
Figure 3. Effect of CDK4 and CDK6 inhibition upon tumour cell-intrinsic and -extrinsic signalling pathways.
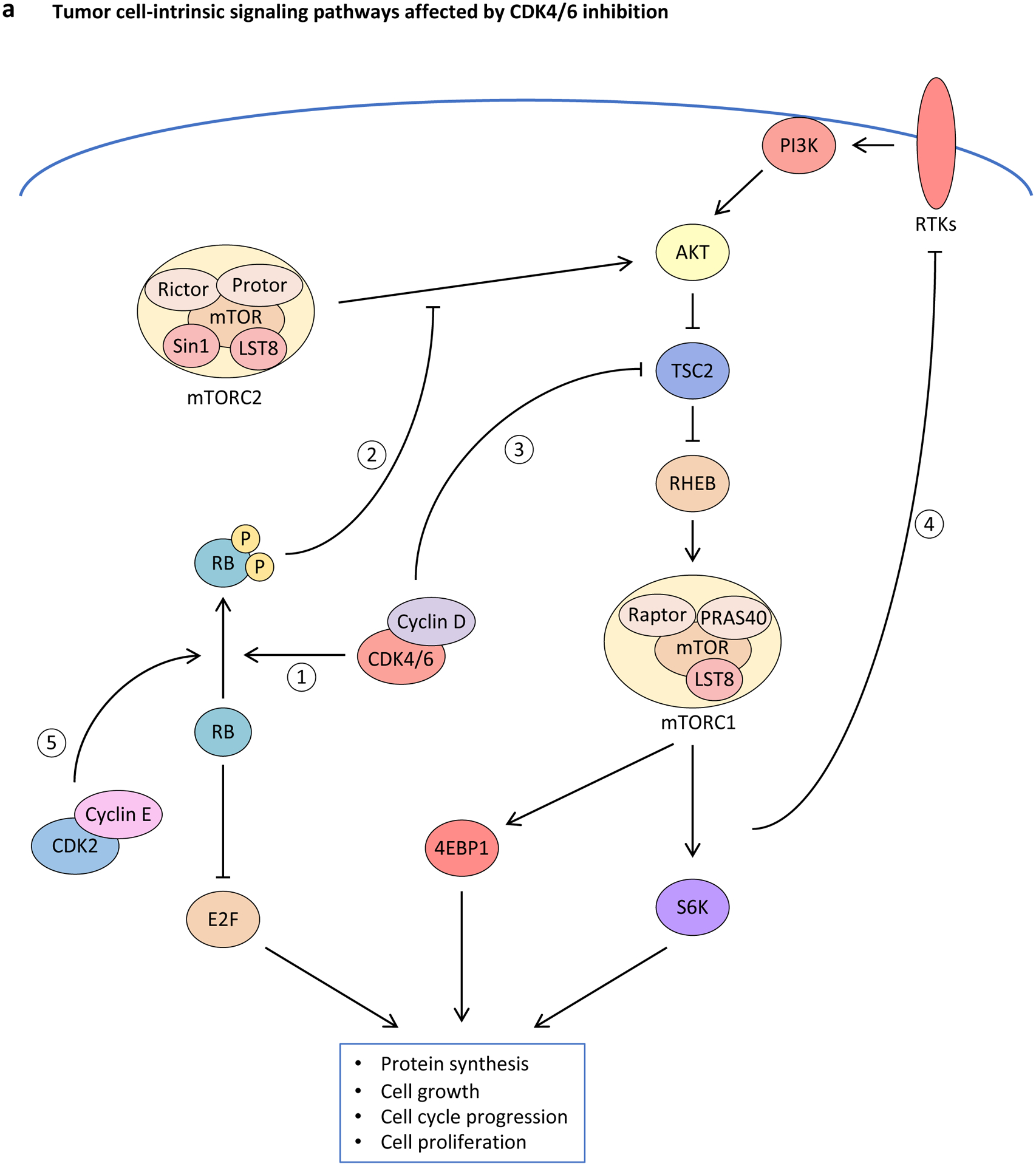
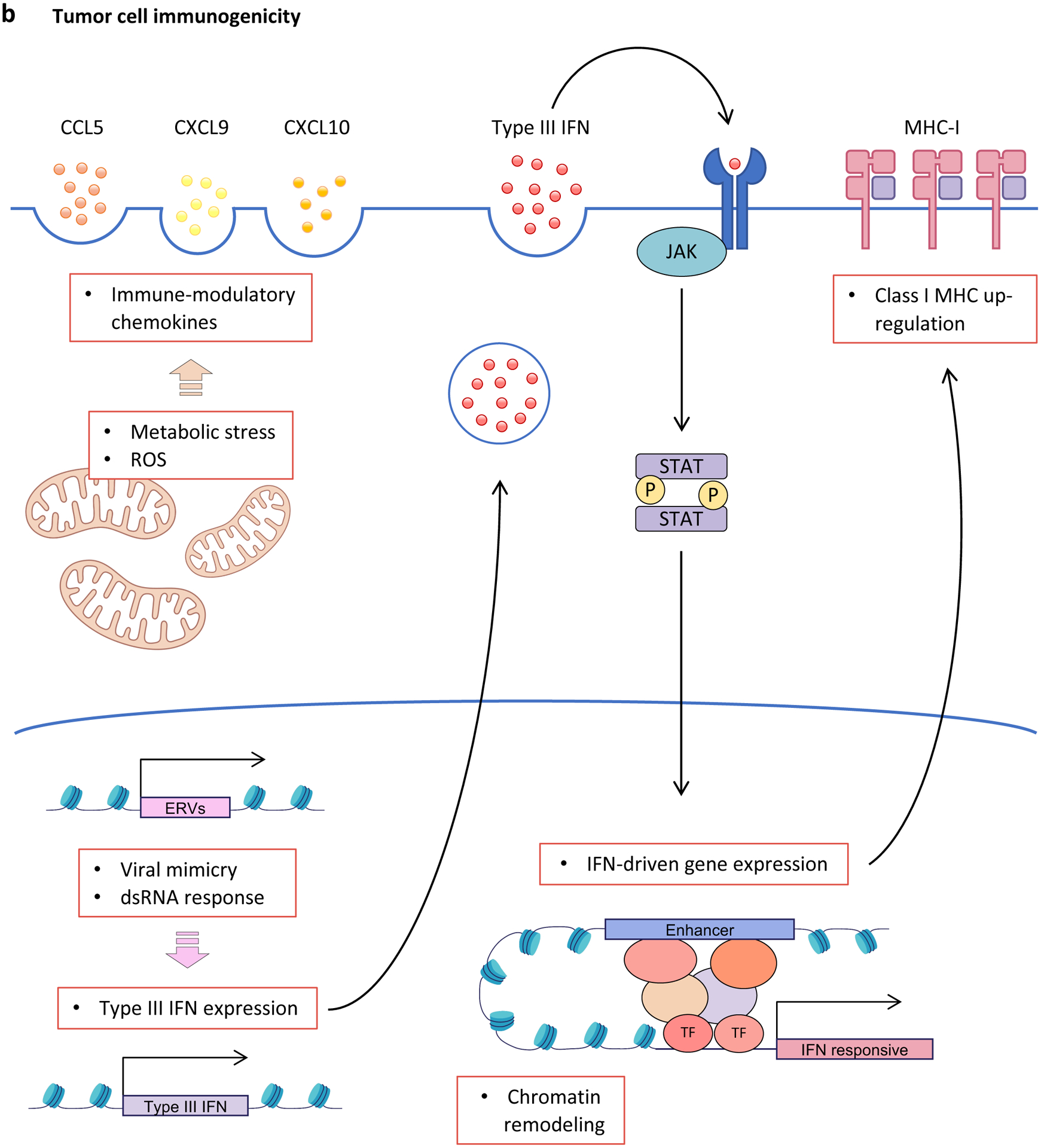
a, Inhibition of cyclin-dependent kinase 4 (CDK4) and CDK6 (CDK4/6) activity induces rewiring of multiple interconnected kinase circuits in tumour cells. First, blocking CDK4/6 inhibits RB phosphorylation (1), thereby preventing E2F-mediated cell cycle progression. However, multiple mechanisms may counteract these effects. For example, decreased phosphorylated-RB levels also lead to enhanced AKT phosphorylation by mTORC complex 2 (mTORC2) (2), which can stimulate cell survival mechanisms. Similarly, decreased CDK4/6 activity can down-regulate mTORC1 signalling via enhanced tuberous sclerosis 2 (TSC2) activity (3), which may lead to loss of negative-feedback regulation of receptor tyrosine kinase (RTK) signalling (4), resulting in up-regulated upstream PI3K–AKT pathway activity. In addition, compensatory CDK2-mediated RB phosphorylation (5) can stimulate cell cycle progression in the absence of CDK4/6 signalling. b, CDK4/6 inhibitors promote tumour cell immunogenicity via multiple mechanisms: (i) Metabolic stress and increased production of reactive oxygen species (ROS) up-regulate secretion of inflammatory chemokines CC-chemokine ligand 5 (CCL5), CXC-chemokine ligand 9 (CXCL9) and CXCL10; (ii) hypomethylation and therefore expression of endogenous retroviruses (ERVs) induces a double-stranded RNA (dsRNA) response, which leads to increased expression and secretion of type III interferon (IFN), activation of Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signalling, enhanced IFN-driven gene expression, and up-regulation of major histocompatibility complex (MHC) class I expression; and (iii) chromatin remodelling facilitates IFN-mediated expression of interferon-responsive genes. 4EBP1; eukaryotic translation initiation factor 4E binding protein 1; IFNLR1, interferon λ receptor 1; IL-10RB, interleukin-10 receptor subunit β; S6K, S6 kinase; TF, transcription factor.