

Abstract
Inflammation has profound but poorly understood effects on metabolism, especially in the context of obesity and nonalcoholic fatty liver disease (NAFLD). Here, we report that hepatic interferon regulatory factor 3 (IRF3) is a direct transcriptional regulator of glucose homeostasis through induction of Ppp2r1b, a component of serine/threonine phosphatase PP2A, and subsequent suppression of glucose production. Global ablation of IRF3 in mice on a high-fat diet protected against both steatosis and dysglycemia, whereas hepatocyte-specific loss of IRF3 affects only dysglycemia. Integration of the IRF3-dependent transcriptome and cistrome in mouse hepatocytes identifies Ppp2r1b as a direct IRF3 target responsible for mediating its metabolic actions on glucose homeostasis. IRF3-mediated induction of Ppp2r1b amplified PP2A activity, with subsequent dephosphorylation of AMPKα and AKT. Furthermore, suppression of hepatic Irf3 expression with antisense oligonucleotides reversed obesity-induced insulin resistance and restored glucose homeostasis in obese mice. Obese humans with NAFLD displayed enhanced activation of liver IRF3, with reversion after bariatric surgery. Hepatic PPP2R1B expression correlated with HgbA1C and was elevated in obese humans with impaired fasting glucose. We therefore identify the hepatic IRF3-PPP2R1B axis as a causal link between obesity-induced inflammation and dysglycemia and suggest an approach for limiting the metabolic dysfunction accompanying obesity-associated NAFLD.
INTRODUCTION
The liver plays a critical role in metabolism, maintaining glucose homeostasis by balancing the storage and release of glucose via glycogenolysis and gluconeogenesis (1, 2). This delicate balance becomes dysregulated during overnutrition, as the liver produces glucose in excess of the body’s needs and begins to accumulate lipids, a condition known as nonalcoholic fatty liver disease (NAFLD). Over time, this can progress from simple steatosis to steatohepatitis and eventually, in some cases, cirrhosis and hepatocellular carcinoma. Although it remains unclear what pathways trigger the initial dysregulation in glucose homeostasis, numerous studies suggest that hepatic inflammation is involved (3–6).
It has been appreciated for decades that obesity triggers a state of chronic inflammation in the liver that is associated with metabolic dysfunction (7–10). The connection between inflammation and metabolism has been suggested to involve the actions of proinflammatory cytokines secreted by infiltrating immune cells (11–13). However, anti-inflammatory strategies targeting cytokines have consistently proven to be ineffective at improving insulin action and metabolic function (14, 15). For example, neutralization of tumor necrosis factor–α (TNFα) had no effect on insulin sensitivity or steatosis in obese diabetic patients (16, 17). Mice lacking either TNFα or TNF receptor 1 demonstrated equivalent steatohepatitis as compared to control animals on high-fat diet (HFD) (18, 19). Furthermore, mice deficient in interleukin-6 (IL-6) display increased obesity, steatohepatitis, and insulin resistance (20). Proinflammatory cytokines generated in obesity are believed to function by activating the transcription factor nuclear factor κB (NF-κB) (21, 22). Many studies have implicated hepatic NF-κB in obesity and fatty liver disease by showing the importance of the upstream kinase inhibitor of NF-κB kinase β (IKKβ) (4), but the interpretation of these studies is complicated by the fact that IKKβ can cause metabolic dysfunction via NF-κB–independent pathways (23). Furthermore, despite displaying a systemic proinflammatory expression pattern, the effect of p65 (the active subunit of NF-κB) overexpression on insulin sensitivity and steatosis varies greatly depending on the target tissue (24–26). Together, these studies suggest that inflammatory transcription factors other than NF-κB must be relevant in the hepatic metabolic dysfunction of obesity.
The interferon (IFN) regulatory factors (IRFs) represent another class of transcription factors with major functions in innate and adaptive immunity. Several of the IRFs have also been linked to metabolism, making them prime candidates to be mediators of immunometabolic changes that occur during obesity (27, 28). IRF3 in particular has been implicated in metabolic processes, including adipogenesis and thermogenesis (28). In the context of cholesterol metabolism, IRF3 was demonstrated to inhibit ABCA1 and other liver X receptor–regulated genes (29, 30).
IRF3 resides in the cytosol until it is activated by pattern recognition receptors (PRRs), such as Cyclic GMP-AMP Synthase (cGAS) and Toll-like receptors (TLRs), which activate the kinases TBK1 (TANK-binding kinase 1), IKKε, and apoptosis signal–regulating kinase 1, triggering IRF3 phosphorylation, dimerization, nuclear translocation, and chromatin binding (31, 32). The best characterized targets of IRF3 in immune cells are a suite of genes collectively called IFN-stimulated genes (ISGs), including Ifnβ, Ifit1, Isg15, and Rsad2 (33). Several studies have shown that activation of the stimulator of interferon genes (STING)/TBK1-IKKε/IRF3 axis is associated with worsening metabolic dysfunction in obesity, though some have implicated the opposite (5, 28, 34–38). Nevertheless, none have identified which downstream targets of IRF3 mediate its metabolic actions. Thus, despite the importance of inflammation in obesity, the mechanism by which inflammatory factors such as IRF3 directly trigger metabolic dysfunction remains an unanswered fundamental biological question. Here, we investigate the effect of hepatic IRF3 activation on metabolism and glucose homeostasis in obesity.
RESULTS
IRF3 is activated in liver tissue and promotes steatohepatitis and insulin resistance
To determine whether hepatic IRF3 plays a role in obesity-induced metabolic dysfunction, we began by assessing hepatic expression of Irf3 and classic IRF3 target genes (ISGs) in HFD-fed mice. Despite the absence of infection in this setting, we detected robust expression of ISG mRNAs (Fig. 1A). In contrast, IRF3-deficient (IRF3KO) mice showed a near complete abrogation of HFD-induced hepatic ISG mRNA expression (fig. S1A). Although IRF3 mRNA and protein expression was similar in HFD-fed and standard chow diet (SCD)–fed mice, we found increased amounts of the IRF3 upstream kinase, TBK1, as well as elevated phosphorylated IRF3 (pIRF3; Ser396) as early as 3 weeks into HFD feeding and persisting until at least 16 weeks (Fig. 1B). Given the cellular heterogeneity of liver, we used the NuTRAP (nuclear tagging and translating ribosome affinity purification) transgenic mouse line to study changes specifically in hepatocytes (39). NuTRAP mice allow isolation of ribosome-bound mRNA and nuclei from specific cell types with Cre-dependent expression of green fluorescent protein (GFP)–tagged 60S ribosomal subunit L10a and mCherry-labeled nuclei, respectively. We crossed NuTRAP mice with hepatocyte-specific albumin-Cre recombinase (Alb-Cre) mice (hereafter called NuTRAPalb) and then isolated hepatocyte mRNA and nuclei from whole liver tissue (fig. S1, B and C). ISG mRNA amounts were elevated in hepatocytes from obese HFD-fed mice compared to SCD-fed (Fig. 1C). Consistent with these findings, nuclear IRF3 protein amounts were also increased in hepatocytes from HFD-fed mice, indicating enhanced IRF3 activation (Fig. 1D).
Fig. 1. IRF3 is activated in liver tissue of obese mice and humans, promoting steatohepatitis and insulin resistance.
