

Abstract
Androgen-producing steroid cell ovarian tumors are rare, comprising less than 1% of ovarian neoplasms, and can present with infertility and rapid virilization. Here we discuss the case of a 28-year-old woman who presented with an unusually insidious 2-year history of infertility, hirsutism, and clitoromegaly who was found to have an elevated serum testosterone and a left ovarian mass. She underwent oophorectomy and pathology revealed a steroid cell tumor, not otherwise specified (NOS), with no malignant features. Following surgery, the patient’s hyperandrogenic symptoms resolved with normalization of testosterone within 6 months, and she was able to conceive spontaneously. In reproductive-aged women with progressive hyperandrogenic symptoms, androgen-producing tumors, including those of ovarian origin, should be suspected. Thorough investigation, including plasma hormone levels and tumor histology, can lead to accurate diagnosis and management. Treatment should be guided by histology and surgical staging, with consideration for future fertility desires. Women who have not completed childbearing can undergo unilateral oophorectomy or tumor resection for benign tumors, with close monitoring of sex hormone levels postoperatively.
Keywords: ovarian steroid cell tumor, androgen-producing tumor, virilization, hirsutism, infertility
Background
Hyperandrogenism is a common condition in adolescent girls and young adults, with a prevalence of 5% to 10% in women of reproductive age. 1 As mild transient hyperandrogenic symptoms are common in early reproductive life, one should have a high index of suspicion to differentiate benign from more serious conditions. Current literature suggests that hormone-producing gynecologic masses should especially be considered in women in their 30s to 40s presenting with rapidly progressive hyperandrogenism. While there are many origins of hyperandrogenism, hirsutism in the setting of elevated 17-hydroxyprogesterone (17-OHP) and testosterone, and normal DHEA-sulfate (DHEA-S), is consistent with steroid-producing ovarian tumors in particular.2-4
We present the case of a young woman who developed virilization due to a hormone-producing ovarian tumor, but whose diagnosis was delayed for nearly 2 years. This insidious presentation is unusual, as generally, tumor-induced hyperandrogenism is rapidly progressive.2,3 Our patient was also much younger than the average age of 43 years, when most women are diagnosed with these tumors. 4
In puberty, as the hypothalamic pituitary ovarian axis activates, luteinizing hormone (LH) pulses increase in amplitude and frequency, leading to a rise in testosterone and androstenedione levels.5,6 The growth hormone/insulin-like growth factor-1 (IGF-1) axis activates in parallel, leading to insulin resistance and a decrease in sex-hormone binding-globulin (SHBG). 5 As a result, early menstrual cycles are anovulatory, with mild hyperandrogenic symptoms. In most cases, normal physiology resumes in 1 to 2 years. 5 If hyperandrogenic symptoms persist or progress, polycystic ovarian syndrome (PCOS), non-classical congenital adrenal hyperplasia (NCCAH), Cushing’s syndrome, and adrenal and ovarian tumors should be considered.
Severe hyperandrogenic features can been seen with any degree of androgen excess, as clitoromegaly is seen in 8% to 20% of girls with NCCAH 5 ; however, androgen-producing adrenal and ovarian tumors are major causes of virilization. 7 Virilization comprises clitoromegaly, alopecia, muscular hypertrophy, breast atrophy, and amenorrhea. 1 A thorough patient history, including inquiry into medications and establishing the onset and duration of symptoms, along with physical examination for signs of virilization, is important.
