fluorescent reporter gene highlights the importance of testing different sgRNA candidates when developing efficient gene therapy strategies.
Lastly, to rule out the possibility of gene editing in reporter cells being caused by soluble non‐EV encapsulated Cas9 protein, mRNA, or DNA co‐isolated with our EV preparations we used detergent treatments of the EV preparations, which disrupt vesicular structures but not protein aggregates. We administered EV preparations from detergent treated and non‐treated conditions to reporter cells and observed that their biological activity was disrupted with detergent treatment supporting that delivery was mediated by vesicular structures. In addition to detergent treatment, we also purified EV preparations using SEC to observe whether non‐vesicular fractions retaining gene editing activity. Indeed, as with detergent controls, we found here that gene editing activity was retained in the EV‐containing SEC fractions as confirmed by NTA counts and the presence of EV markers.
Taking into account the Cas9 EV‐loading efficiencies observed it is possible to estimate the amount of Cas9 delivered by EVs into cells. Considering 25 Cas9 molecules loaded per EV and addition of 5.5 × 104 EVs per cell we can estimate delivering about 1.38 × 106 Cas9 molecules per cell which corresponds to about 0.365pg of Cas9 per cell. For comparison, other strategies such as transfection by lipofectamine or electroporation of Cas9‐sgRNA ribonucleoprotein employ about 8.33pg of Cas9 per cell representing 22.8 times more Cas9 per cell. Nevertheless, it is hard to directly compare distinct delivery methods because of other important considerations such as the degree of Cas9 actually delivered into cell nucleus as opposed to that which may be routed for degradation in lysosomal compartments.
In terms of delivery of Cas9 by EVs there are other studies confirming this to be possible (Chen et al., 2019; Kim et al., 2017; Lin et al., 2018, Montagna et al., 2018; Ye et al., 2020). A complication in directly comparing the findings using these other EV‐based delivery systems to those in the present work is that percentage of gene editing achieved, EV concentration used, or Cas9 protein levels loaded were not reported in all studies. Some studies are focused on the delivery to target cells of Cas9‐encoding plasmids and of sgRNA, loaded into EVs using different strategies. In one case, DNA plasmids encoding Cas9 components were loaded into EVs post isolation via electroporation resulting in 27% gene editing efficiency (Kim et al., 2017). As in our study, another report found that sgRNA expression in producer cells resulted in sgRNA loading into secreted EVs which were fused to liposomes containing Cas9 DNA plasmids via overnight incubation at 37°C (Lin et al., 2018). Other studies have focused on the delivery of Cas9 protein and sgRNA, as in our study. In one report protein engineering was also employed based on arrestin domain containing protein 1 (ARRDC1) to recruit Cas9 and sgRNA into EVs through protein‐protein interactions (Wang et al., 2018). In line with our data, it was also observed that recruitment of Cas9 into EVs via protein‐protein interactions was sufficient to also result in loading of sgRNA and about 12% gene editing efficiency (Wang et al., 2018). In another case the fusogenic envelope glycoprotein of the vesicular stomatitis virus (VSV‐G) was co‐expressed with Cas9 and sgRNA in secreting cells (Montagna et al., 2018). The resulting VSV‐G induced EVs were capable of achieving up to 60% gene editing efficiency (Montagna et al., 2018). Most recently, another report showed that overexpression of Cas9 and sgRNAs in stably expressing cells established with lentiviral particles also resulted in cargo loading into EVs (Chen et al., 2019). Gene editing was also observed after administration of isolated EVs or in mixed co‐cultures of cells expressing the Cas9 constructs with cells that did not express the constructs (Chen et al., 2019).
The tools developed in this study for inducible protein loading into EVs show promising potential as a novel delivery vector for Cas9 and other proteins due to the high levels of functional cargo incorporated, and successfully delivered. In addition to the results presented in this work, the potential of using EVs for therapeutic gene editing is further justified due to their favourable safety profiles compared to other existing delivery methods such as the polymer and lipid‐based transfection reagents used in this study. Indeed, earlier work from our group has already shown EVs to have low toxicity and immunogenic profiles in vivo (Saleh et al., 2019) compared to other delivery methods providing further evidence that EVs could prove to be a useful tool for delivery of therapeutics.