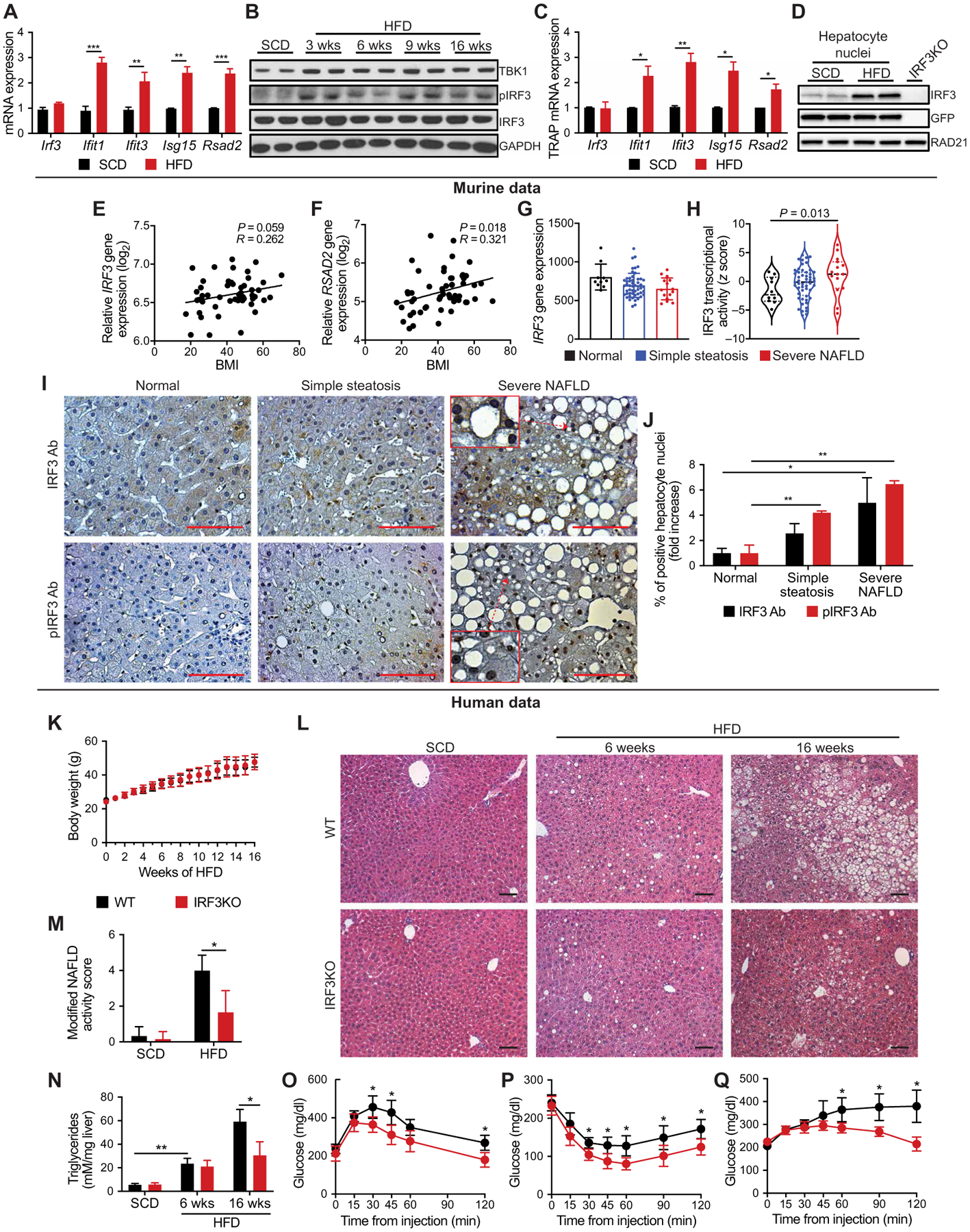
(A) Expression of IFN-stimulated genes (ISGs) in liver tissue from male mice fed SCD or HFD for 6 weeks (n = 4). (B) Immunoblot showing TANK-binding kinase 1 (TBK1), phosphorylated IRF3 (pIRF3; Ser396), and total IRF3 in liver tissue from male mice fed SCD or HFD for 3, 6, 9, and 16 weeks. (C) Expression of hepatocyte ISGs in TRAP liver tissue from male NuTRAPalb mice fed SCD or HFD for 6 weeks (n = 4). (D) Immunoblot showing IRF3 and GFP in hepatocyte nuclei from male NuTRAPalb mice fed SCD or HFD for 6 weeks, as well as total liver cell nuclei from male IRF3KO mice on SCD (last lane). Nuclei from NuTRAPalb mice were sorted by flow cytometry on the basis of hepatocyte-specific nuclear GFP expression. (E) Expression of IRF3 and (F) RSAD2 in liver tissue from healthy lean and obese patients correlated with BMI (n = 54). (G) Expression of IRF3 and (H) transcriptional activity of IRF3 in liver tissue from obese patients without NAFLD (normal), with simple steatosis, or with severe NAFLD (NAFLD activity score > 5; n = 77). (I) Liver tissue from obese patients without NAFLD (normal), with simple steatosis, or with severe NAFLD, stained for pIRF3 (Ser396) or total IRF3 (representative images from n = 14 patients). Ab, antibody. Scale bars, 100 μm. (J) Quantification of hepatocyte nuclei that stained positive for pIRF3 or total IRF3. (K to Q) Male WT and IRF3KO littermate mice were fed HFD for 3 to 16 weeks. (K) Total body weight of WT and IRF3KO mice during HFD feeding (n = 8 to 15). (L) Hematoxylin and eosin (H&E) staining of liver tissue and (M) modified NAFLD activity score of liver histology from WT and IRF3KO mice fed SCD or HFD for 6 or 16 weeks (n = 6–9). Scale bars, 100 μm. (N) Liver triglyceride (TAG) content in WT and IRF3KO mice fed SCD or HFD for 6 or 16 weeks (n = 4 to 9). (O) Glucose (1 g/kg), (P) insulin (1.75 U/kg), and (Q) pyruvate (1.25 g/kg) tolerance tests performed on WT and IRF3KO mice after 13, 14, and 15 weeks on HFD, respectively (n = 8 to 11). Data are presented as means ± SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
We next asked whether IRF3 activity is elevated in the livers of obese patients with NAFLD (fig. S1, D and E) (40, 41). The correlation between hepatic IRF3 expression and body mass index (BMI) was not statistically significant (r value of 0.262, P = 0.059) (Fig. 1E). However, there was a statistically significant correlation (r value of 0.321, P = 0.018) between BMI and expression of RSAD2, a classic IRF3 target gene (Fig. 1F), suggesting increasing hepatic IRF3 activation with obesity. Examining a different cohort of obese patients with NAFLD, we again found no association between hepatic IRF3 expression and NAFLD, but a substantial association existed between hepatic IRF3 transcriptional activity and NAFLD histological severity (Fig. 1, G and H, and fig. S1E) (41). IRF3 transcriptional activity was assessed by analyzing the expression data of an IRF3 target gene set using virtual inference of protein activity by enriched regulon analysis (VIPER) (42, 43), where severe NAFLD was defined as the presence of nonalcoholic steatohepatitis or advanced fibrosis (≥F2). We then evaluated liver samples from this cohort by immunohistochemistry and found progressively increasing hepatocyte nuclear pIRF3 staining with NAFLD severity (Fig. 1, I and J). Together, these data show that hepatic IRF3 phosphorylation and activation of the IRF3 transcriptional program are associated with obesity and NAFLD progression in humans.
To determine the functional significance of obesity-induced IRF3 activation on liver disease and metabolism, we compared indicators of steatohepatitis and insulin sensitivity between wild-type (WT) and IRF3KO mice on HFD (60% fat). On this diet, IRF3KO mice showed no difference in total body weight or food intake (Fig. 1K and fig. S1F). However, prolonged HFD resulted in substantially less histological evidence of hepatic steatosis and inflammation in IRF3KO mice (Fig. 1, L and M, and fig. S1G). Consistent with this, hepatic triglyceride (TAG) accumulation was markedly reduced in IRF3KO mice (Fig. 1N and fig. S1G). Given the known association between steatosis and insulin resistance, we evaluated the effects of IRF3 on glucose homeostasis and insulin action. IRF3KO mice showed reduced fasting insulin after high-fat feeding, with no change in fasting glucose (fig. S1, H and I). Furthermore, IRF3KO mice demonstrated substantially improved glucose and insulin tolerance after 13 and 14 weeks of HFD, as well as enhanced pyruvate tolerance, suggesting decreased hepatic gluconeogenesis (Fig. 1, O to Q, and fig. S1, J to L). Together, these data indicate that global IRF3 deficiency protects against obesity-induced hepatic steatosis and insulin resistance.
Hepatocyte IRF3 deficiency improves obesity-induced insulin resistance and glucose homeostasis
Given the cellular heterogeneity of liver, we sought to determine which hepatic cell type is responsible for the metabolic actions of IRF3. We therefore created an allele in which Irf3 exons three to six were flanked by loxP sites (Fig. 2A), allowing for Cre recombinase–dependent ablation of Irf3. IRF3 floxed mice (Irf3flox) were crossed with Alb-Cre mice, generating hepatocyte-deficient IRF3 (LI3KO) mice. LI3KO mice showed no IRF3 protein or ISG mRNA expression in hepatocytes, but expression in liver nonparenchymal cells (NPCs) was unperturbed (Fig. 2B and fig. S2A). LI3KO mice showed no difference in total body weight or food intake while on HFD (Fig. 2C and fig. S2B). Unexpectedly, LI3KO mice were not protected from HFD-induced steatosis or triglyceride accumulation but did display decreased macrophage infiltration (Fig. 2, D to G). Despite a comparable degree of steatosis as HFD-fed Irf3flox mice, LI3KO mice were more insulin sensitive, with improved glucose and insulin tolerance (Fig. 2, H and I, and fig. S2, C and D). Similarly, fasting insulin and pancreatic β cell mass were reduced in LI3KO mice on HFD (fig. S2, E to H). Furthermore, whole liver lysates from HFD-fed LI3KO mice showed increased insulin-stimulated AKT phosphorylation (pAKT; Ser473), suggesting improved insulin signaling (Fig. 2J). Although the association between steatosis and insulin resistance in obesity is complex, diacylglycerols (DAGs) and ceramides have emerged as the two most-studied mediators of lipid-induced hepatic insulin resistance (44). We saw no statistically significant differences in liver DAG and ceramide amounts between HFD-fed LI3KO and Irf3flox mice (fig. S2, I to L).
Fig. 2. Hepatocyte deficiency in IRF3 improves obesity-induced insulin sensitivity and glucose homeostasis.
