

Abstract
Ectodomain shedding (ES) is a posttranslational protein modification process that plays key roles in health and disease. Many neuronal and synaptic membrane proteins are known to undergo ES, but the complexity of functions regulated by the shed peptides is only beginning to be unraveled. Here, we provide an overview of emerging evidence demonstrating that synaptic ES can mediate autocrine and paracrine signaling. We also discuss how advances in large-scale proteomic analyses are leading to the identification of novel synaptic proteins undergoing ES, as well as the targets and functions of their soluble ectodomains. Finally, we also provide an overview of how cerebrospinal fluid analyses of shed proteins could be used as a potential source of new biomarkers for neuropsychiatric disorders.
Keywords: sheddome, synapse, autism, neurodegeneration, plasticity, proteomics
Synaptic ectodomain shedding is an emerging mechanism in health and disease.
Membrane proteins play essential roles in health and disease; therefore, their function must be tightly regulated. One of the mechanisms regulating membrane protein properties is Ectodomain (see Glossary) shedding (ES), a process in which a protease cleaves the extracellular portion of transmembrane- and glycosylphosphatidylinositol (GPI)-anchored proteins within 10–35 amino acids from the transmembrane region [1]. In some instances, an intramembrane protease can cleave the protein further in a signaling mechanism called regulated intramembrane proteolysis (RIP). RIP generates a second, shorter extracellular fragment and a cytoplasmic one [1] (Figure 1A). The latter can function in intracellular signaling but will not be discussed here.
Figure 1. Synaptic ectodomain shedding.
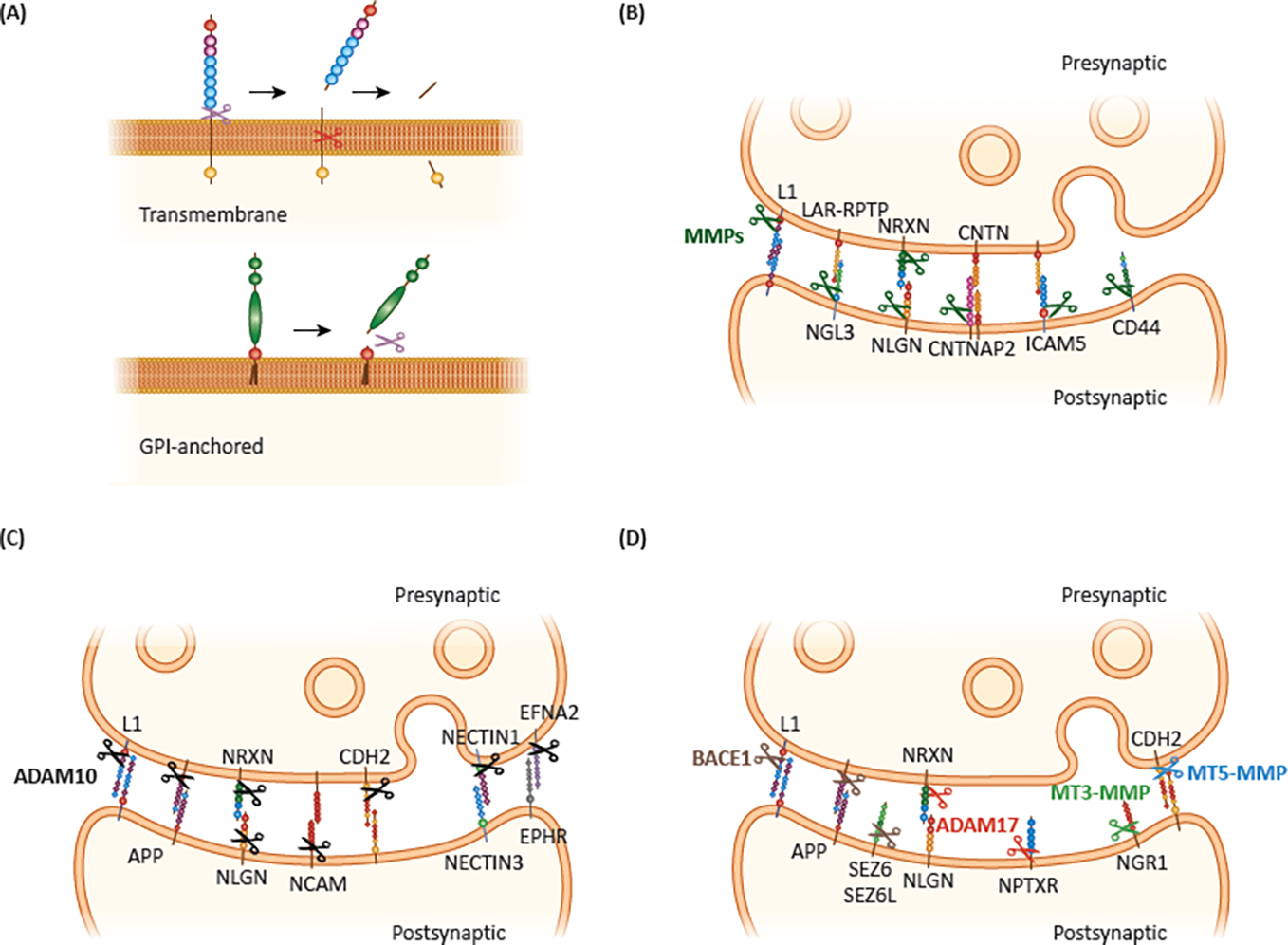
A. Numerous transmembrane and GPI-anchored proteins undergo proteolytic processing, resulting in the release of a soluble extracellular fragment in a process called ES. B-D. ES at the synapse can be mediated by MMPs (B), ADAM10 (C) and BACE1, ADAM17, MT3-MMP and MT5-MMP (D). Cleaved proteins are indicated next to their main protease; however, each substrate can be cleaved by multiple proteases. Of note, for several proteins it is still unclear whether they are found only in one side of the synapse or both.
An important distinction to consider is the one between shed and secreted proteins. Although both can be present in the extracellular space of a given tissue, shed proteins are originally membrane-attached and fully function as membrane proteins but can be proteolytically cleaved. On the other hand, secreted proteins are soluble non-membrane attached proteins, destined to be released into the extracellular environment by the secretory pathway. Examples of neuronal soluble proteins are hormones (i.e., somatostatin) and growth factors (i.e., brain-derived neurotrophic factor, BDNF). Along these lines, the term “secretome” refers to the totality of proteins found in the extracellular milieu of a given biological sample (including secreted and shed proteins), while “sheddome” refers only to the subset of proteins in the secretome that undergo ES.
ES plays widespread roles in development and disease, including in oncogenesis, metastasis, immunity, cardiovascular and kidney disease, and within the central nervous system (CNS) too. In the CNS, ES is involved in a range of processes, from brain development [2], neurogenesis [3], axonal regulation [4], to formation and maintenance of synapses [5, 6]. While ES was initially seen as serving the termination of membrane-bound protein function, it is becoming increasingly clear that it has much broader functions, including autocrine and paracrine signaling. Furthermore, dysregulation of synaptic ES has been linked to a range of neurological disorders, including Alzheimer’s disease (AD) [7] and prion disease [8]. As ectodomains shed within the brain can be detected in the CSF, proteomic analyses of this fluid show the promise of identifying novel (patho)physiological mechanisms, and biomarkers for brain disorders. Here, we provide an overview of synaptic ES, focusing on intercellular signaling exerted by shed ectodomains. We also summarize evidence on altered synaptic ES in CNS disorders, and the use of CSF sheddome analyses to evaluate synaptic (dys)function.
Synaptic ectodomain shedding is catalyzed by sheddases.
A growing body of evidence indicates that many synaptically-localized membrane proteins undergo ES (Figure 1B–D). This is supported by work focusing on individual proteins, such as neuroligins (NLGN) and neurexins (NRXN) [6, 9–11], but also by global analyses of the neuronal sheddome [2, 12–14]. ES is catalyzed by proteases, also known as “sheddases”, which cleave their targets on the extracellular side, near the membrane. Matrix metalloproteases (MMPs), “A disintegrin and metalloproteases” (ADAMs) and “β-site APP cleaving enzymes” (BACE1 and BACE2) are among the most common sheddases in the CNS and thus involved in the cleavage of most synaptic proteins. These proteases have been reviewed in detail before [1], hence we will only introduce them as they are relevant to synaptic ES. While the above mentioned sheddases are relatively well characterized, it is important to note that for most shed proteins, the responsible sheddases are not yet known, and the substrates for only a few sheddases have been catalogued. All sheddases, and likely the synaptically acting ones, exert their functions in manners dependent on developmental stage and tissue, brain region, cell type, subcellular location and cellular activity [9, 15–17]. Moreover, each protein can be cleaved by several sheddases and each sheddase can cleave several proteins, adding to the complexity of ES regulation. For an overview of mechanisms regulating ES, see Box 1.
Box 1. Mechanisms of shedding susceptibility.