CONFLICT OF INTEREST
All authors are or were employed in AstraZeneca R&D.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
The authors would like to acknowledge Graham Belfield (Translational Genomics, AstraZeneca) for project support, and Mario Soriano (Centro Investigácion Príncipe Felipe, Valencia, Spain) for the TEM analysis of EV samples. X.O, E.L.I, N.H, O.S, A.M.S, and A.M are or were fellows of the AstraZeneca postdoc program. Part of this work was supported by the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No 739593.
Osteikoetxea, X. , Silva, A. , Lázaro‐Ibáñez, E. , Salmond, N. , Shatnyeva, O. , Stein, J. , Schick, J. , Wren, S. , Lindgren, J. , Firth, M. , Madsen, A. , Mayr, L. M. , Overman, R. , Davies, R. , & Dekker, N. (2022). Engineered Cas9 extracellular vesicles as a novel gene editing tool. Journal of Extracellular Vesicles, 11, e12225. 10.1002/jev2.12225
Current addresses: Xabier Osteikoetxea, HCEMM‐SE Extracellular Vesicle Research Group, and Department of Genetics, Cell and Immunobiology, Semmelweis University, Budapest, Hungary. Nikki Salmond, Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, Canada. Olga Shatnyeva, Cell Therapy, Evotec SE, Göttingen, Germany. Josia Stein, Institute of Molecular Virology, Ulm University Medical Center, Ulm, Germany. Lorenz M. Mayr, Vector BioPharma AG, Aeschenvorstadt 36, 4051 Basel, Switzerland.
REFERENCES
- Adli, M. (2018). The CRISPR tool kit for genome editing and beyond. Nature Communications, 9, 1911. 10.1038/s41467-018-04252-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone, A. V. , Randolph, P. B. , Davis, J. R. , Sousa, A. A. , Koblan, L. W. , Levy, J. M. , Chen, P. J. , Wilson, C. , Newby, G. A. , Raguram, A. , & Liu, D. R. (2019). Search‐and‐replace genome editing without double‐strand breaks or donor DNA. Nature, 576, 149–157. 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman, E. K. , Chen, T. , Amendola, M. , & Van Steensel, B. (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research, 42, e168. 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H. H. Y. , Pannunzio, N. R. , Adachi, N. , & Lieber, M. R. (2017). Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nature Reviews Molecular Cell Biology, 18, 495–506. 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R. , Huang, H. , Liu, H. , Xi, J. , Ning, J. , Zeng, W. , Shen, C. , Zhang, T. , Yu, G. , Xu, Q. , Chen, X. , Wang, J. , & Lu, F. (2019). Friend or foe? Evidence indicates endogenous exosomes can deliver functional gRNA and Cas9 protein. Small, 15, 1902686. 10.1002/smll.201902686 [DOI] [PubMed] [Google Scholar]
- Choi, H. , Kim, Y. , Mirzaaghasi, A. , Heo, J. , Kim, Y. N. , Shin, J. H. , Kim, S. , Kim, N. H. , Cho, E. S. , In Yook, J. , Yoo, T. ‐. H. , Song, E. , Kim, P. , Shin, E. ‐. C. , Chung, K. , Choi, K. , & Choi, C. (2020). Exosome‐based delivery of super‐repressor IkappaBalpha relieves sepsis‐associated organ damage and mortality. Science Advances, 6, eaaz6980. 10.1126/sciadv.aaz6980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong, O. G. , Murphy, D. E. , Mäger, I. , Willms, E. , Garcia‐Guerra, A. , Gitz‐Francois, J. J. , Lefferts, J. , Gupta, D. , Steenbeek, S. C. , Van Rheenen, J. , El Andaloussi, S. , Schiffelers, R. M. , Wood, M. J. A. , & Vader, P. (2020). A CRISPR‐Cas9‐based reporter system for single‐cell detection of extracellular vesicle‐mediated functional transfer of RNA. Nature Communications, 11, 1113. 