
Figure 1.
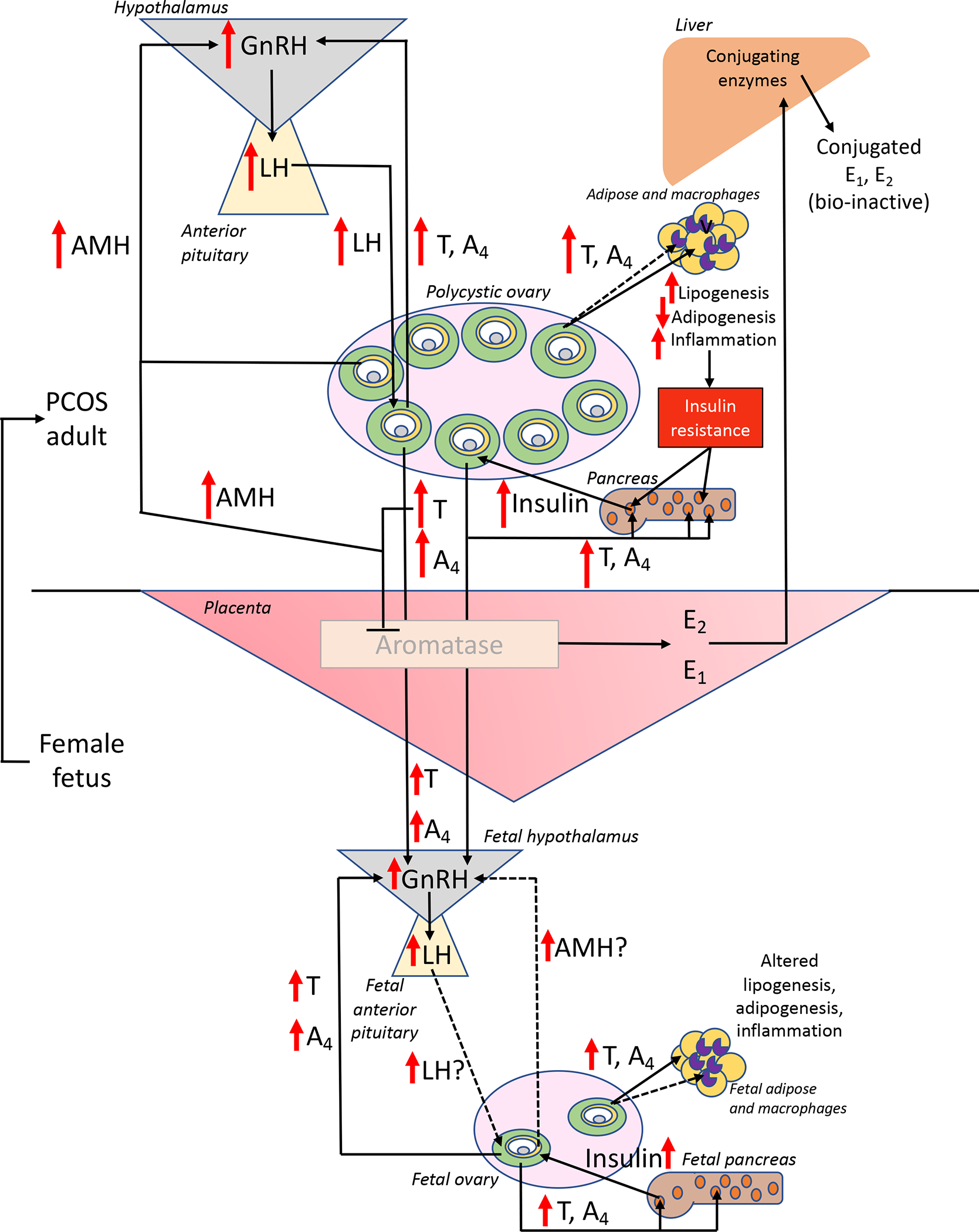
Hypothetical feed-forward mechanisms for transgenerational hyperandrogenic epigenomic transmission of PCOS in genetically susceptible daughters of women with this disorder. In response to elevated maternal AMH levels, LH- and insulin-enhanced hyperandrogenism from polycystic ovaries impairs ovarian steroid negative feedback inhibition of hypothalamic GnRH release and promotes pancreatic beta cell hyperinsulinemia. Compensatory pancreatic beta cell hyperinsulinemia in response to insulin resistance in multiple organ systems is a consequence of hyperandrogenism-induced lipogenic, adipogenic and inflammation dysfunction. During gestation, hyperandrogenism inhibits placental aromatase and alters placental structure-function, permitting trans-placental access of maternal hyperandrogenism to a female fetus. If placental aromatase activity remains sufficient, extensive maternal hepatic conjugation of estrogens (rendering them bio-inactive and favored for excretion) maintains maternal estrogenic homeostasis. If not, androgen excess in the mid-gestation female fetus (1) impairs ovarian steroid negative feedback inhibition of hypothalamic GnRH release to promote LH-stimulated hyperandrogenism, (2) induces pancreatic beta cell hyperinsulinism, and (3) alters adipose and macrophage function, establishing an antecedent susceptibility that favors development into a PCOS phenotype during adolescence. Solid lines and arrows indicate functions identified previously, while dashed arrows and lines are currently hypothetical. Within ovarian antral follicles, green = theca cells, yellow = granulosa cells, gray = oocyte, white = follicular fluid in the antrum; within pancreatic islets, orange represents beta cells; within adipose and macrophages, beige = white adipocytes, purple = macrophages.