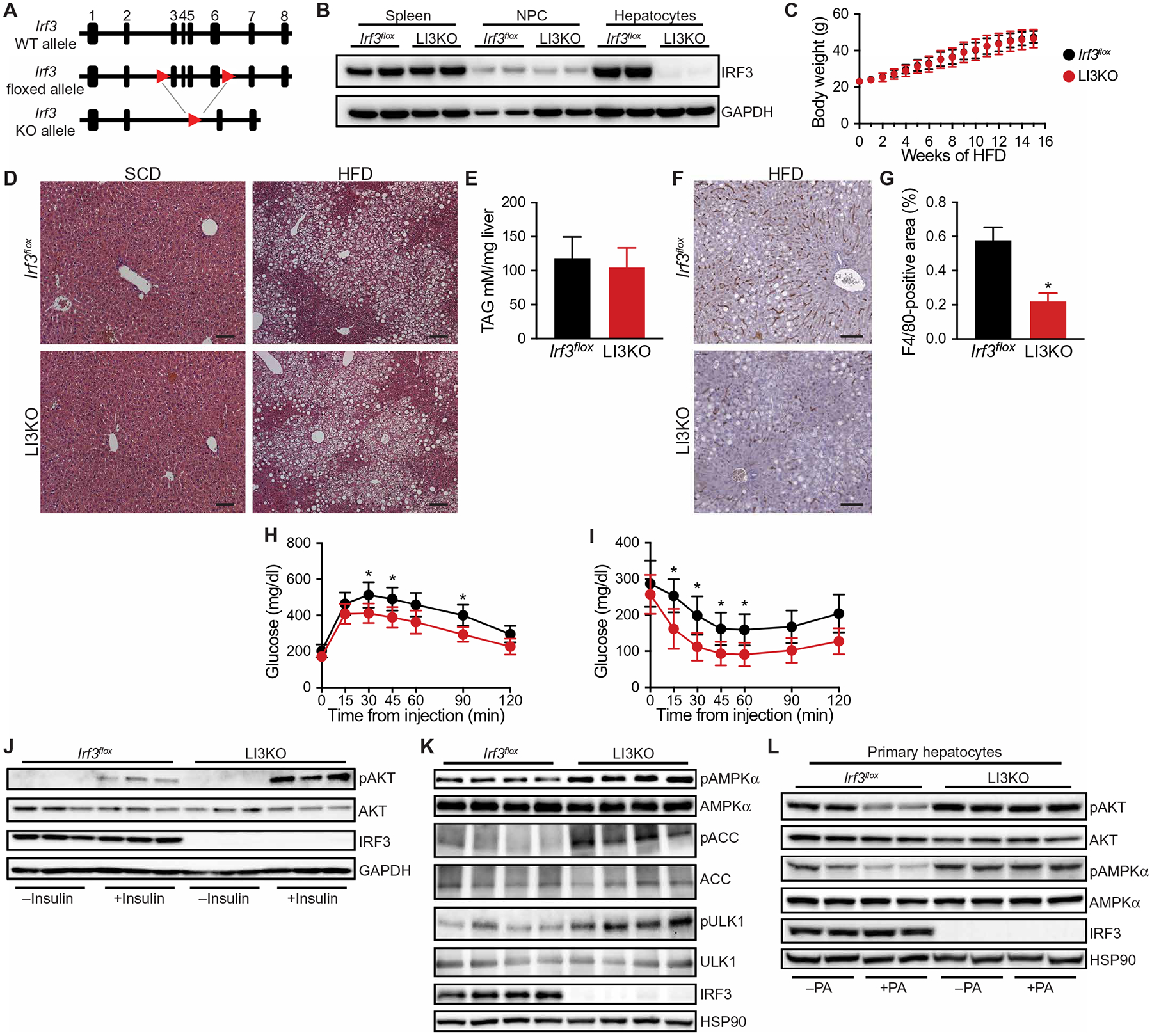
(A) Schematic of floxed Irf3 allele. (B) Immunoblot showing IRF3 in the spleen, liver nonparenchymal cells (NPCs), and hepatocytes from 6-week-old male Irf3flox and LI3KO mice. (C) Total body weight of male Irf3flox and LI3KO littermate mice during HFD feeding (n = 12 to 14). (D) H&E staining of liver tissue from male Irf3flox and LI3KO mice fed SCD or HFD for 20 weeks. Scale bars, 100 μm. (E) Liver triglyceride (TAG) content in male Irf3flox and LI3KO mice fed SCD or HFD for 20 weeks (n = 10 to 12). (F) F4/80 immunostaining of liver tissue from male Irf3flox and LI3KO mice fed HFD for 20 weeks. Scale bars, 100 μm. (G) Quantification of F4/80+ cells (n = 10). (H) Glucose (1.25 g/kg) and (I) insulin (1.5 U/kg) tolerance tests performed on male Irf3flox and LI3KO mice fed HFD for 18 or 19 weeks, respectively (n = 10 to 12). (J) Immunoblot showing insulin-stimulated phosphorylated AKT (pAKT; Ser473) and total AKT in liver tissue from male Irf3flox and LI3KO mice fed HFD for 20 weeks. Mice were fasted for 5 hours, injected intraperitoneally with insulin (5 U/kg), and then euthanized 5 min later. Liver tissue was harvested within 90 s. (K) Immunoblot showing phosphorylated AMPKα (pAMPKα; Thr172) and total AMPKα, phosphorylated ACC (pACC; S79) and total ACC, and phosphorylated ULK1 (pULK1; S555) and total ULK1 in liver tissue from male Irf3flox and LI3KO mice fed HFD for 20 weeks. Mice were fasted for 5 hours before euthanasia. (L) Immunoblot showing pAKT, AKT, pAMPKα, and AMPKα in palmitic acid (PA; 500 μM)–treated primary hepatocytes isolated from 6-week-old male Irf3flox and LI3KO mice. Data are presented as means ± SD; *P < 0.05.
In addition to the insulin-AKT pathway, another major regulator of hepatic glucose production is adenosine 5′-monophosphate (AMP)–activated protein kinase (AMPK) (45–47). AMPK is composed of three subunits, one of which (AMPKα) is activated by phosphorylation at Thr172, triggering inhibition of hepatic gluconeogenesis (47–49). We found increased hepatic AMPKα phosphorylation (pAMPKα; Thr172), as well as acetyl–coenzyme A (CoA) carboxylase (ACC) (pACC; Ser79) and Unc-51 Like Autophagy Activating Kinase 1 (ULK1) (pULK1; Ser555) phosphorylation, two targets of AMPK, in HFD-fed LI3KO mice, suggesting another reason for their improved glucose homeostasis (Fig. 2K). To determine whether the improved insulin signaling and glucose homeostasis in the livers of LI3KO mice were cell-autonomous features, we isolated primary hepatocytes from Irf3flox and LI3KO mice and stimulated them with palmitic acid (PA) to render them insulin resistant (50). Although both pAKT and pAMPKα were reduced in PA-treated Irf3flox hepatocytes, they were increased in LI3KO hepatocytes (Fig. 2L). Collectively, these data suggest that, within hepatocytes, IRF3 mediates HFD-induced insulin resistance and dysregulation of glucose homeostasis, but not steatosis.
Hepatocyte IRF3 drives insulin resistance and glucose production and inhibits AMPKα phosphorylation
We next asked whether activated IRF3 in hepatocytes is sufficient to reduce insulin sensitivity and promote dysglycemia. For these gain-of-function studies, we took advantage of a phosphomimetic construct that we previously created in which Ser388 and Ser390 of murine IRF3 are mutated to aspartic acid, resulting in a constitutively active allele called IRF3-2D (28). Once expressed, IRF3-2D does not require an external stimulus for activation. This allele was FLAG-tagged and placed within the Rosa26 locus downstream of a loxP-stop-loxP cassette, allowing Cre-dependent expression of IRF3-2D (IRF3LSL-2D; Fig. 3A). IRF3LSL-2D mice were injected with adeno-associated virus 8 (AAV8) expressing Cre recombinase under the control of the hepatocyte-specific thyroxine-binding globulin promoter (AAV8-TBG-Cre), enabling selective expression of IRF3-2D in hepatocytes with temporal control (51). Hepatocytes from mice injected with AAV8-TBG-Cre expressed the IRF3-2D allele and displayed elevated ISG mRNAs when compared to liver NPCs (Fig. 3B and fig. S3A). IRF3LSL-2D mice placed on HFD and then injected with AAV8-TBG-Cre showed worsened glucose tolerance (Fig. 3C and fig. S3B) and impaired hepatic insulin signaling with decreased AKT phosphorylation in the liver (Fig. 3D), despite no change in body weight compared to AAV8-TBG-GFP–injected mice (fig. S3C). Furthermore, hepatocytes isolated from IRF3LSL-2D mice transduced ex vivo with a Cre recombinase–expressing adenovirus (Ad-Cre) demonstrated decreased insulin signaling, as measured by insulin-stimulated AKT phosphorylation (Fig. 3E). Although AKT phosphorylation can be used as a proxy for insulin signaling, the true functional measure for insulin sensitivity in hepatocytes is suppression of glucose production. IRF3LSL-2D hepatocytes were transduced ex vivo with Ad-Cre and stimulated with cyclic AMP to drive gluconeogenesis in the presence or absence of insulin. Expression of IRF3-2D blunted the ability of insulin to inhibit glucose production (Fig. 3F). IRF3-2D also increased glucose production in the absence of insulin, suggesting that the effect of IRF3 on hepatic glucose production was not exclusively dependent on its ability to impair insulin signaling through AKT.
Fig. 3. Activation of hepatocyte IRF3 drives insulin resistance and glucose production, and inhibits AMPKα phosphorylation.