10.1038/s41467-020-14977-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Y. , Wu, N. , Gan, X. , Yan, W. , Morrell, J. C. , & Gould, S. J. (2007). Higher‐Order Oligomerization Targets Plasma Membrane Proteins and HIV Gag to Exosomes. Plos Biology, 5, e158. 10.1371/journal.pbio.0050158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann, G. , Herrmann, I. K. , & Stevens, M. M. (2015). Cell‐derived vesicles for drug therapy and diagnostics: Opportunities and challenges. Nano Today, 10, 397–409. 10.1016/j.nantod.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennemark, P. , Walter, K. , Clemmensen, N. , Rekić, D. , Nilsson, C. A. M. , Knöchel, J. , Hölttä, M. , Wernevik, L. , Rosengren, B. , Kakol‐Palm, D. , Wang, Y. , Yu, R. Z. , Geary, R. S. , Riney, S. J. , Monia, B. P. , Isaksson, R. , Jansson‐Löfmark, R. , Rocha, C. S. J. , Lindén, D. , … Davies, N. (2021). An oral antisense oligonucleotide for PCSK9 inhibition. Science Translational Medicine, 13, 10.1126/scitranslmed.abe9117 [DOI] [PubMed] [Google Scholar]
- Heath, N. , Osteikoetxea, X. , De Oliveria, T. M. , Lázaro‐Ibáñez, E. , Shatnyeva, O. , Schindler, C. , Tigue, N. , Mayr, L. M. , Dekker, N. , Overman, R. , & Davies, R. (2019). Endosomal escape enhancing compounds facilitate functional delivery of extracellular vesicle cargo. Nanomedicine (Lond), 14, 2799–2814. 10.2217/nnm-2019-0061 [DOI] [PubMed] [Google Scholar]
- Höijer, I. , Emmanouilidou, A. , Östlund, R. , Van Schendel, R. , Bozorgpana, S. , Tijsterman, M. , Feuk, L. , Gyllensten, U. , Den Hoed, M. , & Ameur, A. (2022). CRISPR‐Cas9 induces large structural variants at on‐target and off‐target sites in vivo that segregate across generations. Nature Communications, 13, 627. 10.1038/s41467-022-28244-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, K. K. , Pan, H. , Schlesinger, P. H. , & Wickline, S. A. (2015). Wickline SA. A role for peptides in overcoming endosomal entrapment in siRNA delivery ‐ A focus on melittin. Biotechnology Advances, 33, 931–940. 10.1016/j.biotechadv.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, S. C. , Kim, O. Y. , Yoon, C. M. , Choi, D. ‐. S. , Roh, T. ‐. Y. , Park, J. , Nilsson, J. , Lötvall, J. , Kim, Y. ‐. K. , & Gho, Y. S. (2013). Bioinspired exosome‐mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano, 7, 7698–7710. 10.1021/nn402232g [DOI] [PubMed] [Google Scholar]
- Kamerkar, S. , Lebleu, V. S. , Sugimoto, H. , Yang, S. , Ruivo, C. F. , Melo, S. A. , Lee, J. J. , & Kalluri, R. (2017). Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature, 546, 498–503. 10.1038/nature22341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano, F. , Suzuki, H. , Furuya, A. , & Sato, M. (2015). Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nature communications, 6, 6256. 10.1038/ncomms7256 [DOI] [PubMed] [Google Scholar]
- Kennedy, M. J. , Hughes, R. M. , Peteya, L. A. , Schwartz, J. W. , Ehlers, M. D. , & Tucker, C. L. (2010). Rapid blue‐light‐mediated induction of protein interactions in living cells. Nature Methods, 7, 973–975. 10.1038/nmeth.1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. M. , Yang, Y. , Oh, S. J. , Hong, Y. , Seo, M. , & Jang, M. (2017). Cancer‐derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism‐dependent targeting. Journal of Controlled Release: Official Journal of the Controlled Release Society, 266, 8–16. 10.1016/j.jconrel.2017.09.013 [DOI] [PubMed] [Google Scholar]