The observation that one sheddase can cleave numerous substrates (Table 1), leads to the question of how ES is fine-tuned to maintain the delicate balance between health and disease. The molecular mechanisms regulating shedding susceptibility remain largely unknown. These mechanisms include phosphorylation, glycosylation, alternative splicing, protein trafficking, and interaction of the sheddase or the sheddable protein with regulatory proteins.
Alternative splicing and O-glycosylation occurring near the cleavage site are two mechanisms associated with shedding regulation [105, 106]. Both modulate ES of the synaptic cell adhesion molecule SynCAM1 [107, 108]. O-glycosylations have been reported to modulate APP ES, thereby decreasing Aβ production [107]. Particular N-glycosylation patterns regulate intracellular trafficking of sortilin-related receptor (SORLA), affecting its shedding susceptibility [109]. Neuronal activity-dependent SIRPα phosphorylation is necessary for releasing its ectodomain, which leads to synapse maturation [104]. Another mechanism regulating ES, is the expression of sheddase regulatory subunits that form active sheddase complexes. As an example, expression of rhomboid 1 is necessary for ADAM17-dependent ES of ‘multiple epidermal growth factor- like domains protein 10’ (MEGF10) [110]. Interestingly, sheddable membrane proteins can avoid ES by binding to certain proteins, as described for ADAM17-mediated Neogenin ES [111]. Finally, the presence of polysialic groups determines which sheddase can cleave NCAM; while polysialylated NCAM can be cleaved by ADAM10 and ADAM17, absence of this glycan group makes it only cleavable by ADAM17 [24, 49].
Several mechanisms could lead to alterations in ES regulation, potentially contributing to disease pathogenesis. Mutations can increase the affinity of the substrate for one sheddase over the others. For instance, the APP Swedish mutation increases its processing by BACE1, accelerating Aβ deposition. In contrast, the APP Icelandic mutation, also located near the BACE1 cleavage site, protects from developing dementia and presents with decreased amyloidogenic processing of APP [112]. A premature stop codon in the CNTNAP2 gene has been found in several Old Order Amish children suffering from Cortical Dysplasia Focal Epilepsy (CDFE) syndrome. The mutation creates a frameshift, resulting in the loss of the transmembrane and the cytosolic domains of CNTNAP2 [113]. This would result in the translation of a soluble version of CNTNAP2, very similar to the shed ectodomain of CNTNAP2. However, the CDFE mutant ectodomain would be released by the secretory pathway, while the wild-type ectodomain is processed by activity-dependent ES [20]. It seems reasonable to speculate that many other disease-associated mutations of sheddable membrane proteins could be altering these proteins’ regulation by ES, thereby contributing to disease pathogenesis.
MMP substrates include proteins that play important roles in synaptogenesis and synaptic plasticity, such as NLGNs [18], intercellular adhesion molecule 5 (ICAM5) [19], and CNTNAP2 [20] (Figure 1B). ADAM10 plays important roles in synapse formation and maintenance, as well as in axonal development, by cleaving synaptic cell adhesion molecules (CAMs), including cadherins and the neurexin–neuroligin complex (Figure 1C). Other sheddases have also been described to process synaptic proteins (Figure 1D). ADAM17 regulates neuronal development and plasticity by catalyzing the shedding of several proteins, including neuronal pentraxin receptor (NPTXR) [21], p75 neurotrophin receptor (p75NTR) [22], L1 cell adhesion molecule (L1CAM) [23], and neural cell adhesion molecule 1 (NCAM or NCAM1) [24]. BACE1 is abundant at synapses and sheds several important synaptic proteins, including amyloid precursor protein (APP), seizure related 6 homolog (SEZ6), SEZ6L, SEZ6L2, close homolog of L1 (CHL1), L1CAM, and NCAM1 and 2 [12, 15]. BACE1-mediated shedding has been shown to affect synapse formation, function, plasticity, and behavior in mice [25]. Less studied metalloproteases, such as MT3-MMP and MT5-MMP also regulate ES of synaptic proteins, including N-cadherin (CDH2) [26] and Nogo-66 receptor 1 (NGR1) [27] (Figure 1D). γ-secretase is an intramembrane protease that also plays important roles at synapses by cleaving mainly substrates with long extracellular domains, and frequently requires ES by sheddases before γ-secretase cleaves its target protein within the transmembrane domain [28]. A different mode of membrane protein processing is catalyzed by GPI phospholipases or GPI-phosphodiesterase 2, which cleave GPI-anchored proteins releasing their extracellular domains. This mechanism is less discussed in the literature, but is essential for neurogenesis, and modulates aging and cognition [3, 29].
Several neurophysiological processes are known to regulate sheddase activity and, thus, synaptic ES. Neuronal activity, long-term potentiation (LTP), and memory formation during Morris water maze learning increase MMP9 activity [30]. Fear conditioning, a model of aversive associative learning, increases MMP9 and MMP2 activity in rodents. Moreover, seizure induction by kainate [31] and pilocarpine administration [32, 33] increase MMP activity in rodents. Psychostimulants such as methamphetamine [34] or cocaine [35] are also known to increase protease activity. Other inducers of MMPs include chronic stress [36] and ischemia [37]. Interestingly, several proinflammatory factors (LPS, IL-1, TNFα) stimulate MMP9 and MMP2 expression [38], while anti-inflammatory stimuli negatively regulate them [39].
Important synaptic proteins undergo ectodomain shedding.
Several synaptic proteins have been shown to undergo ES. Below, we discuss some of the most prominent examples (Table 1).
Table 1.
Examples of synaptic proteins known to undergo ectodomain shedding, including the cleaving proteases and regulating stimuli.
| Synaptic protein | Protease | Stimuli | References |
|---|---|---|---|
| APP | ADAM10 BACE1 γ-secretase* |
Neuronal activity Nicotinic receptor activation NMDAR activation |
[41,42] |
| CD44 | MMP9 | 5-HT7R activation | [56] |
| CDH2 | MT5-MMP ADAM10 |
Bicuculline-induced neuronal activity NMDA-induced neuronal activity |
[26, 50] |
| CHL1 | BACE1 γ-secretase* |
N.D. | [12, 57] |
| CNTNAP2 | MMP9 | cLTP | [20] |
| EFNA2 | ADAM10 | EFNA2/EPHA3 binding | [4] |
| ICAM5 | MMP9 | NMDAR and AMPAR activation | [19, 51] |
| L1CAM | MMPs ADAM10 BACE1 |
PMA-induced PKC signaling NMDA incubation |
[23, 53, 55] |
| NCAM | ADAM10 ADAM17 BACE1 |
EFNA5/EPHA3 binding | [15, 24, 49] |
| Nectin1 | ADAM10 | Constitutive NMDAR activation |
[52] |
| NGL3 | MMPs γ-secretase* |
LTD induced by NMDA Low frequency stimulation (LTD) |
[58] |
| NGR1 | MT3-MMP | N.D. | [27, 103] |
| NLGN1 | MMP9 ADAM10 |
NMDA+glutamate Interaction with sNRXN Pilocarpine-induced epileptic seizures |
[5, 6, 18] |
| NLGN2 | MMP9 | NMDA+glutamate | [18] |
| NLGN3 | ADAM10 MMP9 |
NMDA+glutamate | [18, 48] |
| NOTCH | ADAM10 | Binding to Delta ligands | [66] |
| NPTXR | ADAM17/TACE | mGluR1/5-dependent LTD | [21] |
| NRG1 | BACE1 ADAM10 ADAM17 |
NMDA Kainate |
[62, 63] |
| NRXNs | MMP γ-secretase |
KCl-induced neuronal activity | [45] |
| NRXN3(3 | ADAM10 ADAM17/TACE α-secretase γ-secretase* |
N.D. | [46, 47] |
| SEZ6 SEZ6L |
BACE1 | N.D. | [59] |
| SIRPα | MMPs | KCl Activity-induced tyrosine phosphorylation |
[70, 104] |
N.D. not determined.
Not sheddase, intramembrane cleavage.
APP is a single-pass transmembrane protein abundant at synapses. Its improper proteolytic cleavage is an emblematic example of ES contributing to a brain disorder. The amyloidogenic pathway is mediated by the activity of BACE1 followed by γ-secretase, which generates the neurotoxic amyloid-β (Aβ). Processing of APP by anti-amyloidogenic α-secretases (e.g., ADAM10) followed by γ-secretase, results in the production of the neuroprotective soluble APPα (sAPPα) [40], limiting the production of toxic Aβ. APP proteolytic processing has been described to be activity- and nicotinic receptor-dependent [41, 42]. Interestingly, sublethal NMDA receptor activation in cultured cortical neurons causes a shift from α-secretase to β-secretase APP processing, promoting Aβ production [17]. Proteolytic cleavage of APP has been reviewed extensively, hence we will not discuss it in detail [43, 44].
