

Abstract
Objective
To facilitate patient disease subset and risk factor identification by constructing a pipeline which is generalizable, provides easily interpretable results, and allows replication by overcoming electronic health records (EHRs) batch effects.
Material and Methods
We used 1872 billing codes in EHRs of 102 880 patients from 12 healthcare systems. Using tools borrowed from single-cell omics, we mitigated center-specific batch effects and performed clustering to identify patients with highly similar medical history patterns across the various centers. Our visualization method (PheSpec) depicts the phenotypic profile of clusters, applies a novel filtering of noninformative codes (Ranked Scope Pervasion), and indicates the most distinguishing features.
Results
We observed 114 clinically meaningful profiles, for example, linking prostate hyperplasia with cancer and diabetes with cardiovascular problems and grouping pediatric developmental disorders. Our framework identified disease subsets, exemplified by 6 “other headache” clusters, where phenotypic profiles suggested different underlying mechanisms: migraine, convulsion, injury, eye problems, joint pain, and pituitary gland disorders. Phenotypic patterns replicated well, with high correlations of ≥0.75 to an average of 6 (2–8) of the 12 different cohorts, demonstrating the consistency with which our method discovers disease history profiles.
Discussion
Costly clinical research ventures should be based on solid hypotheses. We repurpose methods from single-cell omics to build these hypotheses from observational EHR data, distilling useful information from complex data.
Conclusion
We establish a generalizable pipeline for the identification and replication of clinically meaningful (sub)phenotypes from widely available high-dimensional billing codes. This approach overcomes datatype problems and produces comprehensive visualizations of validation-ready phenotypes.
Keywords: electronic health records, clustering, electronic medical records, ICD, PhenoGraph, eMERGE
BACKGROUND AND SIGNIFICANCE
In many diseases, patients exhibit a wide variety of symptoms, treatment responses, and disease outcomes. A tantalizing idea is that this clinical heterogeneity might phenotypically cluster in meaningful ways. Knowledge of these latent disease populations could enable enhanced classification of patients into clinically relevant groups, thus facilitating research into their specific etiology, risk factors, prognosis, and treatment. This in turn could uncover opportunities for the repurposing of medication, tailored comorbidity monitoring, or enhanced population screening.
The increasing availability of electronic health records (EHR)1,2 creates a unique opportunity to discover these disease clusters, along with their defining risk factors. This can be done in a data-driven manner, therefore not requiring prior knowledge of the existence of these subgroups or their characteristics. Databases of EHRs consist of longitudinal, observational data on a large number of patients. EHR data encompassing entire hospital systems can comprise millions of individuals each with thousands of clinical features.
Progress in many subdomains of the life sciences is propelled by the combination of an ever-increasing data availability, closely followed by the development of novel algorithms to analyze and interpret these data. A prime example is the subfield of single-cell analyses that aims to understand cellular heterogeneity, by analyzing genomics data (genetic, gene expression, etc) acquired at a single-cell level. Algorithms for graph construction, for example, K-Nearest Neighbors (kNN),3 in conjunction with clustering algorithms, for example, Louvain community detection,4,5 have been instrumental for exploring cell-to-cell heterogeneity.4–6 Such analytic methods could be equally useful in the EHR field, in which we are similarly interested in exploring the heterogeneity between samples with large numbers of often strongly correlated variables.
It has previously been shown that data from EHR systems can be used to support clinical research across the field. However, these past ventures have largely focused on integrating as many EHR components as feasible in order to answer questions pertaining to 1 specific disease.7–13 These algorithms rely on data that are not universally available, which hampers replication. More in general, replication of clustering across EHR systems is in our view underrepresented in available methods. Replication is challenging by the complexity and data gluttony of the general frameworks, but in the end crucial for unsupervised methodologies. Finally, clear visualization remains a challenge in the field of clustering. Across the various approaches, we observe a high prevalence of main findings reported in tabular format and the use of network graphs. The latter is in our view difficult to interpret, because important numerical values are encoded in visual components such as edges. Interactive visualization is sometimes used as solution, but they suffer from great loss of information when presented statically instead of interactively.9,11,14–16 We aspired to develop a pipeline for the identification of subgroups within diseases which is replicable across different data sources and different diseases. In order to intuitively show the value and applicability of such a pipeline, clear and modular visualization is essential.