- Lai, C. P. , Kim, E. Y. , Badr, C. E. , Weissleder, R. , Mempel, T. R. , Tannous, B. A. , & Breakefield, X. O. (2015). Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nature Communications, 6, 7029. 10.1038/ncomms8029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya, A. , Weiner, O. D. , Lim, W. A. , & Voigt, C. A. (2009). Spatiotemporal control of cell signalling using a light‐switchable protein interaction. Nature, 461, 997–1001. 10.1038/nature08446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. , & Todo, T. (2005). The cryptochromes. Genome Biology, 6, 220. 10.1186/gb-2005-6-5-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Wu, J. , Gu, W. , Huang, Y. , Tong, Z. , Huang, L. , & Tan, J. (2018). Exosome‐liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Advances in Science (Weinh), 5, 1700611. 10.1002/advs.201700611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. ‐. C. , Boone, M. , Meuris, L. , Lemmens, I. , Van Roy, N. , Soete, A. , Reumers, J. , Moisse, M. , Plaisance, S. , Drmanac, R. , Chen, J. , Speleman, F. , Lambrechts, D. , Van De Peer, Y. , Tavernier, J. , & Callewaert, N. (2014). Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nature Communications, 5, 4767. 10.1038/ncomms5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , & Su, C. (2019). Design strategies and application progress of therapeutic exosomes. Theranostics, 9, 1015–1028. 10.7150/thno.30853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther, D. C. , Lee, Y. W. , Nagaraj, H. , Scaletti, F. , & Rotello, V. M. (2018). Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opinion on Drug Delivery, 15, 905–913. 10.1080/17425247.2018.1517746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc, T. , & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27, 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Lage, M. , Puig‐Serra, P. , Menendez, P. , Torres‐Ruiz, R. , & Rodriguez‐Perales, S. (2018). CRISPR/Cas9 for cancer therapy: Hopes and challenges. Biomedicines, 6, 105. 10.3390/biomedicines6040105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrak, A. , Bolukbasi, M. F. , Ozdener, G. B. , Brenner, G. J. , Madlener, S. , Erkan, E. P. , Ströbel, T. , Breakefield, X. O. , & Saydam, O. (2013). Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Molecular Therapy: the Journal of the American Society of Gene Therapy, 21, 101–108. 10.1038/mt.2012.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna, C. , Petris, G. , Casini, A. , Maule, G. , Franceschini, G. M. , Zanella, I. , Conti, L. , Arnoldi, F. , Burrone, O. R. , Zentilin, L. , Zacchigna, S. , Giacca, M. , & Cereseto, A. (2018). VSV‐G‐enveloped vesicles for traceless delivery of CRISPR‐Cas9. Molecular Therapy: Nucleic Acids, 12, 453–462. 10.1016/j.omtn.2018.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, K. , Engesser, R. , Timmer, J. , Nagy, F. , Zurbriggen, M. D. , & Weber, W. (2013). Synthesis of phycocyanobilin in mammalian cells. Chemical Communications (Cambridge, England), 49, 8970–8972. 10.1039/c3cc45065a [DOI] [PubMed] [Google Scholar]
- Musunuru, K. , Chadwick, A. C. , Mizoguchi, T. , Garcia, S. P. , Denizio, J. E. , Reiss, C. W. , Wang, K. , Iyer, S. , Dutta, C. , Clendaniel, V. , Amaonye, M. , Beach, A. , Berth, K. , Biswas, S. , Braun, M. C. , Chen, H. ‐. M. , Colace, T. V. , Ganey, J. D. , Gangopadhyay, S. A. , … Kathiresan, S. (2021). In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature, 593, 429–434. 10.1038/s41586-021-03534-y [DOI] [PubMed] [Google Scholar]