Neurexins (NRXN1-3) are a family of transmembrane proteins involved in the development and maturation of synapses. In the CNS they are located mainly pre-synaptically. NRXNs undergo KCl-induced ES regulated by MMPs and then they are further cleaved by γ-secretase [45]. NRXN3β undergoes shedding mediated by ADAM10/17 [46] and α- and γ-secretases [47]. CNTNAP2 is a CAM located at synapses and is a prominent neurodevelopmental disorder risk factor. Proteomic profiling of the neuronal sheddome in vitro and human CSF together with super resolution imaging techniques revealed that CNTNAP2 undergoes activity-dependent shedding mediated by MMP9 [20].
Neuroligins (NLGN1-4) are a family of postsynaptic proteins that interact with presynaptic neurexins through their extracellular domains. Neuroligins promote the maturation of presynaptic terminals and regulate synaptic plasticity. Activity-dependent shedding of NLGN1, 2 and 3 has been reported so far [18]. NLGN1 is cleaved by MMP9 and ADAM10 upon activation of NMDA-R in vitro and by pilocarpine-induced seizures in vivo [5, 6, 18]. NLGN2 undergoes activity-dependent shedding mediated by MMP9 [18]. NLGN3 is cleaved by ADAM10 and MMP9 also in an activity-dependent manner [18, 48].
Classical synaptic CAMs also undergo ES. NCAM is an adhesion molecule that controls axon and dendrite development, as well as synaptic plasticity. NCAM ES is catalyzed by ADAM10 via ephrinA5 (EFNA5)/EphA3 interaction [49]. ADAM17-mediated ES of NCAM regulates neurite outgrowth in vitro [24]. Interestingly, NCAM1 and NCAM2 are differentially processed by BACE1 in vivo, depending on the brain region and developmental stage. A study in mice examined the cleavage of NCAM1 and NCAM2 by BACE1 at different developmental timepoints, including postnatal day 10, and at 4 and 12 months of age. NCAM1 and NCAM2 ES was observed at all ages in the olfactory bulb. In the hippocampus, however, NCAM1 ES was observed only at postnatal day 10, and not during adulthood, while NCAM2 was not processed by ES in this brain region at any age [15]. At synapses, CDH2 is cleaved by MT5-MMP and ADAM10 upon increased neuronal activity induced by bicuculline or NMDA treatment [26, 50]. The neuron-specific ICAM5 is present in filopodia and immature dendritic spines and has an important role in higher-order cognitive functions. ICAM5 shedding is regulated by NMDA-R and AMPA-R in an MMP-dependent manner [19, 51]. Nectin1 is a CAM that localizes pre- and post-synaptically and plays important roles in synapse maturation. It undergoes ADAM10-dependent ES, in the pre- and post-synaptic compartments, both constitutively and upon NMDA-R activation [52]. L1CAM plays significant roles in synaptic function and neurodevelopment, including processes such as neurite outgrowth and myelination. L1CAM ES is regulated by MMPs, ADAM10, ADAM17 and BACE1, and it is stimulated by PKC activation and NMDA incubation. ES of L1CAM is involved in cell adhesion, migration, and neurite outgrowth [12, 53, 54]. KO of l1cam in zebrafish was shown to cause axonal growth abnormalities and hydrocephalus; however, L1cam ES plays a role only in hydrocephalus and not in axonal outgrowth, as shown in rescue experiments with soluble and uncleavable forms of L1cam in zebrafish [55]. CD44 is cleaved by MMP9 upon serotonin receptor 5-HT7R activation inducing synapse remodeling [56]. CHL1 is a cell adhesion molecule that undergoes BACE1-dependent ES, and posterior γ-secretase cleavage. This modulates semaphoring-3A-mediated growth cone collapse. CHL1 ES by BACE1, induces growth cone collapse which is stopped when the membrane-bound CHL1 fragment is further cleaved by γ-secretase [57]. NGL3 (netrin-G ligand-3), a postsynaptic CAM that trans-synaptically interacts with the leukocyte antigen-related (LAR) family of receptor tyrosine phosphatases, undergoes proteolytic cleavage upon NMDA-induced long-term depression (LTD) and low-frequency stimulation, catalyzed by MMPs and γ-secretase/presenilin [58]. Experiments in mice indicated that the cleavage of SEZ6 and SEZ6L, which plays roles in synaptic connectivity and motor coordination, is catalyzed by BACE1 in the brain [59].
Ephrins and Eph receptors are another class of synaptic proteins that undergo shedding. EphA and -B receptors are the largest subfamily of receptor tyrosine kinases and their ligands, ephrins, are transmembrane (ephrin-B, EFNB) or GPI-anchored (ephrin-A, EFNA) proteins localized on opposite cells. Signaling by Eph-ephrin interactions regulate diverse processes including repulsive and attractive axon guidance, dendritic spine remodeling, and synaptic plasticity [4, 60]. Eph-induced axonal retraction requires proteolytic cleavage of EFNA2 by ADAM10 following binding to its receptor EphA3 in vitro [4].
Several other molecules that do not belong to the families discussed above, also undergo ES. NPTXRs are localized to excitatory synapses, where they bind to AMPA-R and regulate AMPA-R dependent plasticity. ADAM17-mediated NPTXR shedding is induced by mGluR1/5 dependent LTD-like stimuli [21]. NGR1 mediates axonal growth inhibition, regulates axonal regeneration, and negatively regulates plasticity. NGR1 undergoes ES catalyzed by MT3-MMP, promoting excitatory synapse formation [27]. Moreover, NGR1 ectodomain administered in vivo promoted the erasure of fear memories in mice [61]. Finally, neuregulin1 (NRG1) is a neurotrophic protein that binds to ERBB3 and ERBB4, and plays important roles in neurodevelopment. NRG1 is shed by BACE1, ADAM10 and ADAM17, processes which in zebrafish have also been shown to regulate myelination [62]. In rats, NRG1 ES is activated by kainate-induced seizures [63]. Interestingly, zebrafish expressing a NRG1 probe fused with mCherry and GFP to the extracellular and intracellular domains, respectively, showed that its ES occurs preferentially at axonal locations despite being expressed also in somas [16].
Physiological functions of synaptic ectodomain shedding.
Many of the physiological functions of ES are only beginning to be uncovered, and the function of most shed ectodomains is currently unknown. However, several biological roles for synaptic ES can already be distinguished.
Termination of activity
For synaptic CAMs ES often causes loss of cell-cell contacts and synapse weakening, as removal of trans-synaptic adhesive protein-protein interactions reduces bidirectional synaptic signaling necessary for synaptic assembly/maturation [5, 6, 64]. NLGN1 ES inhibits synaptic maturation and decreases neurotransmitter release. It also attenuates excitatory postsynaptic potential (EPSC) frequency and amplitude, by terminating the interaction between NLGN1 and presynaptic NRXN [5, 6]. Proteolytic cleavage of NLGN3 also reduces synaptic strength: re-expression of NLGN3 in NLGN1-3 tripleknockdown organotypic hippocampal slices enhanced inhibitory and excitatory synaptic transmission. This effect was abrogated by PMA-induced NLGN3 ES [48]. In cultured cortical neurons, activity-dependent proteolytic cleavage of NLGN1 decreased vesicle release probability and mEPSC frequency by destabilizing its interaction with NRXN1 [5]. ADAM10-dependent CDH2 cleavage negatively regulates dendritic spine morphology in vitro [65].
ES of synaptic receptors may lead to the termination of receptor effects and may even give rise to decoy receptors. For example, NOTCH ES creates an extracellular inert product that is trans-endocytosed and degraded [66]. Eph/ephrin-mediated cell repulsion require termination of the high-affinity Eph-ephrin interaction by either endocytosis or ectodomain cleavage [4].
Consistent with synapse weakening effects, ES is sometimes associated with LTD. For example, NPTXR cleavage is induced by LTD-like stimuli and promotes AMPA-R internalization [21], while the shed ectodomain of NLGN1 decreases synaptic activity by activating presynaptic mGluR2 [67].
Interestingly, in some instances, ectodomain cleavage is a mechanism to remove an inhibitory signal. NGR1 ES by MT3-MMP removes a break on synaptogenesis, as either NGR1 ES or application of sNGR1 promote excitatory synapse formation [27].
Intercellular signaling by synaptic ectodomain shedding.
Growing evidence shows that soluble ectodomains function as intercellular signals to activate downstream pathways in synapse development, plasticity, and brain circuit function. The complexity of effects exerted by shed ectodomains involves autocrine and paracrine signaling, causing diverse effects on various cell types and during different developmental stages (Table 2, Key Table). As such, the same ectodomain may have different, sometimes even opposite roles in different situations, or the shed ectodomain may have opposite roles to the full-length protein. Of note, the technical approach most often used to study the effects of shed ectodomains has been the exogenous application of the peptides. Thus, in some instances, it cannot be specified whether the effects described for these ectodomains are due to paracrine or autocrine signaling. Moreover, paracrine effects have been most frequently studied, probably due to technical reasons. Below we discuss the most prominent examples of autocrine/paracrine effects exerted by shed ectodomains of synaptic proteins.