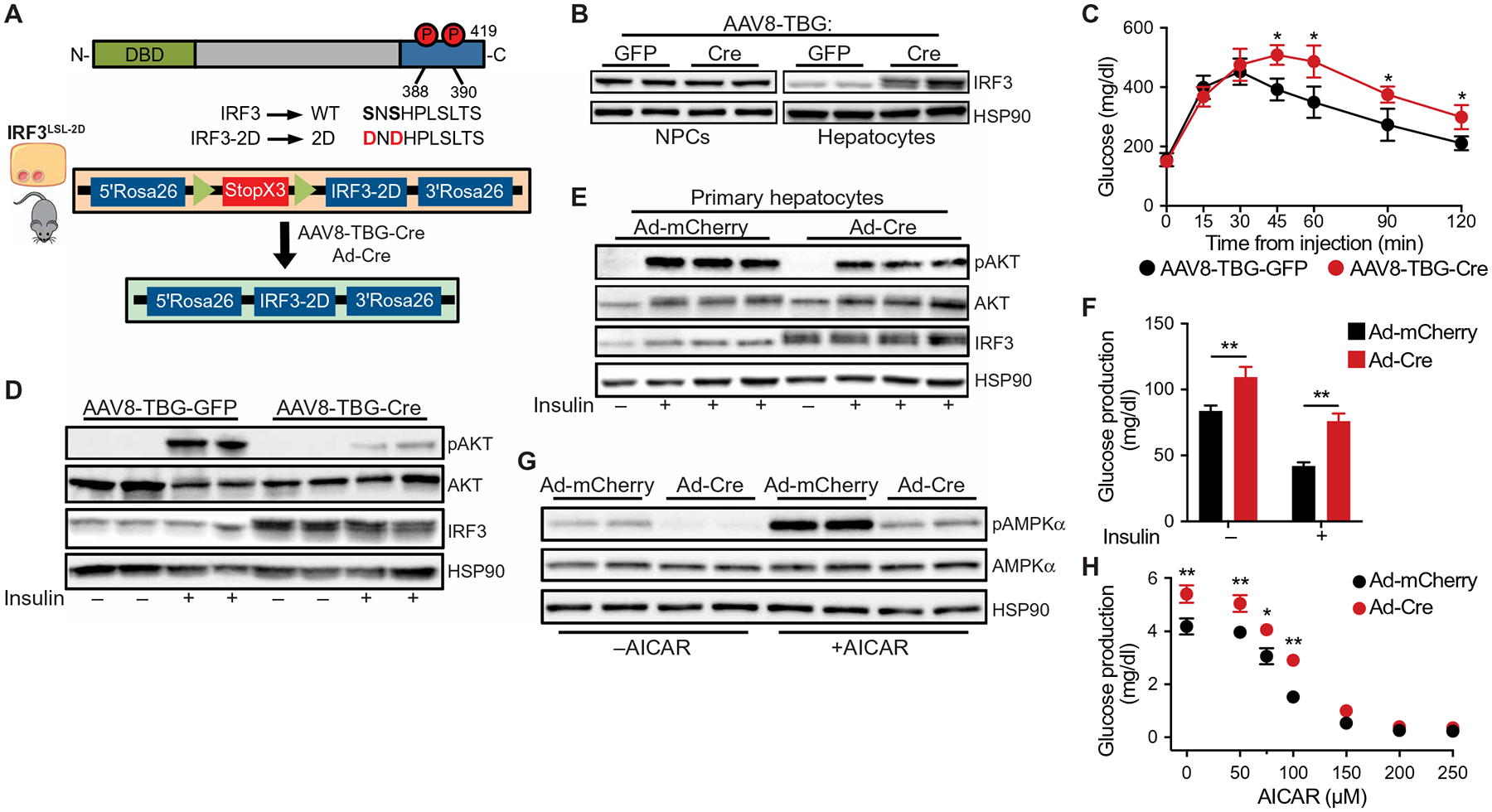
(A) Schematic of IRF3-2D allele and AAV8-TBG-Cre infection strategy. (B) Immunoblot showing IRF3 in liver NPCs and hepatocytes isolated from 6-week-old male IRF3LSL-2D mice tail vein–injected with AAV8-TBG-GFP or AAV8-TBG-Cre. (C) Glucose tolerance test (1 g/kg) in male IRF3LSL-2D mice fed HFD for 10 weeks with tail vein injection of AAV8-TBG-GFP or AAV8-TBG-Cre on week 8 of HFD (n = 6 to 7). (D) Immunoblot showing insulin-stimulated pAKT (Ser473), total AKT, and IRF3 in liver tissue from 10-week-old male IRF3LSL-2D mice on SCD, tail vein–injected with AAV8-TBG-GFP or AAV8-TBG-Cre 2 weeks prior. Mice were fasted for 5 hours, injected intraperitoneally with insulin (5 U/kg), and euthanized 5 min later. Liver tissue was harvested within 90 s. (E) Immunoblot showing insulin-stimulated pAKT (Ser473), total AKT, and IRF3 in hepatocytes isolated from 6-week-old male IRF3LSL-2D mice and transduced ex vivo with adenovirus expressing Cre recombinase (Ad-Cre) or mCherry (Ad-mCherry). Hepatocytes were stimulated with 5 nM insulin in serum-free William’s E medium for 5 min. (F) Glucose production in hepatocytes isolated from 6-week-old male IRF3LSL-2D mice, transduced ex vivo with Ad-Cre or Ad-mCherry, and treated with 8-CPT–cyclic AMP in the presence or absence of insulin (5 nM; n = 4). (G) Immunoblot showing AICAR-stimulated pAMPKα (Thr172) and total AMPKα in hepatocytes isolated from 6-week-old male IRF3LSL-2D mice and transduced ex vivo with Ad-Cre or Ad-mCherry. Hepatocytes were stimulated with 100 μM AICAR in glucose production media for 6 hours. (H) Glucose production in hepatocytes isolated from 6-week-old male IRF3LSL-2D mice, transduced ex vivo with Ad-Cre or Ad-mCherry, and treated with various dosing of AICAR for 6 hours in glucose production media (n = 4). Data are presented as means ± SD; *P < 0.05, **P < 0.01.
We next asked whether IRF3 activation promotes AMPKα phosphorylation independent of insulin signaling. For these studies, we first utilized 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), a known activator of AMPKα and suppressor of gluconeogenesis in hepatocytes, albeit independently of AMPKα (52). AICAR stimulation of hepatocytes induced AMPKα phosphorylation in a dose-dependent fashion (fig. S3D). However, when IRF3LSL-2D hepatocytes were transduced ex vivo with Ad-Cre, the resulting IRF3-2D expression decreased AMPKα phosphorylation, even in the presence of AICAR (Fig. 3G). Concomitantly, IRF3-2D increased basal glucose production and further inhibited the ability of AICAR to suppress glucose production (Fig. 3H).
There are, however, critical limitations in using AICAR as an AMPKα activator for suppressing hepatic glucose production. Namely, AICAR has been shown to decrease glucose production independently of AMPKα (53, 54). Thus, we used mutant AMPK alleles to directly modulate AMPK activity, in the setting of IRF3 activation, and study its effects on glucose production. Primary hepatocytes were isolated from IRF3LSL-2D mice, transfected with either a control plasmid expressing GFP (pGFP) or a plasmid expressing constitutively active AMPKα (pAMPK-Δ312X; fig. S3E) (55). Hepatocytes were then transduced with Ad-Cre to activate IRF3-2D. We found that expressing constitutively active AMPK prevented the ability of IRF3 to increase glucose production (fig. S3F). We also isolated primary hepatocytes from Irf3flox and LI3KO mice, transfected them with either pGFP or a plasmid expressing a dominant negative allele of AMPKα (pAMPK-K45R) (55). Hepatocytes were then treated with PA to stimulate HFD conditions in vitro. Although IRF3 deficiency decreased glucose production, when the dominant negative AMPK mutant was expressed, no glucose suppression was observed (fig. S3G). These results support the notion that AMPK plays a role in mediating the effects of IRF3 on glucose production in a cell-autonomous manner.
Transcriptionally active IRF3 is necessary for its effects on insulin and AMPK signaling
Although IRF3 is best known as a transcription factor, it has also been shown to have nontranscriptional activity, such as triggering apoptosis by translocating to the mitochondria and activating the proapoptotic factor Bcl-2-associated X (BAX) (56, 57). Thus, we ascertained whether the metabolic actions of IRF3 in hepatocytes require its transcriptional function. The nuclear localization signal for IRF3 contains two clusters of basic amino acids that lie within the N-terminal DNA binding domain. Replacing two of these basic amino acids with neutral amino acids renders IRF3 incapable of nuclear translocation (58). We therefore created an alternative IRF3-2D allele in which Arg86 and Lys87 were mutated to Leu and Gln, respectively (IRF3-2D-ΔNLS; fig. S4A). Hepatocytes transduced with IRF3-2D–expressing adenovirus (Ad-IRF3-2D) showed substantial nuclear localization of IRF3 and increased mRNA expression of ISGs (Fig. 4, A and B, and fig. S4B). In comparison, hepatocytes transduced with IRF3-2D-ΔNLS–expressing adenovirus (Ad-IRF3-2D-ΔNLS) showed substantially less nuclear localization of IRF3 and mRNA expression of ISGs (Fig. 4, A and B, and fig. S4B). As before, expression of IRF3-2D inhibited AKT and AMPKα phosphorylation, but IRF3-2D-ΔNLS had no such effect (Fig. 4C). Furthermore, whereas expression of IRF3-2D increased basal glucose production, expression of IRF3-2D-ΔNLS failed to do so. IRF3-2D-ΔNLS also failed to inhibit AICAR-mediated suppression of glucose production (Fig. 4D). Together, these results demonstrate that the metabolic actions of IRF3 on insulin signaling and glucose production are likely to be mediated by its transcriptional activity.
Fig. 4. Transcriptionally active IRF3 is necessary for its effects on hepatic insulin and AMPK signaling.