Objective
The aim of this study was to provide a generalizable framework for employing longitudinally collected EHRs to classify heterogeneous patient populations on disease diagnostic histories requiring only billing codes. Through this framework, we aim to identify phenotypic clusters and their risk factors, in order to facilitate research and treatment tailored to more homogenous disease populations. By developing a concise and attractive visualization method (PheSpec) along with a visual prevalence-based filtering, ranked scope pervasion (RSP), we aspire to a comprehensive comparison of the >1800 phenotypic codes comprising the diagnostic profiles. Using 2 illustrative examples of prostate cancer and headache complaints, we strive to show both the clinical relevance as well as the cross-cohort replicability of our framework.
MATERIALS AND METHODS
Patient data
We used data from 12 healthcare institutions included in the eMERGE consortium of biobanks linked with EHR data. All data are available on request via the eMERGE network. The data were divided into 12 separate cohorts containing the de-identified disease diagnosis histories of 102 880 patients gathered between June 1987 and June 2017 in the United States, shared in a secure data repository at Vanderbilt University Medical Center. Events were coded using International Classification of Disease (ICD) codes, which we translate to a higher hierarchical grouping, namely Phenotype Codes (PheCodes).17–19 PheCodes combine similar individual ICD codes into higher level phenotypic categories using a structured ontology developed for use in Phenotype-Wide Association Studies (PheWAS). These codes contain the full codified medical history of a patient interacting with a connected network of care providers. Aggregation of ICD into PheCodes is important, as ICD codes were recorded using both the 9th and the 10th revision (thus resulting in codes with overlapping indications) as well as the fact that the 47 574 ICD codes represented a granularity, or specificity, which conveys limited additional information for our purposes and leads to a highly sparse dataset. PheCodes were both more distinct and more informative, making them more suited to our purposes.
We construct a code frequency matrix of the 1872 PheCodes by counting the number of times they appear during an individual’s follow-up time, resulting in a sparse (median 0.98 (0.75–1) proportion unobserved codes per individual) quantitative matrix of 102 880 by 1872.
The data collection was approved by the local institutional review boards. We obtained approval from the eMERGE consortium to use the data for the current study.
Mitigating center-specific batch effects
In order to prevent cohort-specific differences such as coding practice, referral preference, or diagnostic procedure from driving the downstream analysis more than the clinical differences we are interested in, we employ a powerful batch correction method called Harmony.20 This tool models the contribution of unwanted sources of variation, such as clinical center, to the data and uses mixture regression modeling to remove this confounding variation during subsequent clustering. Instead of modeling the 1872 codes directly, Harmony projects the records into a linear low-dimensional space with principal components analysis (PCA). We employed the first 500 principal components, explaining 0.49 of the variance in the data, and ran Harmony with default parameters, as described in Korsunsky et al.20
To visualize our data, we utilize a nonlinear technique called t-distributed Stochastic Neighbor Embedding (t-SNE).21 t-SNE is capable of projecting the local structure of the high-dimensional data onto a 2-dimensional plane without an assumption of linearity. It is often employed in the omics field, for example, to visualize similarities between thousands of samples on the basis of a large number of cell markers.22 The effect of Harmony’s harmonization is depicted by the Local Inverse Simpson’s Index (LISI) scores across t-SNE space.20 The LISI is a measure of local diversity regarding a specific marker, here center name, for example, how many centers are represented across the 2D visualization. Successful removal of batch effect should increase LISI, reflecting that clinically similar records from different centers are joined together in the Harmonized space.
Clustering on diagnosis events
After harmonization, we want to partition patients based on clinical similarity. Based on the knowledge that the pan-hospital data should contain a large number of distinct phenotypes but without a clear a priori idea of an exact number, we decided to use the PhenoGraph algorithm with default parameters (k = 30), which uses k-Nearest Neighbors (kNN) for graph construction and Louvain community detection for optimization of the cluster definition (modularity optimization).23 For each patient, the algorithm finds and connects the k nearest neighbors in the high-dimensional space (>1800), thus constructing a graph of all patients based on their expression of the full span of PheCodes. By subsequently identifying regions of the graph which are strongly connected, communities, or clusters, of phenotypically similar patients can be extracted.
Clustered data are once more visualized using t-SNE. No optimization of the cluster parameters or clustering was performed based on the 2D representation of the data, as we expected to find the most relevant structure in the higher dimension and perfect clustering on this plane cannot be assumed to be reflected on the reduced dimensionality embedding or vice versa.