While several ovarian tumors can produce androgens, most are benign. Androgen-producing ovarian tumors are categorized under broader terms of luteinized thecomas, dysgerminomas, gonadoblastomas, and Sertoli-stromal tumors, with the latter further classified as Sertoli, Leydig, and Sertoli-Leydig cell tumors, which comprise <1% of all ovarian tumors. Although most of these tumors are androgenic, they can also be hormonally inactive in women and feminizing in men. Rarely, metastatic lesions from the gastrointestinal tract, such as gonadotropin-producing tumors of pancreas, can also produce androgens. 8
Case Presentation
A 28-year-old nulliparous female presented to the endocrinology clinic with hyperandrogenic symptoms and menstrual irregularity. Menarche was at 15 years of age, and her menstrual cycles were regular until age 25. Attempts at pregnancy had been unsuccessful and she had been prescribed a combined estrogen/progesterone oral contraception pill for dysfunctional uterine bleeding. She had also noticed increased facial hair growth involving her upper lip, chin, arms, and back, as well as clitoral enlargement over the past 2 years. She denied significant weight gain, abdominal striations, proximal muscle weakness, and galactorrhea. She had no family history of infertility or cancer. On physical exam, her body mass index was 31.38 kg/m2 and she had a Ferriman Gallwey score of 9. Pelvic exam confirmed clitoromegaly.
Laboratory analysis showed significantly elevated total testosterone of 222.8 ng/dL [8-48 ng/dL], high-normal androstenedione of 236 ng/dL [41-262 ng/dL], and a normal DHEA-S of 322.8 ng/dL [84.8–378 ng/dL]. She also had an elevated 17-OHP of 964 ng/dL [15-70 ng/dL]. On 250 µg adrenocorticotropic hormone (ACTH) stimulation testing, her basal and stimulated 17-OHP were normal at 102 ng/dL and 113 ng/dL, respectively, and stimulated cortisol was 38.4 µg/dL. Her follicle-stimulating hormone (FSH) and LH were normal. Based on the stimulation test results, which yielded normal 17-OHP, and the normal DHEA-S, an adrenal etiology for her hyperandrogenic symptoms was ruled out. Because her clinical presentation was not consistent with hypercortisolism, laboratory evaluation along these lines was not indicated.
Transvaginal ultrasound revealed an enlarged left ovary with an isoechoic mass measuring 4.4 × 2.8 × 3.8 cm without focal densities. The right ovary was slightly enlarged with findings consistent with a polycystic ovary. Multidisciplinary discussion with gynecology led to a consensus for surgical management of the left ovarian mass, the suspected culprit for her hyperandrogenism.
Exploratory laparoscopy revealed an enlarged left ovary and normal-appearing right ovary (Figure 1). The fallopian tubes, uterus, and intraabdominal viscera appeared normal. Dissection of the left ovarian tumor from normal ovary was attempted but could not be achieved due to lack of a clear tissue plane (Figure 2). The tumor was not consistent with a classic dermoid, endometrioma, or fibroma on surgical examination. For these reasons, a left oophorectomy was performed. Surgical pathology revealed a clear cell neoplasm consistent with steroid cell tumor (SCT), not otherwise specified (NOS). Tumor cells exhibited staining with antibodies directed against inhibin and were negative for antibodies directed against PAX-8, GATA-3, RCC, CD10, and cytokeratin AE1/AE3. A reticulin stain confirmed the presence of reticulin surrounding small aggregates of the clear cells. The immunohistochemical and histologic staining pattern was felt to be most consistent with an SCT. There were no malignant features.
Figure 1.
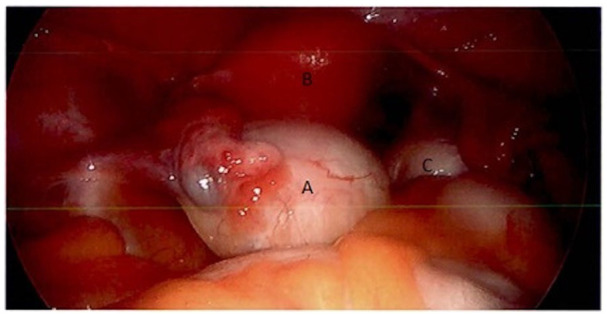
Laparoscopic image of an enlarged left ovary (A), uterus (B), and right ovary (C).
Figure 2.