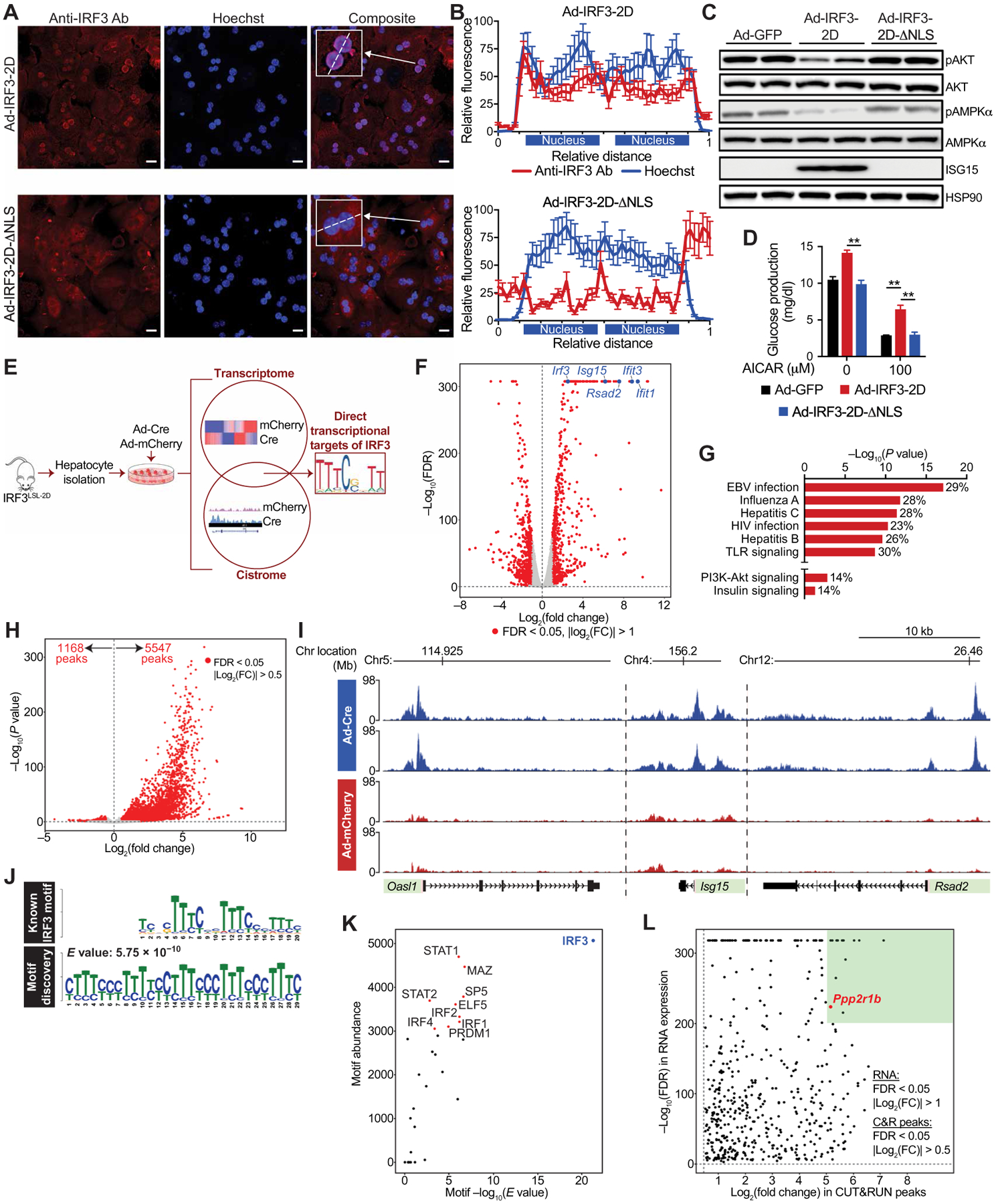
(A) Representative confocal fluorescence images of WT primary hepatocytes transduced ex vivo with adenovirus expressing IRF3-2D (Ad-IRF3-2D) or IRF3-2D-ΔNLS (Ad-IRF3-2D-ΔNLS). Scale bars, 15 μm. Cells were stained with anti-IRF3 antibody (red) and Hoechst nuclear stain (blue), and images were overlaid (right). (B) Quantification of anti-IRF3 antibody and Hoechst stain [inset in (A)] along the line scan (dashed line) at the level of each binucleated hepatocyte. The x axis shows relative distance along the line scan, and the y axis shows arbitrary fluorescence units. Each nucleus along the line scan is labeled on the x axis. (C) Immunoblot showing pAKT (Ser473), total AKT, pAMPKα (Thr172), total AMPKα, and ISG15 in WT primary hepatocytes transduced ex vivo with Ad-GFP, Ad-IRF3-2D, or Ad-IRF3-2D-ΔNLS. (D) Glucose production in Ad-GFP–, Ad-IRF3-2D–, and Ad-IRF3-2D-ΔNLS–transduced hepatocytes treated with AICAR for 6 hours in glucose production media (n = 4). (E) Experimental strategy to identify to complete IRF3 transcriptome and cistrome in hepatocytes. Hepatocytes from 6-week-old male IRF3LSL-2D mice were isolated and transduced ex vivo with Ad-Cre to activate the IRF3-2D allele or Ad-mCherry as a control. (F) Volcano plot of RNA-seq expression data from Ad-Cre– and Ad-mCherry–transduced hepatocytes. Statistically significant [false discovery rate (FDR) < 0.05] differentially expressed genes are shown in red. Several highly differentially expressed ISGs and Irf3 are shown in blue. (G) Kyoto Encyclopedia of Genes and Genomes pathway analysis of RNA-seq data from Ad-Cre– and Ad-mCherry–transduced hepatocytes. Percentage represents percent of affected genes within that pathway. (H) Volcano plot of IRF3 CUT&RUN peaks from Ad-Cre– and Ad-mCherry–transduced hepatocytes. Statistically significant (FDR < 0.05) differentially expressed genes are shown in red. Fold change is represented as Ad-Cre/Ad-mCherry. (I) IRF3 CUT&RUN profiles near the transcription start sites (pink) of several ISGs. Each track displays a different sample (n = 2 for Ad-Cre and n = 2 for Ad-mCherry). The x axis represents the genomic distance, and the y axis represents the relative alignment units. (J) The top motif found by de novo motif discovery analysis of IRF3 CUT&RUN up-regulated peaks (bottom) and the known IRF3 binding sequence (top). Motifs are shown as position weight matrices. E value is shown as reported by MEME. (K) Scatterplot of all transcription factor–matched motifs within the IRF3 CUT&RUN up-regulated peaks. The x axis indicates motif enrichment, and the y axis indicates motif abundance. Top 10 transcription factor–matched motifs within the IRF3 CUT&RUN up-regulated peaks are annotated. (L) Scatterplot of peak-gene associations between IRF3 motif–containing up-regulated CUT&RUN peaks and potentially nearby regulated genes. The x axis indicates fold change in IRF3 CUT&RUN peak expression, and the y axis indicates the FDR of RNA-seq differential gene expression analysis (Ad-Cre/Ad-mCherry). Peak-gene associations with the highest fold change in peak expression (log2FC > 5) and most significant change in gene expression (−log10FDR > 200) are in the green region. Ppp2r1b is annotated in red. Data are presented as means ± SD; **P < 0.01.
To determine which downstream targets of IRF3 mediate its metabolic actions, we sought to define the complete IRF3-dependent transcriptome and cistrome in hepatocytes. Hepatocytes were isolated from IRF3LSL-2D mice and transduced ex vivo with Ad-Cre to activate expression of IRF3-2D (versus Ad-mCherry–transduced cells as a control; Fig. 4E). We conducted genome-wide expression profiling of Ad-Cre– and Ad-mCherry–transduced hepatocytes by RNA sequencing (RNA-seq) and identified 1301 differentially expressed genes (Fig. 4F and fig. S4, C and D). Of these, 583 were up-regulated and 718 were down-regulated. As expected, canonical ISGs such as Isg15, Rsad2, and Ifit3 were among the most highly induced genes (Fig. 4F). Gene set enrichment analysis of up-regulated genes revealed that pathways involved in the antiviral and innate immune response were the most enriched, again consistent with the known function of IRF3 (Fig. 4G). Of particular interest, however, multiple metabolic pathways were also up-regulated, including the PI3K (phosphatidylinositol 3-kinase)–AKT and insulin signaling pathways (Fig. 4G).
IRF3-dependent gene expression changes can be direct (due to binding and transactivation of IRF3 at the locus in question) or indirect (due to a secondary effect of IRF3, such as induced by cytokines that are themselves direct targets of IRF3). To determine which of the observed gene expression changes were due to the direct transcriptional actions of IRF3, we characterized the IRF3 cistrome in hepatocytes using an epigenomic technique called Cleavage Under Targets and Release Using Nuclease (CUT&RUN), which, similar to ChIP-seq, enables mapping of transcription factor–chromatin interactions (59, 60). IRF3 CUT&RUN was performed on IRF3LSL-2D hepatocytes transduced with Ad-Cre to activate IRF3-2D; IRF3LSL-2D hepatocytes transduced with Ad-mCherry were again used as negative controls. A total of 6715 differential peaks were called, of which 83% were up-regulated in Ad-Cre–transduced hepatocytes (Fig. 4H and fig. S4E). Strong IRF3 peaks were observed in the promoter regions of numerous ISGs, including Isg15, Ifit3, and Oasl1 (Fig. 4I). Analysis of genomic peak distribution showed that peaks with an IRF3 motif were most abundant within promoters (25%), introns (35%), and distal intergenic regions (34%; fig. S4F). Unbiased de novo motif discovery of IRF3 CUT&RUN peaks yielded the known IRF3 recognition sequence as the best match with an E value of 5.75 × 10−10 (Fig. 4J). Closely related motifs for STAT1 (signal transducers and activators of transcription 1) and other IRF family members were also identified but were much less enriched and less abundant than the IRF3 motif itself (Fig. 4K).