Table 2.
Autocrine/paracrine effects of shed synaptic ectodomains.
| Synaptic protein | Type of effect | Cell type/system | Developmental stage | Effect | Reference |
|---|---|---|---|---|---|
| APP (Aβ) | Autocrine and paracrine | Cultured hippocampal slices (rat) | Postnatal day 6–7 | ↓Spine density and plasticity | [41] |
| CNTNAP 2 | Paracrine | Cultured cortical neurons (mouse) | Mature neurons | ↓Neuronal synchrony | [20] |
| ICAM5 | Bath application. Autocrine? | Cultured hippocampal neurons (mouse and rat) | Mature neurons | Filopodia elongation, fexcitatory transmission | [51, 69] |
| L1CAM | Autocrine/paracrine | CHO cells | N.A. | Cell migration | [53] |
| NCAM | Paracrine | Neocortex and hippocampus (mouse) | Adult | ↓Synapses in amygdala | [68] |
| NRG1 | Paracrine | Swann cells (zebrafish) | Larva | Peripheral nervous system myelination | [62] |
| NRXN1β | Acute bath application. Paracrine? | Cultured hippocampal neurons (rat) | Immature neurons | Neurite outgrowth | [9] |
| Acute bath incubation. Paracrine? | Autaptic hippocampal neurons (rat) | Mature neurons | ↑Synaptic transmission | ||
| Prolonged bath incubation. Paracrine? | Autaptic hippocampal neurons (rat) | Mature neurons | ↓Synaptic transmission | ||
| NRXN3β | Paracrine | Granule cell to newborn neurons (mouse) | Adult | Spine maturation | [46] |
| Autocrine | Newborn neurons (mouse) | Adult | Axonal development | ||
| SIRPα | Paracrine | Cultured hippocampal neurons (mouse) | Immature neurons | Presynaptic maturation | [70] |
| αNRXN | Paracrine | Neuromuscular junction (C. eleaans) | Adult | ↓ACh release | [10] |
Several autocrine/paracrine roles have been reported for neurexins and neuroligins. In C. elegans, ES of postsynaptic neurexin inhibits acetylcholine release by binding to and inhibiting presynaptic α2δ subunits associated with CaV2 channels at the neuromuscular junction, thereby regulating synaptic transmission [10] (Figure 2A). This interaction is evolutionarily conserved despite different synaptic orientations of neurexin in C. elegans and mouse. Soluble NRXN3β (sNRXN3β) secreted by mature granule cells in the adult mouse hippocampus, increased spine density on hippocampal newborn neurons in a paracrine manner. In contrast, sNRXN3β secretion by newborn neurons had no effect on their own spine density but induced axonal maturation in an autocrine manner [46]. In cultured hippocampal neurons before synaptogenesis, acute bath application of sNRXN1β increased Ca2+ influx via N-type Ca2+ channels, and it increased neuritogenesis; in mature neurons, the Ca2+ influx elicited by sNRXN1β was NMDAR dependent instead [9]. These effects were probably paracrine, because NRXNs are mostly presynaptic, and the effects were NLGN dependent, which is mostly postsynaptic. On the other hand, while an acute exposure to sNRXN1β increased glutamatergic synaptic transmission in mature neurons, a prolonged incubation inhibited it [9].
Figure 2. Examples of paracrine signaling by soluble ectodomains shed from synaptic proteins.
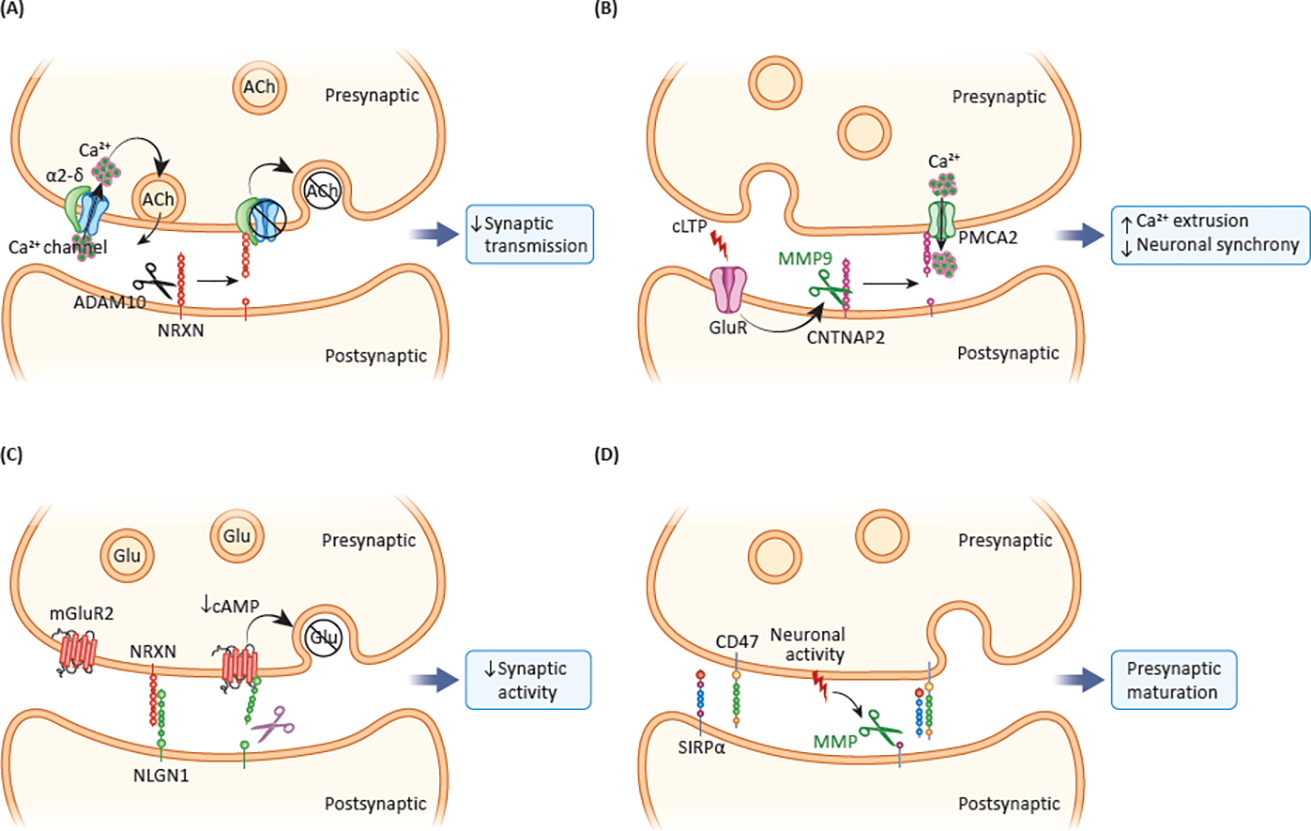
Proteins of origin, sheddases, regulation of shedding, soluble ectodomains, protein targets and biological actions are indicated for the ES of NRXN1 [10] (A), CNTNAP2 [20] (B), NLGN1 [67] (C) and SIRPα [70] (D).
Another member of the neurexin superfamily, CNTNAP2, also undergoes ES [20]. CNTNAP2 ectodomain decreased neuronal network activity and enhanced Ca2+ extrusion in hippocampal slices in a PMCA2-dependent manner. Interestingly, colocalization of shed CNTNAP2 with the presynaptic marker VGLUT1 increased upon chemical LTP (cLTP). We hypothesize that upon activity, shed CNTNAP2 binds to and activates presynaptic PMCA2. This increases presynaptic Ca2+ extrusion, which in turn acts as a feedback inhibitor of neuronal activity [20] (Figure 2B). sNLGN1 activates presynaptic metabotropic glutamate receptor 2 (mGluR2) in a paracrine manner, decreasing glutamate release from mossy fibers, and synaptic activity [67] (Figure 2C).
The soluble ectodomains of several CAMs also have autocrine/paracrine functions. A transgenic mouse expressing the NCAM ectodomain in the neocortex and the hippocampus exhibited a paracrine reduction of synapses in the amygdala and the cingulate and frontal association cortices, but not in the hippocampus, which demonstrates a lack of autocrine effect [68]. Early studies showed that L1CAM ES promotes CHO cell migration by autocrine/paracrine stimulation via αvβ5 integrin receptors [53]. ES of ICAM5 is an interesting example of bidirectional regulation of synaptic development; the full-length protein inhibits dendritic spine maturation, while the shed ectodomain promotes it [51]. The effect of sICAM5 is lost in ICAM5 deficient hippocampal cultures, which could be pointing to a dominant negative effect of sICAM5. This observation also indicates that the effect of sICAM5 might be autocrine, as ICAM5 is mostly expressed postsynaptically. Moreover, bath application of sICAM5 increased excitatory transmission, and insertion of GluA1 in the neuronal surface in cultured hippocampal neurons [69].