- Osteikoetxea, X. , Balogh, A. , Szabó‐Taylor, K. , Németh, A. , Szabó, T. G. , Pálóczi, K. , Sódar, B. , Kittel, Á. , György, B. , Pállinger, É. , Matkó, J. , & Buzás, E. I. (2015). Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. Plos One, 10, e0121184. 10.1371/journal.pone.0121184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteikoetxea, X. , Németh, A. , Sódar, B. W. , Vukman, K. V. , & Buzás, E. I. (2016). Extracellular vesicles in cardiovascular diseases, are they Jedi or Sith? The Journal of Physiology, 594, 2881–2894. 10.1113/JP271336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteikoetxea, X. , Sódar, B. , Németh, A. , Szabó‐Taylor, K. , Pálóczi, K. , Vukman, K. V. , Tamási, V. , Balogh, A. , Kittel, Á. , Pállinger, É. , & Buzás, E. I. (2015). Differential detergent sensitivity of extracellular vesicle subpopulations. Organic & Biomolecular Chemistry, 13, 9775–9782. 10.1039/C5OB01451D [DOI] [PubMed] [Google Scholar]
- Putyrski, M. , & Schultz, C. (2012). Protein translocation as a tool: The current rapamycin story. FEBS Letters, 586, 2097–2105. 10.1016/j.febslet.2012.04.061 [DOI] [PubMed] [Google Scholar]
- Rehman, K. , Sajid Hamid Akash, M. , Akhtar, B. , Tariq, M. , Mahmood, A. , & Ibrahim, M. (2016). Delivery of Therapeutic Proteins: Challenges and Strategies. Current Drug Targets, 17, 1172–1188. 10.2174/1389450117666151209120139 [DOI] [PubMed] [Google Scholar]
- Rothgangl, T. , Dennis, M. K. , Lin, P. J. C. , Oka, R. , Witzigmann, D. , Villiger, L. , Qi, W. , Hruzova, M. , Kissling, L. , Lenggenhager, D. , Borrelli, C. , Egli, S. , Frey, N. , Bakker, N. , Walker, J. A. , Kadina, A. P. , Victorov, D. V. , Pacesa, M. , Kreutzer, S. , … Schwank, G. (2021). In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nature Biotechnology, 39, 949–957. 10.1038/s41587-021-00933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saari, H. , Lázaro‐Ibáñez, E. , Viitala, T. , Vuorimaa‐Laukkanen, E. , Siljander, P. , & Yliperttula, M. (2015). Microvesicle‐ and exosome‐mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. Journal of Controlled Release: Official Journal of the Controlled Release Society, 220, 727–737. 10.1016/j.jconrel.2015.09.031 [DOI] [PubMed] [Google Scholar]
- Saleh, A. F. , Lázaro‐Ibáñez, E. , Forsgard, M. A.‐M. , Shatnyeva, O. , Osteikoetxea, X. , Karlsson, F. , Heath, N. , Ingelsten, M. , Rose, J. , Harris, J. , Mairesse, M. , Bates, S. M. , Clausen, M. , Etal, D. , Leonard, E. , Fellows, M. D. , Dekker, N. , & Edmunds, N. (2019). Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale, 11, 6990–7001. 10.1039/C8NR08720B [DOI] [PubMed] [Google Scholar]
- Schindler, C. , Collinson, A. , Matthews, C. , Pointon, A. , Jenkinson, L. , Minter, R. R. , Vaughan, T. J. , & Tigue, N. J. (2019). Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency. Plos One, 14, e0214545. 10.1371/journal.pone.0214545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, B. , Wu, N. , Yang, J.‐M. , & Gould, S. J. (2011). Protein targeting to exosomes/microvesicles by plasma membrane anchors. The Journal of Biological Chemistry, 286, 14383–14395. 10.1074/jbc.M110.208660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria, F. N. , Pampliega, O. , Bourdenx, M. , Meissner, W. G. , Bezard, E. , & Dehay, B. (2017). Exosomes, an unmasked culprit in neurodegenerative diseases. Frontiers in Neuroscience, 11, 26. 10.3389/fnins.2017.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbichler, T. B. , Dudás, J. , Riechelmann, H. , & Skvortsova, I. ‐. I. (2017). The role of exosomes in cancer metastasis. Seminars in Cancer Biology, 44, 170–181. 10.1016/j.semcancer.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. ‐. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , …, & Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7, 1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda, Y. , Goto, Y. , Oda, S. , Kohchi, T. , Matsuda, M. , & Aoki, K. (2017). Efficient synthesis of phycocyanobilin in mammalian cells for optogenetic control of cell signaling. Proceedings of the National Academy of Sciences of the United States of America, 114, 11962–11967. 10.1073/pnas.1707190114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Zhang, C. , Wang, B. , Li, B. , Wang, Q. , Liu, D. , Wang, H. , Zhou, Y. , Shi, L. , Lan, F. , & Wang, Y. (2019). Optimized CRISPR guide RNA design for two high‐fidelity Cas9 variants by deep learning. Nature Communications, 10, 4284. 10.1038/s41467-019-12281-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Breton, C. , Warzecha, C. C. , Bell, P. , Yan, H. , He, Z. , White, J. , Zhu, Y. , Li, M. , Buza, E. L. , Jantz, D. , & Wilson, J. M. (2021). Long‐term stable reduction of low‐density lipoprotein in nonhuman primates following in vivo genome editing of PCSK9. Molecular Therapy: the Journal of the American Society of Gene Therapy, 29, 2019–2029. 10.1016/j.ymthe.2021.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Yu, J. , Kadungure, T. , Beyene, J. , Zhang, H. , & Lu, Q. (2018). ARMMs as a versatile platform for intracellular delivery of macromolecules. Nature Communications, 9, 960. 10.1038/s41467-018-03390-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H. , Xiao, T. , Chen, C. ‐. H. , Li, W. , Meyer, C. A. , Wu, Q. , Wu, D. , Cong, L. , Zhang, F. , Liu, J. S. , Brown, M. , & Liu, X. S. (2015). Sequence determinants of improved CRISPR sgRNA design. Genome Research, 25, 1147–1157. 10.1101/gr.191452.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez‐Mó, M. , Siljander, P. R.‐M. , Andreu, Z. , Bedina Zavec, A. , Borràs, F. E. , Buzas, E. I. , Buzas, K. , Casal, E. , Cappello, F. , Carvalho, J. , Colás, E. , Cordeiro‐Da Silva, A. , Fais, S. , Falcon‐Perez, J. M. , Ghobrial, I. M. , Giebel, B. , Gimona, M. , Graner, M. , Gursel, I. , … De Wever, O. (2015). Biological properties of extracellular vesicles and their physiological functions. Journal of Extracellular Vesicles, 4, 27066. 10.3402/jev.v4.27066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmolinsky, M. , & Hoess, R. (2015). The legacy of Nat Sternberg: The genesis of cre‐lox technology. Annual Review of Virology, 2, 25–40. 10.1146/annurev-virology-100114-054930 [DOI] [PubMed] [Google Scholar]
- Ye, Y. , Zhang, X. , Xie, F. , Xu, B. , Xie, P. , Yang, T. , Shi, Q. , Zhang, C.‐Y. , Zhang, Y. , Chen, J. , Jiang, X. , & Li, J. (2020). An engineered exosome for delivering sgRNA:Cas9 ribonucleoprotein complex and genome editing in recipient cells. Biomaterials Science, 8, 2966–2976. 10.1039/D0BM00427H [DOI] [PubMed] [Google Scholar]
- Yim, N. , Ryu, S. ‐. W. , Choi, K. , Lee, K. R. , Lee, S. , Choi, H. , Kim, J. , Shaker, M. R. , Sun, W. , Park, J.‐H. , Kim, D. , Heo, W. D. , & Choi, C. (2016). Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein‐protein interaction module. Nature Communications, 7, 12277. 10.1038/ncomms12277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You, L. , Tong, R. , Li, M. , Liu, Y. , Xue, J. , & Lu, Y. (2019). Advancements and obstacles of CRISPR‐Cas9 technology in translational research. Molecular Therapy – Methods & Clinical Development, 13, 359–370. 10.1016/j.omtm.2019.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information