We next used genomic distance to transcription start sites to form unique peak-gene associations between IRF3 motif-containing peaks and potentially regulated genes. Assuming that functionally important peak-gene associations must trigger increases in gene expression, we integrated these data with our RNA-seq dataset and compiled a list of direct transcriptional targets of IRF3 in hepatocytes (data file S1). Peak-gene associations with the highest fold change in peak expression (log2FC > 5) and most significant change in gene expression (−log10FDR > 200) were prioritized (Fig. 4L).
PPP2R1B is a transcriptional target of IRF3 and associates with dysglycemia
Integration of the IRF3 transcriptome and cistrome datasets revealed multiple potential mediators of IRF3’s metabolic action in hepatocytes. Among the top hits, we were immediately drawn to the Protein Phosphatase 2 Scaffold Subunit A beta (Ppp2r1b) gene, which encodes a subunit of the heterotrimeric serine/threonine protein phosphatase 2 (PP2A). PP2A is a negative regulator of both the AKT and AMPK signaling pathways (61, 62), which we showed are markedly affected by IRF3 activation. PP2A consists of a dimeric core enzyme composed of the structural A and catalytic C subunits, along with a regulatory B subunit (63). The evolutionarily conserved A subunit is the scaffold required for formation of the active complex. In humans and mice, structural subunit A is encoded by two different genes, Ppp2r1a and Ppp2r1b (64). Whereas PPP2R1A is expressed throughout the human body, PPP2R1B expression is largely restricted to the liver (fig. S5A). We found strong intronic IRF3 CUT&RUN peaks within the Ppp2r1b gene in Ad-Cre–transduced IRF3LSL-2D hepatocytes, showing that, when IRF3 is activated, it binds to chromatin within the Ppp2r1b gene (Fig. 5A). Concordant with the binding data, Ppp2r1b expression was substantially up-regulated in Ad-Cre–transduced hepatocytes, both at the transcript and protein levels (Fig. 5, B and C). This up-regulation was not seen when the transcriptionally silent IRF3-2D-ΔNLS allele was overexpressed (Fig. 5D). Furthermore, Ad-Cre–transduced IRF3LSL-2D hepatocytes demonstrated substantially increased PP2A phosphatase activity toward an independent substrate (Fig. 5E). Conversely, hepatocytes isolated from LI3KO mice showed decreased PP2A phosphatase activity compared to WT hepatocytes from Irf3flox mice (Fig. 5F).
Fig. 5. PPP2R1B is a transcriptional target of IRF3 and associates with dysglycemia.
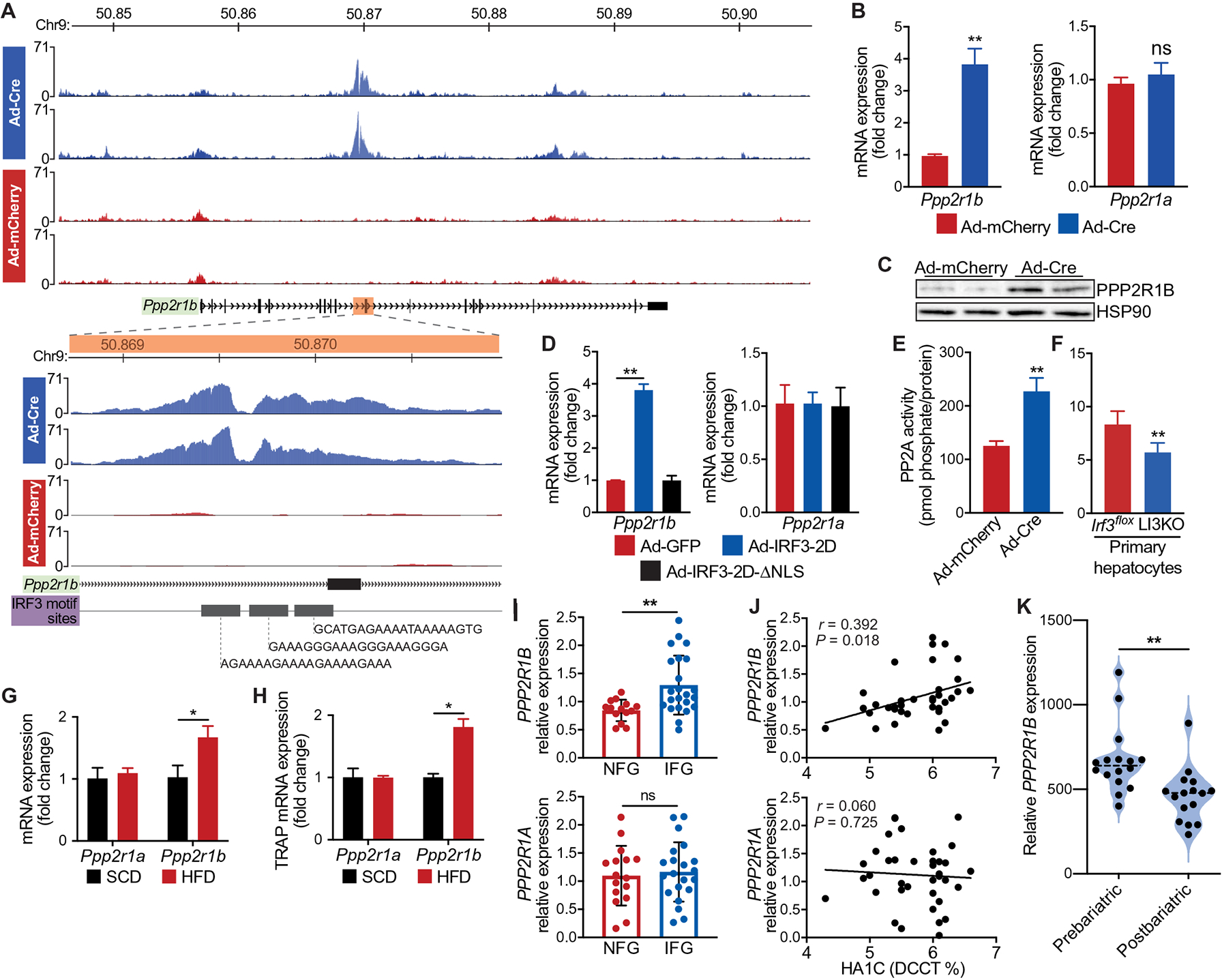
Hepatocytes from 6-week-old male IRF3LSL-2D mice were isolated, transduced ex vivo with Ad-Cre to activate IRF3-2D or Ad-mCherry as a control, and then subjected to IRF3 CUT&RUN. (A) IRF3 CUT&RUN peaks near Ppp2r1b. Each track displays a different sample (n = 2 for Ad-Cre and Ad-mCherry). The x axis represents the genomic distance, and the y axis represents the relative alignment units. The inset shows a zoomed-in view of each track, centered on the intronic Ppp2r1b peaks. (B) Expression of Ppp2r1b and Ppp2r1a in IRF3LSL-2D hepatocytes transduced ex vivo with Ad-Cre or Ad-mCherry (n = 4). (C) Immunoblot showing PPP2R1B in IRF3LSL-2D hepatocytes transduced ex vivo with Ad-Cre or Ad-mCherry (n = 4). (D) Expression of Ppp2r1b and Ppp2r1a in WT hepatocytes transduced ex vivo with adenovirus expressing GFP (Ad-GFP), IRF3-2D (Ad-IRF3-2D), or IRF3-2D-ΔNLS (Ad-IRF3-2D-ΔNLS) (n = 4). (E) PP2A phosphatase activity in IRF3LSL-2D hepatocytes transduced ex vivo with Ad-Cre or Ad-mCherry (n = 4). (F) PP2A phosphatase activity in LI3KO and Irf3flox hepatocytes (n = 6). (G) Expression of Ppp2r1b and Ppp2r1a in liver tissue from male mice fed HFD for 18 weeks (n = 8). (H) Expression of hepatocyte Ppp2r1b and Ppp2r1a in TRAP liver tissue mRNA from male NuTRAPalb mice fed SCD or HFD for 18 weeks (n = 4). (I) Expression of PPP2R1B and PPP2R1A in liver tissue from obese humans with fatty liver disease, with normal (NFG) or impaired (IFG) fasting glucose (n = 14 to 23). (J) Association between HgbA1C and hepatic PPP2R1B or PPP2R1A expression in obese humans with fatty liver disease (n = 36 to 37). (K) Liver microarray data from a cohort of patients with NAFLD and diabetes (n = 16) who underwent weight loss surgery and had a liver biopsy taken at the time of surgery and then taken again 5 to 9 months later. Relative expression of hepatic PPP2R1B before (prebariatric) and after (postbariatric) weight loss surgery. Data are presented as means ± SD; *P < 0.05, **P < 0.01. ns, not significant.