Identification of residual structure
Obtained patient clusters may still show interesting heterogeneity after the initial clustering, which one might wish to explore further. For this purpose, we included an additional, optional step in our pipeline which takes all members of a previously identified cluster and performs another cycle of the graph clustering using just these patients.
Calculation of top 10 distinguishing features
In order to enhance the explainability of the clustering algorithm, top 10 codes most predictive for cluster membership are determined per cluster through 5 times repeated 5-fold cross validated elastic net regularization. Counts of all 1873 codes are used as possible predictors and the binary status of being in a particular cluster as the outcome for each individual. We take the 10 codes with the absolute largest estimated coefficients and represent their importance through the within-cluster prevalence, as coefficients from a model derived using a regularization model are prone to misinterpretation when taken out of the context of the full model. The decision to visualize 10 codes is based on a need for clear visualization but is otherwise arbitrary and does not impact the clustering itself, it merely provides a comprehensive explanation of the clustering “choices” made by PhenoGraph.
Visualizing phenotypic patterns and defining features: PheSpecs
To comprehensively visualize the phenotypic patterns that characterize the clusters, we developed PheSpec’s (Phenotype Spectrograph). These graphs depict the proportion of patients in the clusters labeled with each PheCode. To ensure readability, only the most prevalent top 500 codes of each cluster are included. Peaks in the main graph are colored based on ICD chapter, see Supplementary Material 1.
The PheSpecs of the separate datasets also indicate the Pearson correlation of the dataset-specific peak pattern with the peak pattern of all datasets combined. This was done to investigate to what extent the overall phenotype of identified patient clusters generalizes between individual participating centers. We similarly calculate the Pearson correlation among the individual centers in order to assess similarity in their specific patient populations. Both correlation measures represent the level of correlation between the proportion of patients expressing each PheCode in the compared sets; a high correlation coefficient indicates that patients in the 2 groups have a highly similar pattern of previous events in their medical histories and can therefore be considered as representing the same clinical phenotype. The comparison of individual sites to the combined PheSpec therefore shows how individual sites have contributed to the complete cluster and correlations among clinical sites shows how consistent this phenotype is observed across our datasets.
The full phenotypic figure consists of 4 distinct graphs: the PheSpec of the entire cluster, the PheSpecs of the independent datasets, a heatmap showing correlation of the phenotypic patterns between datasets, and a cluster position graph. It also includes a complementary table summarizing the cluster’s top 10 codes most predictive of cluster membership and their within-cluster prevalences. A white background color indicates a positive regularization coefficient, gray a negative one. Combined, these graphics show the general phenotype (peak pattern), degree of replication of this phenotype across hospitals (miniatures and heatmap), a measure of cluster homogeneity (2D visualization), as well as the distinguishing features of the phenotype when compared to other patients in our data (table).
Ranked scope pervasion
While our hypothesis-free approach means we avoid filtering of features prior to the clustering, our initial visualization showed an overrepresentation of certain codes across clusters, which obscured relevant differences between clusters. We therefore developed a code score which can be used to filter at the visualization level without affecting the actual clustering, by removing codes that are abundant but not discriminatory. Ranked Scope Pervasion (RSP) scores are fully dependent on the scope which one intends to visualize (eg, top 10 most prevalent PheCodes in a cluster) and are calculated for those codes which occur at least once within this prespecified scope across all clusters. The scores and upper limit used for filtering are derived using the following set of functions:
where is the number of clusters in the dataset, the size of the scope of interest, an arbitrary cluster index, and the position of the specific PheCode in the ranked scope of cluster . These calculations translate to scores which increase the more often a code is observed to rank highly within a cluster and which are thus inversely proportional to the cluster specificity of the code. The RSP score maxes out for a hypothetical code which ranks first across all clusters at a value of and reaches its minimum value of when a code is observed merely once at the lowest scope rank. The calculation of the upper bound we use to determine which codes ought to be filtered out of our visualizations has been constructed such that any code more pervasive than a hypothetical code which takes the lowest within-scope position for all clusters is excluded. This results in the removal of 9 codes from the prevalence-based PheSpec graphs. Importantly, RSP filtering was applied to the graphs and thus not performed on the table component of the PheSpec compositions.