Shed ectodomains of several other types of proteins also function as paracrine signals. For example, activity-dependent ES of signal regulatory protein-α (SIRPα) is a trans-synaptic mechanism that promotes presynaptic maturation by the interaction of sSIRPα and the presynaptic CD47 in a paracrine manner [70] (Figure 2D). The soluble EGF-like domain of NRG1 is released by BACE1 and ADAM17 cleavage, and rescues myelination abnormalities in BACE1 KO zebrafish in a paracrine manner [62]. The effects of the proteolytic processing of APP to generate Aβ have been described to be autocrine and paracrine. Aβ production was sensitive to neuronal activity and nicotinic receptor blockade, and reduced the number and plasticity of dendric spines in an autocrine and paracrine manner [41].
Collectively, these findings illustrate the richness of effects exerted by shed ectodomains depending on the developmental stage, cell type, directionality of effect (autocrine/paracrine), or exposure time. The mechanisms differentiating between autocrine vs paracrine signaling remain to be elucidated, although different affinities for presynaptic or postsynaptic binding partners, or competition with the full-length protein probably play a role. In some instances, both the shed ectodomain and the full-length protein are known to bind the same target. Two known examples are the sCNTNAP2-PMCA2 [20] and sAPP-integrin [71] interactions. However, it is still unknown whether both protein species bind their targets with the same affinity. Moreover, soluble ectodomains with the same binding profiles as the full-length protein, may play dominant negative effects. This may occur when binding of the full-length protein to its target induces an intracellular cell signaling cascade, which would be impeded by the soluble version due to the lack of the transmembrane and the cytosolic domains.
Targets of shed synaptic ectodomains
To date, few studies have identified binding partners for synaptically shed ectodomains or examined the functional consequences of this interaction. In one of the studies addressing these issues, the authors used affinity pull-down assays of recombinant sAPP interacting with synaptosomal proteins, coupled with mass spectrometry, and showed that sAPP binds to GABAB1A receptors, which modulated synaptic transmission and plasticity [72]. Earlier studies have shown that sNRXN1 binds CaV2 channels reducing acetylcholine release at the C. elegans and mammalian neuromuscular junction [10]. Lastly, in a recent study from our group, sCNTNAP2, which interacts with and activates the extrusion pump PMCA2, was shown to decrease cytosolic Ca2+ [20]. These emerging reports underscore the potential of identifying novel targets of shed ectodomains in revealing processes controlled by autocrine/paracrine signals.
Global analyses of neuronal ectodomain shedding and the CSF
While most of the reports address ES of individual proteins, a growing number of studies have examined the sheddome globally, facilitated by recent advances in proteomics. These studies typically involve the analysis of conditioned media from cultured neurons, sometimes followed by validation in mice. They led to the identification of novel substrates for known sheddases such as BACE1 or ADAM10 [2, 12], but also new biological functions of ES [20]. Several methods have been developed to analyze neuronal sheddomes, such as “secretome protein enrichment with click sugars” (SPECS), or its recent version “high-performance SPECS” (hiSPECS) [13]. They have been used in conjunction with sheddase inhibition or knockouts to identify substrates. These studies allowed the generation of comprehensive lists and databases of shed proteins. One such searchable database, SheddomeDB, compiled experimentally validated shed membrane proteins [73] (available at http://120.126.86.226:8080/).
In most cases, these investigations were not specifically designed to address the synaptic sheddome. To gain insight into global patterns of synaptic ES, a recent study from our group analyzed datasets from SPECS and liquid chromatography tandem mass spectrometry (LC-MS/MS) analyses of media from cultured neurons [20]. Based on a bioinformatic analysis, the most enriched biological process was found to be ‘positive regulation of synapse assembly’. It was also found that the neuronal sheddome is enriched in risk factors for neurodevelopmental disorders. Interestingly, this unbiased analysis underscored the role of ES in synapse regulation and synaptopathologies, which is supported by previous information on individual protein’s ES.
The CSF is the most readily accessible source of proteins derived from the brain in living humans, hence it has the potential to offer a readout of brain function. Several proteomic analyses have been performed to detect brain-enriched proteins in the CSF, including in human CSF extracted by lumbar puncture. Studies in mice show that the genetic or pharmacological manipulation of sheddases such as BACE1 [74] and ADAM10 [75] modulates the levels of ectodomains in the CSF. There is a notable overlap between the in vitro neuronal and CSF sheddome datasets, suggesting that the CSF sheddome mirrors the neuronal sheddome [20]. Moreover, autism spectrum disorder (ASD) and schizophrenia (SZ) risk gene-encoded proteins were enriched in the cleaved, but not in the secreted CSF fraction. While requiring further investigation, this supports the idea that proteomic analysis of the CSF might be useful to study synapse pathophysiology. Indeed, the human CSF (hCSF) has been used to understand synapse pathology, as discussed later.
Implications of synaptic ectodomain shedding for neuropsychiatric disorders
Altered ES, including of synaptic proteins, has been associated with CNS diseases. This suggests broader roles of synaptic ectodomains in pathogenic processes or as biomarkers, which remain to be explored. The most studied example of deleterious ES is the amyloidogenic APP cleavage in AD. However, ES has also been implicated in other pathologies including glioma proliferation [11] or microglial activation [76]. As discussed next, both global proteomic ES analyses and investigations of specific synaptic ectodomains are helping advance current understanding of how synaptic ES contributes to neuropsychiatric disorders.
Individual synaptic ectodomain alterations in specific CNS diseases.
Altered levels of individual soluble ectodomains of synaptic proteins have been detected in the CSF of patients with numerous neurological and psychiatric disorders (Table 3). There could be several reasons for such alterations in shed ectodomains in the CSF, including altered expression levels or transport of the cleavable proteins, genetic variants that affect the cleavage, aggregation of pathogenic cleaved ectodomains, altered expression levels or targeting of sheddases, altered neuronal activity levels which modulate shedding, altered number of synapses, or other yet unknown mechanisms.
Table 3.
Synaptic ectodomains detected in hCSF in CNS diseases.
| Protein | Disorder | Levels | Reference |
|---|---|---|---|
|
| |||
| sCNTNAP2 | ASD | Decreased | [20] |
|
| |||
| sICAM5 | Acute encephalitis | Increased | [87] |
| MS | Decreased | [89] | |
| Temporal lobe epilepsy | Increased | [88] | |
|
| |||
| sLICAM | SZ | Decreased | [77] |
| Dementia, AD | Increased | [78] | |
| Glioblastoma | Increased | [90] | |
|
| |||
| sNCAM | SZ | Increased | [77] |
| MS | Decreased | [86] | |
|
| |||
| NRXN2and 3 | Preclinical AD | Decreased | [96] |
| Prodromal AD | Increased | ||
| AD | Increased | ||
|
| |||
| sp75NTR | AD | Decreased | [79] |
Increased levels of sNCAM have been detected in the CSF of patients with SZ [77], and a transgenic mouse overexpressing sNCAM displayed SZ-related abnormal behaviors including deficits in sensory gating and emotional memory, as well as decreased dendritic spine density [68]. Decreased sL1CAM has also been found in SZ CSF [77], while elevated sL1CAM has been found in the CSF of dementia patients, including AD [78]. Decreased levels of p75NTR ectodomain have also been detected in the CSF of AD patients [79].
As for autism, proteins encoding ASD susceptibility genes were found to be enriched within the sheddome, but not in the secreted components of the secretome, both in neurons in vitro and in the human CSF [20]. Moreover, decreased levels of sCNTNAP2 in the CSF were observed in a group of individuals with ASD [20]. This points to a potential role of ES in ASD, a hypothesis that needs further validation.
While a role for ES in epilepsy has not yet been extensively investigated, a number of studies suggest it. Several sheddases including MMP9 and ADAM10 are altered in epilepsy patients [80–82], and their manipulations modify seizure phenotypes in mice [83–85].
Altered levels of soluble ectodomains have also been reported in several other neurological disorders. In multiple sclerosis (MS) patients, sNCAM levels were lower when compared to healthy controls, while treatment with disease-modifying therapies increased them [86]. Whether this reversal in the levels of sNCAM is a reflection of neuroplasticity/neurorepair mechanisms remains to be elucidated. sICAM5 levels in the CSF also change in pathophysiological conditions: they are increased in acute encephalitis [87] and temporal-lobe epilepsy [88], and decreased in MS [89]. On the other hand, treatment of a mouse model of MS with sICAM5, ameliorated the disease’s symptoms [89]. Finally, patients with glioblastoma and brain metastasis present increased levels of sL1CAM in the CSF [90].