In agreement with these findings, we found increased hepatic expression of Ppp2r1b in HFD-fed mice (Fig. 5G). Using NuTRAPalb, we confirmed that this diet-induced increase in Ppp2r1b expression occurred specifically within hepatocytes (Fig. 5H). We next sought to determine whether hepatic PPP2R1B expression was elevated in obese humans with fatty liver disease (fig. S5B). In liver tissue, PPP2R1B mRNA was correlated with increasing hemoglobin A1c (HgbA1C) and elevated in patients with impaired fasting glucose (Fig. 5, I and J). No such trend was observed with PPP2R1A mRNA (Fig. 5, I and J). Furthermore, we analyzed liver biopsy microarray data from a cohort of patients with NAFLD and diabetes who underwent weight loss surgery (fig. S5C). This cohort consisted of 16 patients who had a liver biopsy taken at the time of weight loss surgery and then taken again 5 to 9 months later. Concurrent with an improvement in glycemic control, we found that patients had decreased hepatic expression of PPP2R1B after weight loss surgery relative to preoperative amounts (Fig. 5K). Together, these data demonstrate that liver PPP2R1B expression associates with poor glycemic control in patients with NAFLD and that IRF3 is a transcriptional regulator of Ppp2r1b expression.
The metabolic actions of hepatocyte IRF3 are mediated by PPP2R1B
We next asked whether Ppp2r1b contributes causally to the metabolic actions of IRF3 by knocking down Ppp2r1b expression in hepatocytes (fig. S6, A and B). IRF3LSL-2D hepatocytes were transduced ex vivo with Ad-Cre to activate IRF3-2D, in the presence of Ppp2r1b or control small interfering RNA (siRNA). When Ppp2r1b expression was inhibited by siRNA, IRF3-2D failed to suppress AKT and AMPKα phosphorylation (Fig. 6A). As before, expression of IRF3-2D substantially increased basal glucose production, whereas knockdown of Ppp2r1b prevented this, and also blocked the ability of IRF3-2D to inhibit AICAR-mediated suppression of glucose production in hepatocytes (Fig. 6B). Concordantly, overexpression of Ppp2r1b in LI3KO hepatocytes resulted in decreased AMPKα phosphorylation and increased basal glucose production (Fig. 6, C and D). We then used AAV8 expressing Ppp2r1b under the control of hepatocyte-specific TBG promoter (AAV8-TBG-Ppp2r1b) to selectively express Ppp2r1b in hepatocytes in vivo (fig. S6C). LI3KO mice placed on HFD and then injected with AAV8-TBG-Ppp2r1b showed worsened glucose and insulin tolerance compared to those injected with AAV8-TBG-GFP (Fig. 6, E and F, and fig. S6, D and E). Furthermore, whole liver lysate from HFD-fed LI3KO mice injected with AAV8-TBG-Ppp2r1b showed decreased insulin-stimulated AKT and AMPKα phosphorylation (Fig. 6, G and H). Together, these data demonstrate that the metabolic effects of IRF3 activation on glucose homeostasis are mediated by its transcriptional action on Ppp2r1b and PP2A activity.
Fig. 6. The metabolic actions of hepatocyte IRF3 are mediated by PPP2R1B.
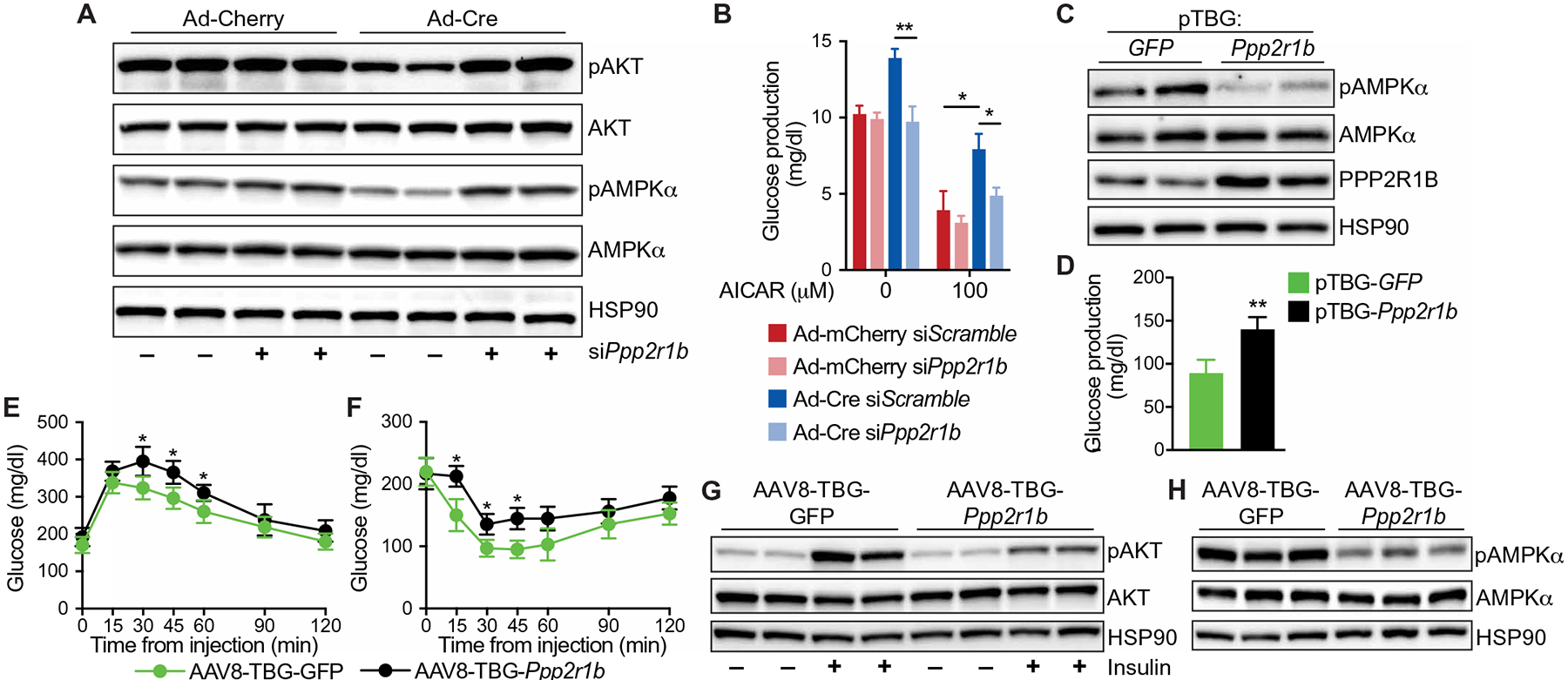
(A) Immunoblot showing pAKT (Ser473), total AKT, pAMPKα (Thr172), and total AMPKα in IRF3LSL-2D hepatocytes transduced ex vivo with Ad-Cre or Ad-mCherry and treated with scramble siRNA (siScramble) or siRNA targeting Ppp2r1b (siPpp2r1b). (B) Glucose production in IRF3LSL-2D hepatocytes transduced ex vivo with Ad-Cre or Ad-mCherry and treated with siScramble or siPpp2r1b for 24 hours, followed by AICAR stimulation for 6 hours in glucose production media (n = 4). (C) Immunoblot showing pAMPKα (Thr172), total AMPKα, and PPP2R1B in LI3KO hepatocytes transfected with a plasmid expressing Ppp2r1b (pTBG-Ppp2r1b) or GFP (pTBG-GFP) under the control of thyroxine-binding globulin (TBG) promoter. (D) Glucose production in LI3KO hepatocytes transfected with pTBG-Ppp2r1b or pTBG-GFP for 24 hours (n = 4). (E to H) Six-week-old male LI3KO mice were placed on HFD for 12 weeks and then tail vein–injected with AAV8 expressing Ppp2r1b (AAV8-TBG-Ppp2r1b) or GFP (AAV8-TBG-GFP) under the control of the TBG promoter while remaining on HFD. (E) Glucose (1.25 g/kg) and (F) insulin (1.25 U/kg) tolerance tests performed on LI3KO mice injected with AAV8-TBG-Ppp2r1b or AAV8-TBG-GFP after 14 and 15 weeks on HFD, respectively (n = 6). (G) Immunoblot showing insulin-stimulated pAKT (Ser473) and total AKT in liver tissue from LI3KO mice fed HFD for 16 weeks and injected with AAV8-TBG-Ppp2r1b or AAV8-TBG-GFP 4 weeks prior. Mice were fasted for 5 hours, injected intraperitoneally with insulin (5 U/kg), and then euthanized 5 min later. Liver tissue was harvested and frozen within 90 s. (H) Immunoblot showing pAMPKα (Thr172) and total AMPKα in liver tissue from LI3KO mice fed HFD for 16 weeks and injected with AAV8-TBG-Ppp2r1b or AAV8-TBG-GFP 4 weeks prior. Mice were fasted for 5 hours before euthanasia. Data are presented as means ± SD; *P < 0.05, **P < 0.01.
Acute inhibition of IRF3 reverses obesity-induced glucose dysregulation
To evaluate the therapeutic potential of our findings, we treated 10-week HFD-fed WT mice with antisense oligonucleotides targeting IRF3 (IRF3 ASO; Fig. 7A). IRF3 ASO treatment had no effect on the expression of other IRFs, with the exception of IRF7, a known transcriptional target of IRF3 (fig. S7A) (65). However, IRF3 ASO treatment abrogated IRF3 expression in the liver, specifically in hepatocytes as compared to liver NPCs (Fig. 7, B to D). Irf3 expression in other tissues, such as muscle and white adipose tissue, was not altered with ASO treatment (fig. S7B). IRF3 ASO treatment reduced liver ISG expression, including Ppp2r1b, and improved HFD-induced glucose, insulin, and pyruvate intolerance without affecting body weight (Fig. 7, E to H, and fig. S7, C to F). Similar to the phenotype observed in HFD-fed LI3KO mice, IRF3 ASO treatment had no effect on HFD-induced steatosis (Fig. 7, I and J). However, we found that IRF3 ASO treatment led to substantially decreased HFD-induced liver injury, as estimated by serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Fig. 7K). Together, these data demonstrate that transient inhibition of the IRF3 response in hepatocytes, initiated after HFD-induced metabolic derangements, can improve glucose homeostasis, reverse insulin resistance, and protect against liver injury.