Identification of relevant clusters
In order to facilitate inspection, we wanted to identify clusters characterized by an overexpression of a PheCode of interest, in other words where the PheCode was both part of the top 10 most prevalent codes and more prevalent than in the total dataset. We use “other headache syndromes” as an example.
Clinical expertise validation
We want to assess the level of clinical coherence of our identified clusters, therefore 3 unaffiliated clinical experts reviewed our clustering. They were asked to reduce the top 10’s of all clusters to a “logical” clinical phenotype in a parsimonious manner, requiring as little alteration to the top 10 as possible, by indicating which codes would need to be removed for the phenotype to be unambiguous. To enable us to draw any conclusions from these results, we also included 10 randomly generated top 10 PheCodes, obtained through random sampling without replacement of the full list of possible PheCodes. A coherence measure was derived from the clinician verdicts by taking the proportion of PheCodes left after reduction and averaging this value over all clinicians.
Programs
All analyses and visualizations were performed using “R” version 3.6.3.24 A list of specific packages can be found in the Supplementary Material 2. All scripts are publicly available at https://github.com/MarcMaurits/EHRClustering
RESULTS
Our developed pipeline aims to identify phenotypically similar groups of patients from highly heterogeneous EHR data. Our approach combines several existing algorithms (Harmony, PhenoGraph) with bespoke analysis tools (PheSpecs, RSP) (Figure 1).
Figure 1.
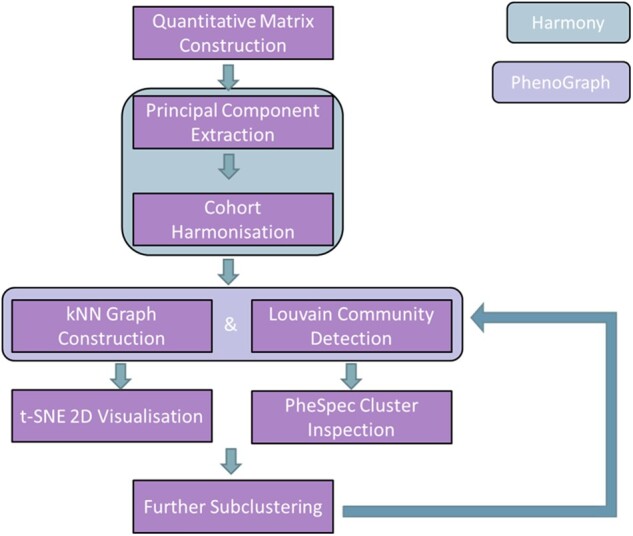
Full pipeline flowchart. Overview of the full pipeline described in this manuscript. As indicated by the legend, steps in the green field are part of Harmony and those in the purple field are part of PhenoGraph.
We applied this pipeline to the EHR data of 12 U.S. healthcare centers of the eMERGE network (demographic and data structure characteristics in Supplementary Table S1).
We demonstrate that with our analytical pipeline can:
Overcome batch effects while maintaining principal differences between datasets;
Obtain clinically meaningful clusters; and
Identify phenotypically divergent subsets of diseases.
Harmonization increases cohort mixing without loss of relevant structure
Due to center-specific conventions concerning EHR usage and treatment decisions, we expect any patient clustering performed on the pooled data of all 12 cohorts to be at least partially confounded by between-study variation. In order to account for such batch effects, we applied Harmony, which stimulates mixing. This mitigated existing batch effects, as illustrated by a mixed pattern of cluster contributions by cohorts (Supplementary Figure S1). The median (range) number of cohorts contributing at least 10% of a single cluster is observed to be 3 (1–5). Conversely, the median (range) number of clusters each cohort contributes at least 10% to is 12.5 (2–96). After applying Harmony, we observe an overall increase in the LISI score, reflecting that patients form different cohorts now have more similar characteristics (Figure 2).
Figure 2.

The effects of dataset harmonization. Overview showing (A) the t-SNE embedding of all 102,880 individuals colored for LISI score prior to harmonization with Harmony and (B) the same post-harmonization, as well as (C) an example showing that relevant structure is maintained by (D) not forcing dataset mixing where local structure is best represented by a small selection of datasets (developmental disorders in children’s hospitals, green arrows in A and B).