Global analyses of CSF in CNS diseases
Global proteomic analyses of CSF have been performed in patients with several neurodegenerative disorders, including AD [91, 92], amyotrophic lateral sclerosis (ALS) [93], and sporadic Creutzfeldt-Jakob disease (sCJD) [94]. In many of the related studies, synaptic proteins have been of particular interest, given the broadly accepted role synapses play in pathogenesis. In ALS, WD repeat-containing protein 63, amyloid-like protein 1, SPARC-like protein 1, and cell adhesion molecule 3 have been proposed as candidate biomarkers [93]. Synaptic proteins like proSAAS, apolipoprotein J, neurosecretory protein VGF, phospholemman, and chromogranin A were proposed as early biomarkers in AD [95]. CSF analyses in different stages of AD demonstrated that certain synaptic proteins are decreased in preclinical patients before CSF markers of neurodegeneration or symptoms are observed [96]. In contrast, in later stages of neurodegenerative disorders including ALS, Parkinson’s and AD, elevated levels of synaptic proteins in the CSF were observed, and these changes are thought to reflect synapse loss [79, 93, 96–98]. Finally, CSF proteome analyses of major depressive disorder patients also showed a decrease in synaptic proteins, including NRXN3 and NPTXR [99], and bipolar disorder 1 patients showed reduced levels of several brain-expressed proteins including testican-1, TNFRSF21, CADM3, and ADAM22 in the CSF [100].
Overall, these studies indicate that synaptic changes might be detected in the CSF and that events occurring at different stages of the pathophysiological cascade could be measured with a relatively low-invasive technique such as a lumbar puncture. Indeed, the CSF has been proposed as a source for biomarkers of synapse function or pathology in the brain [91, 93, 95]. However, it must be considered that changes in the synaptic CSF proteome could be reflective of synapse loss, altered ES, or disease-associated cell death. Of note, there have been some efforts to define the synaptic sheddome by bioinformatically filtering synaptically expressed proteins from the CSF sheddome, however none of the published data addresses the synaptically-originated sheddome. Thus, proteomic studies are needed to shed light on how the synaptic sheddome is altered in CNS disorders.
Soluble ectodomains as potential therapeutics in CNS diseases
In preclinical studies, soluble synaptic ectodomains have also been described to have therapeutic utility. For example, in an AD mouse model, the p75NTR ectodomain was shown to be neuroprotective against amyloid-β toxicity, as it improved memory performance, reduced Aβ levels in the brain, and attenuated inflammation and dendritic spine loss [101]. In hippocampal slices, sAPPα, but not sAPPβ generated by the amyloidogenic pathway, protected against Aβ-associated dendritic spine loss and increased tau phosphorylation [40]. sNRX1β also showed neuroprotective properties in cultured hippocampal neurons exposed to H2O2 and potassium deprivation [9]. An NCAM mimetic peptide, has shown neuroprotective properties in cultured cerebellar and dopaminergic neurons [102]. In addition, treatment of a MS mouse model with sICAM5, ameliorated the disease symptoms [89].
Caveats in studying the synaptic sheddome
There are several important caveats of studies addressing the synaptic sheddome. In experimental studies, the endogenous targets of ectodomains may also interact with the full-length, membrane-attached, non-cleaved proteins. Competition of exogenous ectodomains with endogenous binding of its target or its source protein may be a confound.
There are also caveats inherent to mass spectrometry analyses. Mass spectrometry could miss some proteins due to low peptide counts and results could be influenced by biochemical sample preparation. Proteomic analyses of CSF carry caveats as well, in particular in human subjects. When analyzing human CSF in brain disorders, incorrect diagnosis or comorbidity with other diseases could be an important confound. Other parameters that could influence human CSF studies are medication, age differences, alterations in blood-brain barrier permeability, or contamination with blood during CSF extraction. Replications, independent validation, and consistent methodology are thus essential to ensure reproducibility. Validation of results in larger independent cohorts of patients and longitudinal analyses of the CSF proteome during the different phases of the diseases will be also crucial to determine future reliable biomarkers. Another potential confound could be extracellular vesicles present in the CSF, however, their levels are typically low compared to shed proteins and can be eliminated by ultracentrifugation. Proteins could also be present in the CSF due to cell death or damage. They could also originate from other tissues apart from the brain, as the CSF is derived from the blood plasma.
Concluding remarks and future perspectives
Initially considered as a mechanism that terminates membrane protein function, ES is now considered as a key posttranslational mechanism regulating cell biology, which is supported by its role in the pathogenesis of disorders such as cancer and AD. Shed synaptic ectodomains are emerging as neuronal modulators that function in conjunction with classic neurotransmitters and neuromodulators. They have the potential of exerting autocrine and/or paracrine effects, as they can diffuse away from the cell of origin. Moreover, the effects of shed ectodomains are complex, as they depend on the developmental stage, cell/tissue of origin and the target cell. Despite significant progress over the last years, there are still many questions that need to be addressed, particularly in the context of synaptic ectodomain shedding. These include the comprehensive identification of shed synaptic proteins, their binding partners, their modulatory effects, and the molecular mechanisms regulating their ES (see Outstanding Questions). Moreover, exogenous ectodomains or smaller fragments derived from them could be further explored as therapeutic approaches for brain disorders.
Outstanding Questions Box.
Which synaptic proteins undergo ES? What is the full extent of the synaptic sheddome, and which soluble ectodomains have roles in paracrine and autocrine signaling? How do soluble ectodomains affect the function of individual neurons and neuronal circuits? How do they affect behavior?
How do physiological conditions, such as neuronal activity, neuromodulators, immune signals, alter synaptic ES globally? What individual proteins and pathways are preferentially affected?
What are the targets of shed synaptic ectodomains? How do ectodomains affect the function of these targets and off-target cells?
How does synaptic ectodomain shedding participate in pathogenic processes in the CNS? Can faulty ES give rise to brain disorders or contribute to disease progression? Do mutations in sheddase substrates alter their ES in a manner that could contribute to disease?
How is the synaptic sheddome reflected in the CSF? Can CSF shed ectodomains originating from central synapses be used to monitor biochemical processes in the brain? Can the analysis of the sheddome reflected in the CFS be exploited for biomarkers?
Can exogenous ectodomains or bioactive peptides derived from them be employed therapeutically in CNS disorders?
The evidence for altered synaptic ES in neuropsychiatric disorders is growing. This is perhaps not surprising given the central roles synapses play in brain pathogenesis, and the fact that many membrane proteins undergo ES. However, better understanding of the global function and regulation of synaptic ES in the context of CNS (patho)physiology is needed. In this regard, proteomic analyses of the synaptic sheddome in various conditions could reveal novel pathogenic pathways, biomarkers, and drug targets. As shed synaptic proteins can be detected in the CSF, this fluid has the potential of offering information on synapse (patho)physiology. Yet, the relationship of the synaptic sheddome and the CSF proteome remains to be systematically investigated.
Highlights.
Numerous synaptic proteins undergo ectodomain shedding, often regulated by neuronal activity, and many more await discovery.
Previously thought as a modality for protein inactivation, emerging evidence indicates that ectodomain shedding generates soluble ectodomains which act as paracrine signals to modulate neuronal development, function, plasticity, and circuit properties.
Faulty neuronal ectodomain shedding is associated with a growing number of neurological disorders.
Shed ectodomains originating from central synapses are detectable in the cerebrospinal fluid (CSF), and their monitoring could provide insights into brain processes and potentially serve as CSF biomarkers for brain disorders.
Large-scale proteomic analyses are poised to identify novel synaptic proteins undergoing ectodomain shedding, as well as new functions and targets for soluble ectodomains, raising the possibility that soluble ectodomains could be developed into novel therapeutics.