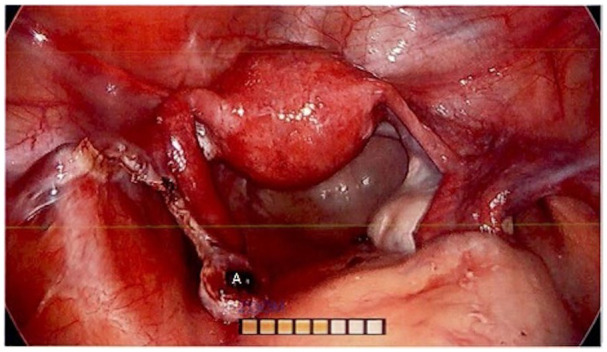
Laparoscopic image of pelvic organs, with site of left ovarian tumor dissection noted (A). Lack of a clear tissue plane between the neoplasm and normal tissue prompted subsequent left oophorectomy.
Six weeks following oophorectomy, our patient’s total testosterone decreased to 65 ng/dL (Table 1). She was able to successfully conceive spontaneously 3 months after surgery, and at her 6-month follow-up, her hyperandrogenic manifestations had improved.
Table 1.
Elevated 17-OHP and Testosterone, and Normal DHEA-S, Are Consistent With an Androgen-Producing Ovarian Neoplasm.
| At presentation | Postoperative | Reference range | |
|---|---|---|---|
| 17-OHP | 964 ng/dL | 15-70 ng/dL | |
| Total testosterone | 222.8 ng/dL | 65 ng/dL | 8-48 ng/dL |
| DHEA-S | 322.8 ng/dL | 84.8-378 ng/dL |
A decrease in testosterone is observed after surgical resection.
Abbreviations: 17-OHP, 17-hydroxyprogesterone; DHEA-S, DHEA-sulfate.
Discussion
Steroid cell tumors have been described as a very rare (<0.1%) class of ovarian neoplasm, with SCT-NOS, comprising between 50% and 60% of the subtypes within this category.9-11 A review of the literature demonstrates that most patients present in their 30s to 40s with rapidly progressive hyperandrogenism leading to virilization.,2-4,9-11 though this tumor has also been described in postmenopausal women. 12 The typical hormone profile of SCT-NOS, has been consistently described as elevated 17-OHP, elevated testosterone, and normal DHEA-S.9,10 Scheker and colleagues have also identified increased expression of enzymes (namely AKR1C3) associated with ovarian steroidogenesis in SCT-NOS, suggesting a potential biomarker and a more specific mechanism for hyperandrogenism with ovarian origins. 13
Clinical Presentation
Nonspecific symptoms associated with ovarian SCT-NOS include ascites, abdominal bloating and distension, and pelvic pain. The clinical presentation is also largely determined by hormones secreted by the tumor. In 56% to 77% of patients with SCT-NOS, testosterone predominates, resulting in virilization. 11 Typically, hormone panels reveal elevated testosterone and androstadiene, indicating an ovarian origin of virilization, and DHEA-S is often normal, ruling out an adrenal etiology. 4
In addition, approximately 6% to 23% of women with SCT-NOS present with menorrhagia and postmenopausal uterine bleeding due to excess estradiol production; 6% to 10% of patients have symptoms and signs of excessive cortisol; and 25% are asymptomatic. 12 Individuals with hypercortisolism may exhibit characteristic deposits of adipose tissue at the posterior neck/mid-upper back, hirsutism, fragile skin, delayed wound healing, and broad purple abdominal striae. Asymptomatic patients are typically found to have an incidental adnexal mass during routine pelvic examination or hysterectomy performed for other reasons.