Relevant structure should be maintained throughout batch correction with Harmony, as some disease populations will invariably be tied to specific datasets, such as children’s hospitals. Harmony is capable of this, as illustrated by the developmental disorders cluster with a median age of 5.15 years old (Figure 2); this cluster is very much dominated by the 2 children’s hospitals in eMERGE, which are correctly kept together and separate from the remaining datasets.
Obtaining clinically meaningful phenotypes
We were presented with 114 clusters with a median (range) number of patients of 657 (88–4817). Reduction of disease diagnosis data from 1872 to 2 dimensions using t-SNE showed distinct clusters of patients (Supplementary Figure S2). With scope of 10, chosen for brevity and clarity, we derive an upper RSP bound of 11.4, above which 9 PheCodes (eg, 1010 “Other tests,” 401.1 “Essential hypertension”) are excluded from visualization (Supplementary Table S2).
The identified clusters represented meaningful clinical phenotypes; of our 114 clusters, 94 (82%) were at least 50% coherent (codes conforming to a conceivable phenotype) according to the averaged clinician opinions. Only 20% of the fake clusters reached this same threshold (Supplementary Figure S3). A further example is the cluster with diabetes and several cardiovascular complications such as coronary atherosclerosis and peripheral vascular disease (Supplementary Figure S4). This shows how clusters highlight the relation between risk factors (eg, diabetes) and adverse health outcomes (eg, cardiovascular problems), without assumptions of causality. We will discuss a few selected examples below (full results in Supplementary File 1).
We further illustrate the results of our framework by discussing a cluster which is characterized by prostate cancer. This cluster is unsurprisingly a male-dominated patient cluster (Figure 3) consisting of 1888 patients (92% male). As expected, female patients with highly similar phenotypes form only a small portion of the cluster. The PheSpec gives us an insight into the phenotype by highlighting the top 3 most prevalent codes: “erectile dysfunction,” “hyperplasia of prostate,” and “cancer of prostate.” This is complemented with a table of the most distinguishing PheCodes; the codes most predictive of cluster membership are consistent with a prostate cancer patient population. A similar phenotypic pattern is seen across 8/12 datasets (correlations range from 0.82 to 0.97), meaning highly similar patient populations are present across various centers.
Figure 3.

Prostate cancer cluster PheSpec composition. PheSpec composition showing the phenotypic profile (= profile of medical events) of one of the identified clusters as captured by the methodology described in this paper. The main PheSpec graph (A) is a representation of the harmonized dataset where the frequency (y-axis) reflects the proportion of total cluster members with each RSP filtered top 500 Phenotypic code (PheCode) (x-axis), the top 3 most prevalent codes are labeled. In the main PheSpec, all PheCodes are grouped and colored by ICD chapter (Supplementary Material 1). Table (B) shows the prevalence of the 10 most predictive codes of cluster membership (selected by elastic net), white background indicates a positive predictor, gray a negative one. The miniatures (C) show the replication of the phenotype cluster of prostate cancer across the separate cohorts, by splitting the cluster into its individual centers. Correlations of the cluster’s phenotypic profile between each centers are shown as a heatmap (D). Localization of the cluster in t-SNE space is shown in purple (E).
Upon a t-SNE-based visual inspection, we noted interesting residual heterogeneity. We therefore reran our pipeline on just the patients included in the original prostate cancer cluster. The newly identified clusters mapped well to the visual heterogeneity detected with the t-SNE. These subsets are indicative of prostate cancer in differing stages of the diagnostic process, ranging from very early problems such as urinary retention to the actual diagnosis of prostate cancer (Supplementary Figure S5).
The elastic net prediction models for all clusters can be found in the Supplementary File 2. We note that these models are built to discriminate between the specific clusters in this particular dataset and should not be interpreted as accurate models for cluster prediction in a different setting.
Identifying phenotypically divergent subsets of diseases
When analyzing the patient clusters, one sees that different clusters could be enriched for the same disease code yet would differ in other co-occurring disease codes. In effect, such patient clusters can be viewed as phenotypically different subgroups of the same disease. To systematically identify such relations between patient clusters, we employed prevalence-rank plots. In the total set, we observed 67 disease terms enriched in more than 1 patient cluster (Supplementary Figure S6).
