
Figure 3.
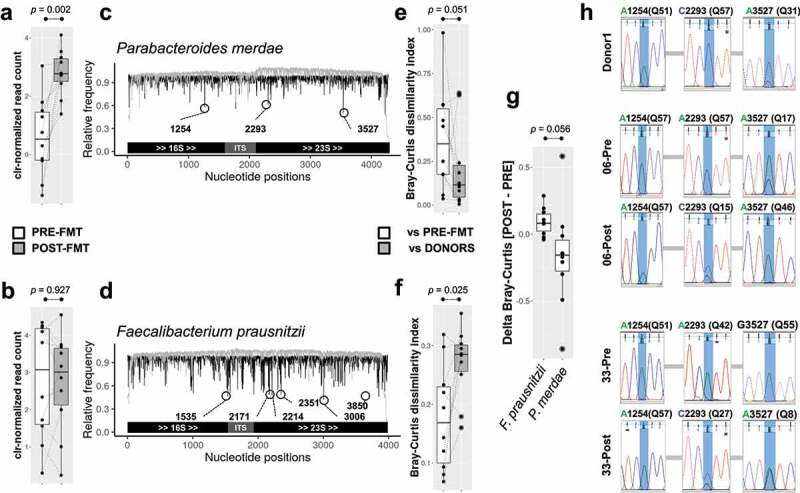
Single-nucleotide variation (SNV) analysis to detect species transfer and engraftment. The species Parabacteroides merdae and Faecalibacterium prausnitzii rrns were used to reveal SNVs within each species and their abundance as a consequence of FMT (a and b panels, respectively). Variation in one unit of clr-normalized counts is roughly equal to a doubling in raw DNA read counts according to data distribution in both cases. c and d – The mapping, indexing, and pileup of thousands of reads of P. merdae and F. prausnitzii rrns allowed us to identify polymorphic sites exhibiting an even frequency for at least two nucleotides. These potential SNVs are highlighted inside open circles, and the position number in the rrn sequence is also indicated. The structural arrangement of rrn is illustrated on the x-axis. The black lines indicate the relative frequency of the dominant allele, whereas the gray lines indicate the relative coverage per site related to the average across the rrn. e and f – The distribution of the Bray-Curtis dissimilarity index of the microbiota between POST-FMT and PRE-FMT samples and the corresponding donors, using the combined nucleotide frequencies of SNVs detected in P. merdae and F. prausnitzii, respectively. The Wilcoxon signed-rank test for paired samples was used to compare the genetic distances of the microbiotas. g – Assessment of delta Bray-Curtis distances computed as POST-FMT vs DONOR samples minus PRE-FMT vs DONOR samples for Faecalibacterium prasunitzii and Parabacteroides merdae species. h – Electropherograms obtained from Sanger sequencing for the strains of P. merdae SNV-3527, SNV-2293, and SNV-1254. The predominant alleles inferred for every sample (based on the Q-score of base calling) supported the hypothesis that the strains were transferred between donor and recipient pairs, as anticipated during the Nanopore-based assessment.