Patients with excessive hormone production must be evaluated for both adrenal and ovarian neoplasms, PCOS, hypercortisolism, and hyperprolactinemia. Early morning assays for total testosterone, DHEA-S, and 17-OHP are a reasonable first approach. For later assays, it is important to test in the follicular phase, as steroid hormone production from the corpus luteum can yield false positive results. 5 Galactorrhea and symptoms of hypercortisolism may point toward hyperprolactinemia and/or Cushing syndrome, respectively. Pubertal growth arrest, loss of bone density, weight gain, and hypertension should increase suspicion for cortisol excess. 6 If NCCAH is suspected based on elevated 17-OHP, ACTH stimulation testing should be performed. Elevated DHEA-S and testosterone should prompt suspicion for adrenal and ovarian tumors, respectively, and adrenal CT and pelvic magnetic resonance imaging should be obtained. 5
Classification of Ovarian Tumors
The differential diagnosis of a unilateral ovarian mass includes both benign and malignant tumors, which may or may not secrete hormones. Most ovarian neoplasms are primary, originating in the ovary, though metastatic spread from another site, for example, breast or colon, is possible. The World Health Organization histologic classification of primary ovarian tumors is based on the likely tissue of origin; this categorization is also used by the National Comprehensive Cancer Network guidelines in the histopathologic diagnosis of malignant ovarian neoplasms. 14 Surface epithelial tumors begin in the outermost tissue covering the ovaries, comprising about 90% of malignant ovarian tumors. Sex cord–stromal tumors comprise about 5% to 6% of ovarian neoplasms and arise from the ovarian tissue that contains hormone-producing cells. The even rarer germ cell tumors (2%-3%) stem from the egg-producing cells (Figure 3).15-17
Figure 3.
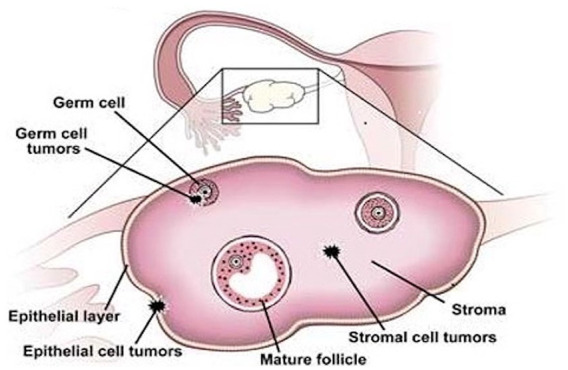
Ovarian tumors are categorized based on their most likely tissue of origin: surface epithelium, the thin outer layer of the ovary; sex cord–stromal, the tissue that contains hormone-producing cells; and germ cell, which is the egg-producing region of the ovary. Reproduced with permission from “Ovarian Cancer: A Review of Current Serum Markers and Their Clinical Applications” by Dr Alicia Algeciras-Schimnich in the March 2013 issue of Clinical Laboratory News.
Within each of the 3 main histologic categories of ovarian tumors, many subtypes exist and are further classified as benign, malignant, or borderline, that is, with atypical proliferation but low potential for malignancy. SCTs fall under the sex cord–stromal tumors and are classified as pure stromal tumors based on their location of origin within the ovary and their histologic appearance (Figure 4). The overall incidence of SCTs of the ovary is rare: They comprise only about 0.1% of all ovarian tumors. 18 Steroid cell tumors, NOS, have no clear cell of origin—unlike, for example, Leydig cell tumors which arise from the Leydig cells in the stromal hilum of the ovary—and comprise the majority of all SCTs. 19
Figure 4.

Schematic representation (noninclusive) of primary ovarian tumors based on the World Health Organization histologic classification, focusing on the sex cord–stromal tumors. Steroid cell tumors originate from the stroma, the ovarian tissue that contains hormone-producing cells, and can be either benign or malignant.
Abbreviation: NOS, not otherwise specified.