We further elaborate on 1 such disease term “other headache syndromes,” as it is an intuitive example of a clinical manifestation with a very heterogeneous etiology. We distinguished 6 groups of patients presenting with headache symptoms (Figure 4). Their PheSpecs showed clearly distinguishable clinical features in each group (Figure 5); clusters were characterized by migraine, swelling of the eye, joint pain, convulsions, pituitary gland disorders, or injury (Supplementary Figure S7). Each of these subpopulations replicated well across multiple datasets, with on average 6 (2–8) individual cohort populations correlating strongly (≥0.75) to the full cluster. Each cluster consisted of at least 2 datasets contributing at least 10% of the total number of cluster members.
Figure 4.

Prevalence–rank plot “other headache syndromes.” Clusters of interest for PheCode “Other headache syndromes.” The Prevalence-rank plot depicts the proportion of patients in the cluster with the code of interest on the y-axis and the prevalence rank of the code within the cluster on the x-axis. The prevalence of PheCode 339 (“Other headache syndromes”) in the entire set was 0.27 (dotted line). We labeled the clusters (arbitrary cluster identifier) where the prevalence of this code was higher than its overall prevalence and where the code was present in the clusters' top 10 most prevalent codes.
Figure 5.

Overview of 6 headache subgroups. N is the number of patients located in the cluster. Location of clusters characterized by “other headache syndromes” in t-SNE space (A) and their corresponding phenotypic profiles (B). The frequency (y-axis) reflects the proportion of total cluster members with each RSP filtered top 500 code (x-axis). The graphs summarize the data from all cohorts together. For complete PheSpec compositions, see Supplementary Figure S5.
DISCUSSION
With this proof-of-principle study we have described our methodology to expose hidden subpopulations in EHR data and to identify additional phenotypes in these populations. We used established single-cell approaches and applied them to the processing and subsequent analysis of complex medical record data. We ascertain that our methods are capable of extracting subpopulations more intricate than 1-on-1 disease associations, without any prior knowledge of the potential existence of such latent structure. As our approach overcomes heterogeneity between medical centers, it also allows replication of these results. Our RSP filtering offers a way to reduce noise by removing noninformative features, which combined with our PheSpecs facilitates visual data exploration for the identification of cluster defining factors. To show the efficacy of our framework, we applied it to 12 different cohorts, which we harmonized into 1 dataset while maintaining the structure inherent in the EHR. Graph-based clustering identifies higher dimensional neighborhoods and is able to extract patient clusters which consist of individuals with high expression similarity of over 1800 PheCodes. From the similar expression patterns observed across multiple contributing cohorts within clusters, we conclude that the stratifications shown here are not the product of random clustering. Moreover, by inspecting several headache associated populations, we highlight the fact that this methodology is capable of differentiating people with similar phenotypic characteristics into clinically coherent and relevant subgroups. The potential to distinguish as-of-yet unknown patient populations in complex diseases seems clear. All 6 clusters are composed of patients presenting with the same symptom (headache); however, we are looking at very different subgroups with regard to medical history. This would, in a hypothesis generating study, provide interesting factors to investigate for an etiological role.
Our pipeline offers a solution to the problem of multiple latent (sub)phenotype, which exists in many complex diseases. Conventional methodologies are ill-suited to this task, as they rely on prior assumptions of linearity (principal component analysis), specific risk factors (controlled cohort study), or both (linear regression analysis). More sophisticated tools tend to combine data sources and require additional individual-level data, such as sex and age.9,11,13,15,25 Our single-cell inspired approach uses only deidentified medical history data to simultaneously identify the subsets as well as important risk events, overcoming the unknown subpopulation paradox; not being able to identify risk factors when the diseases are heterogeneous, while simultaneously not being able to identify disease subsets when independent risk factors are unknown. Previous studies have focused on genetics when aiming for further classification of patients,26 have developed their methodology for the investigation of 1 particular disease,27 or used a very limited number of features.28 Some work has also been performed in the direction of disease prediction from EHR data and investigators have tried a more generalist approach, aiming to improve clustering by incorporating various other variables (eg, lab values, demographics). Finally, investigators have created algorithms to phenotype individual patients based on their EHR. However, our approach provides a more comprehensive method for gaining insight into the underlying structure of illness by clustering patients on purely their phenome-wide disease history. The fact that these phenotype clusters are shown to transcend cohorts, through multicenter replication with Harmony and clinical validation by several experts, is further proof of the relevance of this approach.