Acknowledgements
This work was funded by NIH grant NS100785 (to P.P). Illustrations were created and adapted from BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates
Glossary
- Ectodomain
the extracellular portion of a transmembrane domain or a GPI anchored protein. It could be part of the full-length protein or soluble, upon shedding
- Sheddase
protease that catalyzes the cleavage, and the shedding, of membrane protein ectodomains
- Secretome
all proteins found in the extracellular milieu of a given biological sample (including secreted and shed proteins)
- Sheddome
subset of proteins in the secretome that undergo ES and thus originally possess at least one transmembrane domain or a GPI anchor
Footnotes
Declaration of Interests The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lichtenthaler SF et al. (2018) Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J 37 (15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuhn PH et al. (2016) Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 5, e12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park S et al. (2013) GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 339 (6117), 324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hattori M et al. (2000) Regulated cleavage of a contact-mediated axon repellent. Science 289 (5483), 1360–5. [DOI] [PubMed] [Google Scholar]
- 5.Peixoto RT et al. (2012) Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 76 (2), 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki K et al. (2012) Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76 (2), 410–22. [DOI] [PubMed] [Google Scholar]
- 7.Vassar R et al. (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286 (5440), 735–41. [DOI] [PubMed] [Google Scholar]
- 8.Altmeppen HC et al. (2015) The sheddase ADAM10 is a potent modulator of prion disease. Elife 4, e04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wierda KDB et al. (2020) The soluble neurexin-1β ectodomain causes calcium influx and augments dendritic outgrowth and synaptic transmission. Scientific Reports 10 (1), 18041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong XJ et al. (2017) Retrograde Synaptic Inhibition Is Mediated by alpha-Neurexin Binding to the alpha2delta Subunits of N-Type Calcium Channels. Neuron 95 (2), 326–340 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatesh HS et al. (2015) Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 161 (4), 803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn PH et al. (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 31 (14), 3157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tushaus J et al. (2020) An optimized quantitative proteomics method establishes the cell type-resolved mouse brain secretome. EMBO J 39 (20), e105693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thouvenot E et al. (2012) Quantitative proteomic analysis reveals protein expression changes in the murine neuronal secretome during apoptosis. J Proteomics 77, 394–405. [DOI] [PubMed] [Google Scholar]
- 15.Kim W et al. (2021) Spatiotemporal processing of neural cell adhesion molecules 1 and 2 by BACE1 in vivo. J Biol Chem 296, 100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamezaki A et al. (2016) Visualization of Neuregulin 1 ectodomain shedding reveals its local processing in vitro and in vivo. Scientific Reports 6 (1), 28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesné S et al. (2005) NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci 25 (41), 9367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chmielewska JJ et al. (2019) Neuroligin 1, 2, and 3 Regulation at the Synapse: FMRP-Dependent Translation and Activity-Induced Proteolytic Cleavage. Mol Neurobiol 56 (4), 2741–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conant K et al. (2010) Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience 166 (2), 508–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martín-de-Saavedra MD et al. (2022) Shed CNTNAP2 ectodomain is detectable in CSF and regulates Ca2+ homeostasis and network synchrony via PMCA2/ATP2B2. Neuron 110 (4), 627–643.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho RW et al. (2008) mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron 57 (6), 858–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao XQ et al. (2015) p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Molecular Psychiatry 20 (11), 1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maretzky T et al. (2005) L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol Cell Biol 25 (20), 9040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalus I et al. (2006) Proteolytic cleavage of the neural cell adhesion molecule by ADAM17/TACE is involved in neurite outgrowth. J Neurochem 98 (1), 78–88. [DOI] [PubMed] [Google Scholar]
- 25.Munro KM et al. (2016) Functions of the Alzheimer’s Disease Protease BACE1 at the Synapse in the Central Nervous System. J Mol Neurosci 60 (3), 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monea S et al. (2006) Membrane localization of membrane type 5 matrix metalloproteinase by AMPA receptor binding protein and cleavage of cadherins. J Neurosci 26 (8), 2300–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanz RL et al. (2018) MT3-MMP Promotes Excitatory Synapse Formation by Promoting Nogo-66 Receptor Ectodomain Shedding. J Neurosci 38 (3), 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guner G and Lichtenthaler SF (2020) The substrate repertoire of gamma-secretase/presenilin. Semin Cell Dev Biol 105, 27–42. [DOI] [PubMed] [Google Scholar]
- 29.Horowitz AM et al. (2020) Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 369 (6500), 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ethell IM and Ethell DW (2007) Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res 85 (13), 2813–23. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JW et al. (1998) Regional and differential expression of gelatinases in rat brain after systemic kainic acid or bicuculline administration. Eur J Neurosci 10 (11), 3358–68. [DOI] [PubMed] [Google Scholar]
- 32.Hoehna Y et al. (2012) Matrix metalloproteinase 9 regulates cell death following pilocarpine-induced seizures in the developing brain. Neurobiol Dis 48 (3), 339–47. [DOI] [PubMed] [Google Scholar]
- 33.Dubey D et al. (2017) Increased metalloproteinase activity in the hippocampus following status epilepticus. Epilepsy Res 132, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y et al. (2008) Relationship between methamphetamine exposure and matrix metalloproteinase 9 expression. Neuroreport 19 (14), 1407–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown TE et al. (2007) Role of matrix metalloproteinases in the acquisition and reconsolidation of cocaine-induced conditioned place preference. Learn Mem 14 (3), 214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kucharczyk M et al. (2016) Chronic mild stress influences nerve growth factor through a matrix metalloproteinase-dependent mechanism. Psychoneuroendocrinology 66, 11–21. [DOI] [PubMed] [Google Scholar]
- 37.Planas AM et al. (2001) Expression and activation of matrix metalloproteinase-2 and −9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis 8 (5), 834–46. [DOI] [PubMed] [Google Scholar]
- 38.Gottschall PE and Yu X (1995) Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J Neurochem 64 (4), 1513–20. [DOI] [PubMed] [Google Scholar]
- 39.Corcoran ML et al. (1992) Interleukin 4 inhibition of prostaglandin E2 synthesis blocks interstitial collagenase and 92-kDa type IV collagenase/gelatinase production by human monocytes. J Biol Chem 267 (1), 515–9. [PubMed] [Google Scholar]
- 40.Tackenberg C and Nitsch RM (2019) The secreted APP ectodomain sAPPα, but not sAPPβ, protects neurons against Aβ oligomer-induced dendritic spine loss and increased tau phosphorylation. Mol Brain 12 (1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei W et al. (2010) Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 13 (2), 190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan XZ et al. (2012) Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J Neurosci 32 (11), 3910–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andrew RJ et al. (2016) A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J Biol Chem 291 (37), 19235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lichtenthaler SF et al. (2022) Secretases in Alzheimer’s disease: Novel insights into proteolysis of APP and TREM2. Current Opinion in Neurobiology 72, 101–110. [DOI] [PubMed] [Google Scholar]
- 45.Servián-Morilla E et al. (2018) Proteolytic Processing of Neurexins by Presenilins Sustains Synaptic Vesicle Release. J Neurosci 38 (4), 901–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borcel E et al. (2016) Shedding of neurexin 3beta ectodomain by ADAM10 releases a soluble fragment that affects the development of newborn neurons. Sci Rep 6, 39310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bot N et al. (2011) Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J Biol Chem 286 (4), 2762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bemben MA et al. (2019) Isoform-specific cleavage of neuroligin-3 reduces synapse strength. Mol Psychiatry 24 (1), 145–160. [DOI] [PubMed] [Google Scholar]
- 49.Brennaman LH et al. (2014) EphrinA/EphA-induced ectodomain shedding of neural cell adhesion molecule regulates growth cone repulsion through ADAM10 metalloprotease. J Neurochem 128 (2), 267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malinverno M et al. (2010) Synaptic localization and activity of ADAM10 regulate excitatory synapses through N-cadherin cleavage. J Neurosci 30 (48), 16343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tian L et al. (2007) Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J Cell Biol 178 (4), 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J et al. (2010) Activity-dependent alpha-cleavage of nectin-1 is mediated by a disintegrin and metalloprotease 10 (ADAM10). J Biol Chem 285 (30), 22919–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mechtersheimer S et al. (2001) Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol 155 (4), 661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maretzky T et al. (2005) L1 Is Sequentially Processed by Two Differently Activated Metalloproteases and Presenilin/γ-Secretase and Regulates Neural Cell Adhesion, Cell Migration, and Neurite Outgrowth. Molecular and Cellular Biology 25 (20), 9040–9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linneberg C et al. (2019) L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing. Scientific Reports 9 (1), 3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bijata M et al. (2017) Synaptic Remodeling Depends on Signaling between Serotonin Receptors and the Extracellular Matrix. Cell Rep 19 (9), 1767–1782. [DOI] [PubMed] [Google Scholar]