Diagnosis
Histopathologic examination remains the gold standard for definitive diagnosis of SCT-NOS. The gross appearance is of a well-circumscribed, noncalcified, lobulated solid mass; cross section demonstrates a yellow-orange surface and occasional cystic changes. 20 Under microscopy, cells have a nested appearance, and cytology reveals polygonal cells with clear borders and prominent nuclei, scanty stroma, and abundant eosinophilic cytoplasm. 20 Under microscopy, cells have a characteristic nested appearance, and cytology reveals polygonal cells with clear borders and prominent nuclei, scanty stroma, and abundant eosinophilic cytoplasm. 19
Steroid cell tumors, NOS, can also be differentiated from other types of stromal tumors, such as Leydig cell tumors, by their lack of cytoplasmic Reinke crystals. 18 Immunohistochemical markers for inhibin and calretinin can be sensitive markers for SCT-NOS, and also help differentiate it from other subtypes of SCTs. 21
Although the morphologic appearance of SCTs can appear benign, one must correlate with clinical and pathologic features, as these neoplasms can behave in a malignant way. Hayes and Scully identified that over 35% of patients with SCTs have clinically malignant disease. Five pathologic characteristics are associated with malignancy, including tumor size >7 cm, excessive necrosis and hemorrhage within the tumor, high mitotic rate, and high degree of cytological atypia. 7 As described above, our patient’s tumor did not have any features of malignancy on pathologic analysis.
Management and Follow-up
Treatment of these hormone-producing ovarian tumors should be based on histology and surgical staging, with consideration for the patient’s future fertility desires. For women with benign tumors who have completed childbearing, limited evidence recommends total hysterectomy with salpingo-oophorectomy. 13 For unilateral and benign tumors, unilateral resection and cystectomy are options in women who have not completed childbearing. 22 However, in these women, sex hormone levels must be closely monitored postoperatively. 13
For malignant SCTs of the ovary and/or metastatic disease, cytoreductive surgery sparing the lymph nodes followed by adjuvant chemotherapy is recommended.13,20 While there is no standard of care with regard to systemic chemotherapy regimens for advanced SCTs with malignant features, combination bleomycin, etoposide, and cisplatin is reasonable in the first line.19,23 Surgical resection should always be considered in patients with advanced-stage primary tumors and in those with recurrent disease, as evidence supports relatively favorable progression-free intervals after second cytoreductive surgery. 22
Conclusion
In reproductive-age females with progressive hyperandrogenic symptoms, especially virilization and a unilateral pelvic mass, ovarian SCTs should be considered in the differential diagnosis. These tumors are rare and can be difficult to diagnose. Clinical history, physical exam, laboratory studies including hormone levels, and appropriate imaging and histopathologic evaluation can assist in accurately identifying this neoplasm. The management approach depends on the tumor histology and the patient’s future fertility desires but typically comprises surgical resection, with follow-up including longitudinal monitoring of androgen levels to ensure normalization.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval: Our institution where the patient was managed does not require ethical approval for reporting individual cases or case series.
Informed Consent: Informed consent was obtained from the patient for their anonymized information.
References
- 1. Yildiz BO. Diagnosis of hyperandrogenism: clinical criteria. Best Pract Res Clin Endocrinol Metab. 2006;20(2):167-176. [DOI] [PubMed] [Google Scholar]
- 2. Rachoń D. Differential diagnosis of hyperandrogenism in women with polycystic ovary syndrome. Exp Clin Endocrinol Diabetes. 2012;120(4):205-209. [DOI] [PubMed] [Google Scholar]
- 3. Macut D, Dusan I, Jovanovic AM, et al. Androgen-secreting ovarian tumors. Front Horm Res. 2019;53:100-107. [DOI] [PubMed] [Google Scholar]