Although our approach overcomes several of the current limitations in the field of EHR clustering, several potential limitations exist. First, by harmonizing the separate cohorts we potentially reduce informative patient differences. This does not appear to pose a large problem, as we do not observe forced introduction of members from each cohort in clusters where certain centers are expected to dominate (eg, children development in children’s hospitals). A further complication of the Harmonization is the need to collate all data in 1 digital location, something which could hamper adoption of our pipeline when data cannot be shared. Recently an adaptation to Harmony, called Symphony, was released which is capable of harmonizing data based on a comprehensive atlas, thus only requiring the sharing of a representative sample of data to construct said atlas, after which downstream analyses could be contained to individual sites.29 Second, we utilize elastic net regularization, while we aim to eliminate any assumption of linearity throughout the pipeline, as we are convinced that the disease history associations of interest go beyond linear 1-on-1 connections. We nonetheless chose to rely on EN for the identification of the top 10 most discerning PheCodes of a cluster, as it is but one aspect of the downstream analysis of identified clusters. Linearly identified discerning codes are informative and therefore serve an important supporting explainability role in the visualization. A further subjective decision is the use of RSP filtering of the visualizations. One might argue that highly pervasive codes are still of interest because they are common complaints; however, our approach aims to facilitate the superficial screening of truly distinct phenotypes in order to generate hypotheses. Furthermore, the development of RSP filtering was spurred by screening of preliminary results and is thus a tailored solution which could lead to reduced generalizability. However, by using a subjective, data-driven measure, we believe this issue is greatly mitigated. The pipeline proposed here could guide further studies, in which study-specific factors will likely lead to different parameter selections. The flexibility of the individual components (unsupervised clustering, variable visualization scopes, etc) results in a highly adaptable framework in which parameters such as the k value in PhenoGraph can be tweaked according to prior knowledge. We opt for an “out-of-the-box” approach to avoid overfitting to our specific datasets, for while optimizing might lead to improved results in our specific case, it would surely reduce the generalizability of the pipeline. We have to note that co-occurrence and longitudinal associations between PheCodes could be the result of clinician behavior. We therefore emphasize that our approach is hypothesis-generating and that validation of such hypotheses beyond our 12-cohort replication is important. In applied studies, integration of the extended EHR (eg, lab values, medications, demographics) could further aid in the clarification and validation of any results.
The next step of our research will be to apply the methodology to complex diseases known to be highly heterogeneous, such as rheumatoid arthritis, in order to uncover novel patient subgroups.
CONCLUSION
Our EHR clustering pipeline can identify latent subgroups of patients with the same phenotype but different etiologies, comorbidities, and prognoses together with their corresponding diagnostic events. Lacking stringent dependency upon prior knowledge and researcher assumptions this approach can lead to novel hypotheses in incompletely understood diseases. We overcame replicability and generalizability limitations of conventional cluster methods in EHR data by using widely available high-dimensional billing code data. Through several bespoke visual and computational tools we demonstrated that our method is robust and provides clinically informative results.
FUNDING
This work was supported by ReumaNederland and NIH grants U01 HG008685 and P30 AR070253.
Supplementary Material
AUTHOR CONTRIBUTIONS
MPM designed the study, ran analyses and wrote the paper; EA and RK designed the study supervised the analyses and cowrote the paper; TH and MR provided input on research design; IK and SR provided analytic solutions; JWS, SNM, SW, and EWK collected data, all authors read the manuscript and provided input on content, methodology, and framing.
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
ACKNOWLEGDMENTS
We would like to acknowledge the contribution of the following clinicians in reviewing the phenotypic coherence of the clusters: Willemien Visser, MD, PhD, rheumatologist; Joy vd Pol, MD, general medicine; and Nienke Grotenhuis, MD, PhD, otolaryngologist.
CONFLICTS OF INTERESTS STATEMENT
None declared.
DATA AVAILABILITY STATEMENT
All data used in this study are available on request via the eMerge consortium (https://emerge-network.org/ & https://phekb.org/). The framework developed here, as well as full results are publicly available on GitHub via https://github.com/MarcMaurits/EHRClustering.