- 57.Barão S et al. (2015) Antagonistic Effects of BACE1 and APH1B-γ-Secretase Control Axonal Guidance by Regulating Growth Cone Collapse. Cell Rep 12 (9), 1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee H et al. (2014) Long-term depression-inducing stimuli promote cleavage of the synaptic adhesion molecule NGL-3 through NMDA receptors, matrix metalloproteinases and presenilin/gamma-secretase. Philos Trans R Soc Lond B Biol Sci 369 (1633), 20130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pigoni M et al. (2016) Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol Neurodegener 11 (1), 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hruska M and Dalva MB (2012) Ephrin regulation of synapse formation, function and plasticity. Mol Cell Neurosci 50 (1), 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhagat SM et al. (2016) Erasure of fear memories is prevented by Nogo Receptor 1 in adulthood. Mol Psychiatry 21 (9), 1281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fleck D et al. (2013) Dual cleavage of neuregulin 1 type III by BACE1 and ADAM17 liberates its EGF-like domain and allows paracrine signaling. J Neurosci 33 (18), 7856–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iwakura Y et al. (2017) Glutamate-dependent ectodomain shedding of neuregulin-1 type II precursors in rat forebrain neurons. PLoS One 12 (3), e0174780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lim ST et al. (2012) Ectodomain shedding of nectin-1 regulates the maintenance of dendritic spine density. J Neurochem 120 (5), 741–51. [DOI] [PubMed] [Google Scholar]
- 65.Gardoni F et al. (2012) The neuropeptide PACAP38 induces dendritic spine remodeling through ADAM10-N-cadherin signaling pathway. J Cell Sci 125 (Pt 6), 1401–6. [DOI] [PubMed] [Google Scholar]
- 66.Parks AL et al. (2000) Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development 127 (7), 1373–85. [DOI] [PubMed] [Google Scholar]
- 67.Gjorlund MD et al. (2017) Soluble Ectodomain of Neuroligin 1 Decreases Synaptic Activity by Activating Metabotropic Glutamate Receptor 2. Front Mol Neurosci 10, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pillai-Nair N et al. (2005) Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J Neurosci 25 (18), 4659–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lonskaya I et al. (2013) Soluble ICAM-5, a product of activity dependent proteolysis, increases mEPSC frequency and dendritic expression of GluA1. PLoS One 8 (7), e69136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toth AB et al. (2013) Synapse maturation by activity-dependent ectodomain shedding of SIRPα. Nat Neurosci 16 (10), 1417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Young-Pearse TL et al. (2008) Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Development 3 (1), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rice HC et al. (2019) Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363 (6423). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tien WS et al. (2017) SheddomeDB: the ectodomain shedding database for membrane-bound shed markers. BMC Bioinformatics 18 (Suppl 3), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dislich B et al. (2015) Label-free Quantitative Proteomics of Mouse Cerebrospinal Fluid Detects beta-Site APP Cleaving Enzyme (BACE1) Protease Substrates In Vivo. Mol Cell Proteomics 14 (10), 2550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brummer T et al. (2019) NrCAM is a marker for substrate-selective activation of ADAM10 in Alzheimer’s disease. EMBO Mol Med 11 (4), e9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conant K et al. (2017) Matrix metalloproteinase activity stimulates N-cadherin shedding and the soluble N-cadherin ectodomain promotes classical microglial activation. J Neuroinflammation 14 (1), 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poltorak M et al. (1995) Disturbances in cell recognition molecules (N-CAM and L1 antigen) in the CSF of patients with schizophrenia. Exp Neurol 131 (2), 266–72. [DOI] [PubMed] [Google Scholar]
- 78.Strekalova H et al. (2006) Elevated levels of neural recognition molecule L1 in the cerebrospinal fluid of patients with Alzheimer disease and other dementia syndromes. Neurobiol Aging 27 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- 79.Jiao SS et al. (2015) Differential levels of p75NTR ectodomain in CSF and blood in patients with Alzheimer’s disease: a novel diagnostic marker. Translational Psychiatry 5 (10), e650–e650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tao H et al. (2016) Promoter Variants of the ADAM10 Gene and Their Roles in Temporal Lobe Epilepsy. Frontiers in Neurology 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cudna A et al. (2017) Serum metalloproteinase 9 levels increase after generalized tonic-clonic seizures. Epilepsy Res 129, 33–36. [DOI] [PubMed] [Google Scholar]
- 82.Acar G et al. (2015) Increased Expression of Matrix Metalloproteinase-9 in Patients with Temporal Lobe Epilepsy. Turk Neurosurg 25 (5), 749–56. [DOI] [PubMed] [Google Scholar]
- 83.Murase S et al. (2016) Matrix Metalloproteinase-9 Regulates Neuronal Circuit Development and Excitability. Molecular Neurobiology 53 (5), 3477–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prox J et al. (2013) Postnatal Disruption of the Disintegrin/Metalloproteinase ADAM10 in Brain Causes Epileptic Seizures, Learning Deficits, Altered Spine Morphology, and Defective Synaptic Functions. The Journal of Neuroscience 33 (32), 12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu X et al. (2018) Metalloprotease Adam10 suppresses epilepsy through repression of hippocampal neuroinflammation. Journal of Neuroinflammation 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Axelsson M et al. (2019) Cerebrospinal fluid NCAM levels are modulated by disease-modifying therapies. Acta Neurol Scand 139 (5), 422–427. [DOI] [PubMed] [Google Scholar]
- 87.Lindsberg PJ et al. (2002) Release of soluble ICAM-5, a neuronal adhesion molecule, in acute encephalitis. Neurology 58 (3), 446–51. [DOI] [PubMed] [Google Scholar]
- 88.Rieckmann P et al. (1998) Telencephalin as an indicator for temporal-lobe dysfunction. Lancet 352 (9125), 370–1. [DOI] [PubMed] [Google Scholar]
- 89.Birkner K et al. (2019) Neuronal ICAM-5 Plays a Neuroprotective Role in Progressive Neurodegeneration. Front Neurol 10, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wachowiak R et al. (2018) Increased L1CAM (CD171) levels are associated with glioblastoma and metastatic brain tumors. Medicine (Baltimore) 97 (38), e12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Colom-Cadena M et al. (2020) The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer’s Research & Therapy 12 (1), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Higginbotham L et al. (2020) Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv 6 (43). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Collins MA et al. (2015) Label-Free LC-MS/MS Proteomic Analysis of Cerebrospinal Fluid Identifies Protein/Pathway Alterations and Candidate Biomarkers for Amyotrophic Lateral Sclerosis. J Proteome Res 14 (11), 4486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang C et al. (2017) Proteome Analysis of Potential Synaptic Vesicle Cycle Biomarkers in the Cerebrospinal Fluid of Patients with Sporadic Creutzfeldt-Jakob Disease. Mol Neurobiol 54 (7), 5177–5191. [DOI] [PubMed] [Google Scholar]
- 95.Jahn H et al. (2011) Peptide fingerprinting of Alzheimer’s disease in cerebrospinal fluid: identification and prospective evaluation of new synaptic biomarkers. PLoS One 6 (10), e26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lleo A et al. (2019) Changes in Synaptic Proteins Precede Neurodegeneration Markers in Preclinical Alzheimer’s Disease Cerebrospinal Fluid. Mol Cell Proteomics 18 (3), 546–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bereczki E et al. (2017) Synaptic proteins in CSF relate to Parkinson’s disease stage markers. NPJ Parkinsons Dis 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khoonsari PE et al. (2016) Analysis of the Cerebrospinal Fluid Proteome in Alzheimer’s Disease. PLoS One 11 (3), e0150672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Al Shweiki MHDR et al. (2020) Proteomic analysis reveals a biosignature of decreased synaptic protein in cerebrospinal fluid of major depressive disorder. Translational Psychiatry 10 (1), 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Göteson A et al. (2021) Cerebrospinal fluid proteomics targeted for central nervous system processes in bipolar disorder. Mol Psychiatry, doi.org/10.1038/s41380-021-01236-5. [DOI] [PubMed] [Google Scholar]
- 101.Yao XQ et al. (2015) p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Mol Psychiatry 20 (11), 1301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ditlevsen DK et al. (2003) The role of phosphatidylinositol 3-kinase in neural cell adhesion molecule-mediated neuronal differentiation and survival. J Neurochem 84 (3), 546–56. [DOI] [PubMed] [Google Scholar]
- 103.Ferraro GB et al. (2011) Membrane-type matrix metalloproteinase-3 regulates neuronal responsiveness to myelin through Nogo-66 receptor 1 cleavage. J Biol Chem 286 (36), 31418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nagappan-Chettiar S et al. (2018) Tyrosine phosphorylation of the transmembrane protein SIRPalpha: Sensing synaptic activity and regulating ectodomain cleavage for synapse maturation. J Biol Chem 293 (31), 12026–12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iwagishi R et al. (2020) Negatively charged amino acids in the stalk region of membrane proteins reduce ectodomain shedding. J Biol Chem 295 (35), 12343–12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goth CK et al. (2015) A systematic study of modulation of ADAM-mediated ectodomain shedding by site-specific O-glycosylation. Proc Natl Acad Sci U S A 112 (47), 14623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Akasaka-Manya K et al. (2017) Excess APP O-glycosylation by GalNAc-T6 decreases Aβ production. J Biochem 161 (1), 99–111. [DOI] [PubMed] [Google Scholar]
- 108.Shirakabe K et al. (2017) Mechanistic insights into ectodomain shedding: susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci Rep 7, 46174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Christensen SK et al. (2020) Endosomal trafficking is required for glycosylation and normal maturation of the Alzheimer’s-associated protein sorLA. bioRxiv, doi.org/10.1101/2020.07.12.199885. [Google Scholar]
- 110.Tushaus J et al. (2021) The pseudoprotease iRhom1 controls ectodomain shedding of membrane proteins in the nervous system. FASEB J 35 (11), e21962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van Erp S et al. (2015) Lrig2 Negatively Regulates Ectodomain Shedding of Axon Guidance Receptors by ADAM Proteases. Dev Cell 35 (5), 537–552. [DOI] [PubMed] [Google Scholar]
- 112.Tambini MD et al. (2020) Opposite changes in APP processing and human Aβ levels in rats carrying either a protective or a pathogenic APP mutation. Elife 9, e52612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Strauss KA et al. (2006) Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med 354 (13), 1370–7. [DOI] [PubMed] [Google Scholar]