- 4. Lee J, John VS, Liang SX, D’Agostino CA, Menzin AW. Metastatic malignant ovarian steroid cell tumor: a case report and review of the literature. Case Rep Obstet Gynecol. 2016;2016:6184573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Catteau-Jonard S, Cortet-Rudelli C, Richard-Proust C, et al. Hyperandrogenism in adolescent girls. Endocr Dev. 2012;22:181-193. [DOI] [PubMed] [Google Scholar]
- 6. Stafford DE, Gordon CM. Adolescent androgen abnormalities. Curr Opin Obstet Gynecol. 2002;14(5):445-451. [DOI] [PubMed] [Google Scholar]
- 7. Hayes MC, Scully RE. Ovarian steroid cell tumors (not otherwise specified). A clinicopathological analysis of 63 cases. Am J Surg Pathol. 1987;11(11):835-845. [DOI] [PubMed] [Google Scholar]
- 8. Moore A, Permezel M, Mulvany N, Pepperell RJ. Hilus cell tumour of the ovary in a virilized, premenopausal woman. Aust N Z J Obstet Gynaecol. 1999;39(1):75-78. [DOI] [PubMed] [Google Scholar]
- 9. Wong FCK, Chan AZ, Wong WS, et al. Hyperandrogenism, elevated 17-hydroxyprogesterone and its urinary metabolites in a young woman with ovarian steroid cell tumor, not otherwise specified: case report and review of the literature. Case Reports in Endocrinology. 2019;2019:9237459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faten H, Dorra G, Slim C, et al. Ovarian steroid cell tumor (not otherwise specified): a case report of ovarian hyperandrogenism. Case Rep Oncol Med. 2020;82020:6970823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu AX, Sun J, Shao WQ, et al. Steroid cell tumors, not otherwise specified (NOS), in an accessory ovary: a case report and literature review. Gynecol Oncol. 2005;97(1):260-262. [DOI] [PubMed] [Google Scholar]
- 12. Sood N, Desai N, Chindris AM, et al. Symptomatic ovarian steroid cell tumor not otherwise specified in a post-menopausal woman. Rare Tumors. 2016;8(2):6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheker EV, Kathuria A, Esnakula A, et al. Expression of key androgen-activating enzymes in ovarian steroid cell tumor, not otherwise specified. J Investig Med High Impact Case Rep. 2020;8:2324709620933416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kurman RJ, Carcangiu ML, Herrington CS, Young RH. World Health Organization classification of tumours of the female reproductive organs. International Agency for Research in Cancer, Lyon, 2014. In: National Comprehensive Cancer Network Guidelines, Ovarian Cancer version 1.2020, page OV-E 1 of 2:51/127. NCCN guidelines. Accessed March 28, 2022. bit.ly/NCCNOvarianCA [Google Scholar]
- 15. Algeciras- Schimnich A. Ovarian cancer: a review of current serum markers and their clinical applications. Clinical Laboratory News. 2013. Accessed March 28, 2022. https://www.aacc.org/cln/articles/2013/march/ovarian-cancer
- 16. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017;14(1):9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sankaranarayanan R, Ferlay J. Worldwide burden of gynaecological cancer: the size of the problem. Best Pract Res Clin Obstet Gynaecol. 2006;20(2):207-225. [DOI] [PubMed] [Google Scholar]
- 18. Bhagat R, Bodal VK, Gupta N, Garg P. Steroid cell tumour of ovary—a rare case report. J Clin Diagn Res. 2016;10(9):ED06-ED07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qian L, Shen Z, Zhang X, et al. Ovarian steroid cell tumor, not otherwise specified: a case report and literature review. Mol Clin Oncol. 2016;5(6):839-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan EC, Khong CC, Bhutia K. A rare case of steroid cell tumor, not otherwise specified (NOS), of the ovary in a young woman. Case Rep Obstet Gynecol. 2019:4375839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bettegowda A, Rangaiah N, Prasad N, Channaveeregowda S. Virilizing ovarian steroid cell tumor: a rare case. J Clin Diagn Res. 2015;9(9):QD05- QD06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Homesley HD, Bundy BN, Hurteau JA, Roth LM. Bleomycin, etoposide, and cisplatin combination therapy of ovarian granulosa cell tumors and other stromal malignancies: a Gynecologic Oncology Group study. Gynecol Oncol. 1999;72(2):131-137. [DOI] [PubMed] [Google Scholar]
- 23. Reedy MB, Richards WE, Ueland F, et al. Ovarian steroid cell tumors, not otherwise specified: a case report and literature review. Gynecol Oncol. 1999;75(2):293-297. [DOI] [PubMed] [Google Scholar]