REFERENCES
- 1. Mosley JD, Feng Q, Wells QS, et al. A study paradigm integrating prospective epidemiologic cohorts and electronic health records to identify disease biomarkers. Nat Commun 2018; 9 (1): 3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cowie MR, Blomster JI, Curtis LH, et al. Electronic health records to facilitate clinical research. Clin Res Cardiol 2017; 106 (1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cover T, Hart P.. Nearest neighbor pattern classification. IEEE Trans Inform Theory 1967; 13 (1): 21–7. [Google Scholar]
- 4. Andrews TS, Hemberg M.. Identifying cell populations with scRNASeq. Mol Aspects Med 2018; 59: 114–22. [DOI] [PubMed] [Google Scholar]
- 5. Blondel V, Guillaume J-L, Lambiotte R, Lefebvre E.. Fast unfolding of communities in large networks. J Stat Mech 2008; 2008 (10): P10008. [Google Scholar]
- 6. Luecken MD, Theis FJ.. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 2019; 15 (6): e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, Farooq S, Edwards J, et al. Patterns of symptoms before a diagnosis of first episode psychosis: a latent class analysis of UK primary care electronic health records. BMC Med 2019; 17 (1): 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ibrahim ZM, Wu H, Hamoud A, Stappen L, Dobson RJB, Agarossi A.. On classifying sepsis heterogeneity in the ICU: insight using machine learning. J Am Med Inform Assoc 2020; 27 (3): 437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li L, Cheng W-Y, Glicksberg BS, et al. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci Transl Med 2015; 7 (311): 311ra174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pikoula M, Quint JK, Nissen F, Hemingway H, Smeeth L, Denaxas S.. Identifying clinically important COPD sub-types using data-driven approaches in primary care population based electronic health records. BMC Med Inform Decis Mak 2019; 19 (1): 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu Z, Wang F, Adekkanattu P, et al. Subphenotyping depression using machine learning and electronic health records. Learn Health Syst 2020; 4 (4): e10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X, Chou J, Liang J, et al. Data-driven subtyping of Parkinson’s disease using longitudinal clinical records: a cohort study. Sci Rep 2019; 9 (1): 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Landi I, Glicksberg BS, Lee H-C, et al. Deep representation learning of electronic health records to unlock patient stratification at scale. npj Digit Med 2020; 3 (1): 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doshi-Velez F, Ge Y, Kohane I.. Comorbidity clusters in autism spectrum disorders: an electronic health record time-series analysis. Pediatrics 2014; 133 (1): e54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Warner JL, Denny JC, Kreda DA, Alterovitz G.. Seeing the forest through the trees: uncovering phenomic complexity through interactive network visualization. J Am Med Inform Assoc 2015; 22 (2): 324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang L, Zhang Y, Cai T, et al. Automated grouping of medical codes via multiview banded spectral clustering. J Biomed Inform 2019; 100: 103322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene–disease associations. Bioinformatics 2010; 26 (9): 1205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei W-Q, Bastarache LA, Carroll RJ, et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. PLoS ONE 2017; 12 (7): e0175508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Denny JC, Bastarache L, Ritchie MD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 2013; 31 (12): 1102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 2019; 16 (12): 1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Maaten L, Hinton G.. Viualizing data using t-SNE. J Mach Learn Res 2008; 9: 2579–605. [Google Scholar]
- 22. Kobak D, Berens P.. The art of using t-SNE for single-cell transcriptomics. Nat Commun 2019; 10 (1): 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levine JH, Simonds EF, Bendall SC, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015; 162 (1): 184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2010. [Google Scholar]
- 25. Wang Y, Zhao Y, Therneau TM, et al. Unsupervised machine learning for the discovery of latent disease clusters and patient subgroups using electronic health records. J Biomed Inform 2020; 102: 103364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopez C, Tucker S, Salameh T, Tucker C.. An unsupervised machine learning method for discovering patient clusters based on genetic signatures. J Biomed Inform 2018; 85: 30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamid JS, Meaney C, Crowcroft NS, Granerod J, Beyene J; UK Etiology of Encephalitis Study Group. Cluster analysis for identifying sub-groups and selecting potential discriminatory variables in human encephalitis. BMC Infect Dis 2010; 10 (1): 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018; 6 (5): 361–9. [DOI] [PubMed] [Google Scholar]
- 29. Kang JB, Nathan A, Weinand K, et al. Efficient and precise single-cell reference atlas mapping with Symphony. Nat Commun 2021; 12 (1): 5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study are available on request via the eMerge consortium (https://emerge-network.org/ & https://phekb.org/). The framework developed here, as well as full results are publicly available on GitHub via https://github.com/MarcMaurits/EHRClustering.