Fig. 7. Acute inhibition of IRF3 reverses obesity-induced glucose dysregulation.
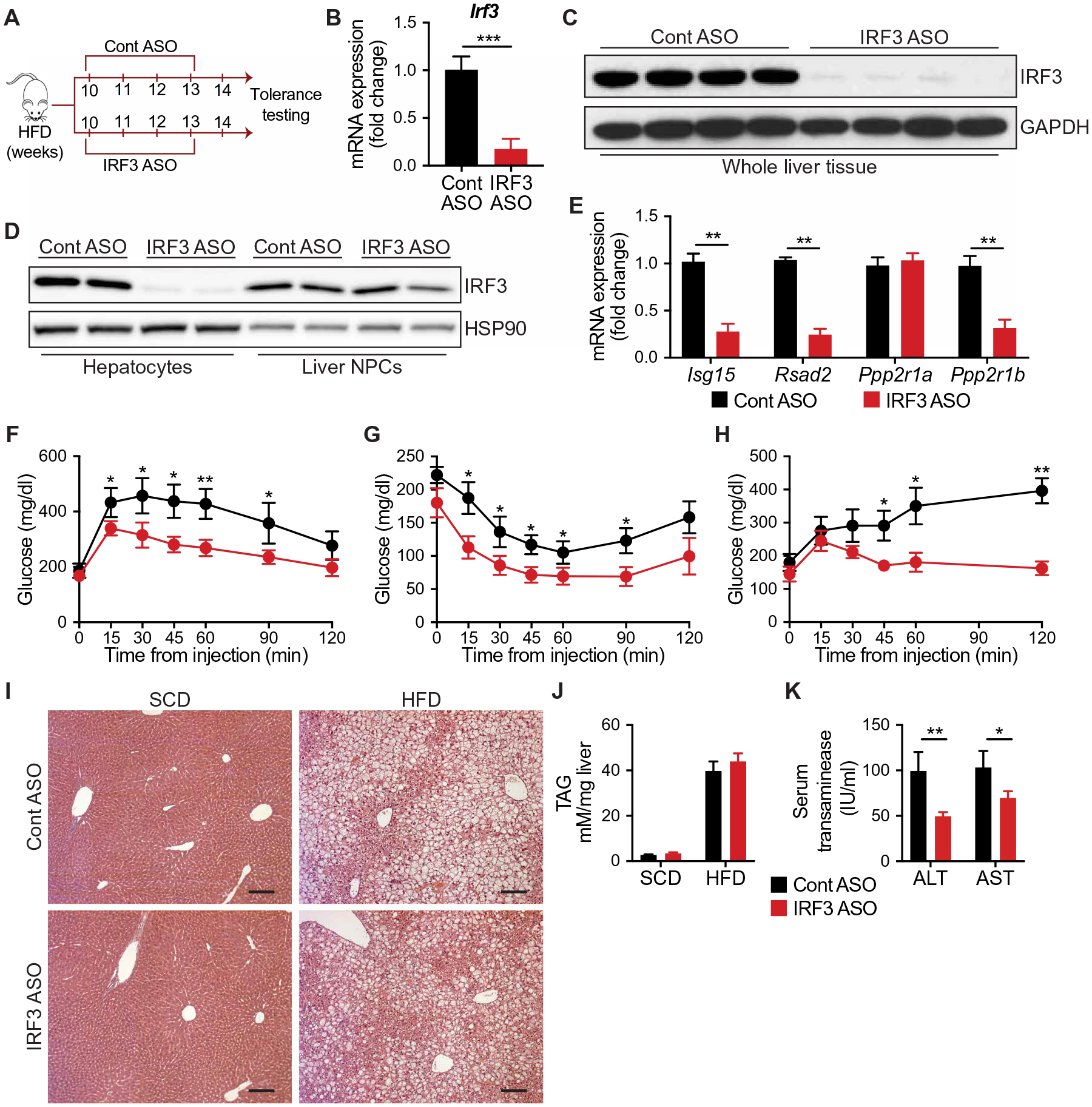
(A) Experimental strategy for antisense oligonucleotide (ASO) therapy. Six-week-old male WT mice were fed HFD for 10 weeks and then randomized to receive four weekly intraperitoneal injections of ASOs targeting IRF3 (IRF3 ASO) or scramble ASO (Cont ASO). Mice were evaluated after a total of 14 weeks of HFD. (B) Expression of Irf3 in liver tissue from Cont ASO– and IRF3 ASO–treated mice (n = 5). (C) Immunoblot showing IRF3 in liver tissue from Cont ASO– and IRF3 ASO–treated mice. (D) Immunoblot showing IRF3 in hepatocytes and liver NPCs isolated from Cont ASO– and IRF3 ASO–treated mice. (E) Expression of Isg15, Rsad2, Ppp2r1a, and Ppp2r1b in liver tissue from HFD-fed mice treated with Cont ASO or IRF3 ASO (n = 4). (F) Glucose (1 g/kg), (G) insulin (1.75 U/kg), and (H) pyruvate (1.25 g/kg) tolerance tests after 14, 15, and 16 weeks on HFD in Cont ASO– or IRF3 ASO–treated mice, respectively (n = 5 to 6). (I) H&E staining of liver tissue from HFD-fed mice treated with Cont ASO or IRF3 ASO. Scale bars, 100 μm. (J) Liver TAG measurement and (K) serum transaminase in HFD-fed mice treated with Cont ASO or IRF3 ASO (n = 6). Data are presented as means ± SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
DISCUSSION
The liver is crucial for maintaining normal glucose homeostasis, contributing more than 80% of endogenous glucose production and serving as a major reservoir of carbohydrate in the form of glycogen (66). This careful balance between glucose storage, production, and release becomes deranged during obesity, due, at least in part, to chronic inflammation in the liver. The transcriptional basis for this response is often attributed to factors such as NF-κB, but their precise role in driving metabolic dysfunction remains unknown. Here, we describe an IRF3-PPP2R1B axis that regulates hepatic glucose metabolism in obesity. The influence of IRF3 on glycemia is not mediated by ill-defined effects on cytokine release, as is commonly supposed, but rather by the direct transcriptional induction of Ppp2r1b, a component of PP2A.
PP2A is one of the most abundant serine/threonine phosphatases in eukaryotic cells and plays an important role in the regulation of many proteins, including metabolic enzymes, hormone receptors, kinase cascades, and growth factors (67). In the liver, PP2A is involved in glucose transport, insulin signaling, triglyceride export, and gluconeogenesis. Hepatocyte-specific ablation of Ppp2ca, the catalytic subunit of PP2A, results in increased insulin-stimulated AKT, FOXO1, and ERK phospho rylation and improves glucose tolerance and insulin sensitivity (68, 69). Inactivation of Ppp2r5c, a regulatory subunit of PP2A, in hepatocytes leads to increased glucose uptake and de novo lipogenesis via regulation of AMPK and SREBP-1 (70). Furthermore, the action of ceramides on hepatic insulin signaling has been suggested to involve PP2A-mediated dephosphorylation of AKT, a pathway referred to as ceramide-activated protein phosphatase (71). In agreement with these prior findings, our work demonstrates the causal role that Ppp2r1b, a structural subunit of PP2A, plays in mediating the metabolic actions of IRF3 on hepatic glucose metabolism during obesity. Whereas most PP2A subunits are ubiquitously expressed, Ppp2r1b is preferentially expressed in the liver (72), suggesting that its role in triggering inflammation-induced metabolic dysfunction might be liver specific.
PP2A controls the phosphorylation status of a wide range of proteins. Although our data show that the IRF3-PPP2R1B axis regulates hepatic glucose production, the exact contributions of AMPK, AKT, or other yet unknown proteins to this regulatory axis remain unknown. The role of AMPK in hepatic glucose metabolism is controversial and complex. There are multiple lines of evidence suggesting that AMPK is unlikely to directly inhibit glucose production (53, 54). Conversely, there are several studies that suggest the opposite, that AMPK activation suppresses hepatic glucose production (45, 46, 49). Our data show that AMPK is a part of the mechanism connecting IRF3 activation to changes in glucose metabolism, but this is undoubtedly not the only mechanism. As a key regulator of cellular energy status, AMPK is activated by phosphorylation when the cellular AMP/ATP ratio is increased (73). In obese diabetic mice and humans, the hepatic energy state has been reported to be attenuated, suggesting that high AMP/ATP ratios are part of the metabolic disease milieu (49, 74). This reduced energetic state would be predicted to enhance activation of AMPK. However, multiple studies have shown that despite low ATP, phosphorylation of hepatic AMPKα is inhibited in obesity, contributing to unrestrained gluconeogenesis in the setting of hyperglycemia (75, 76). The mechanistic basis for this paradoxical finding has been unclear. Our work reveals a direct, unexpected link between inflammation and inhibition of AMPK activity.
In addition to dysregulated glucose homeostasis, steatosis, is a hallmark of NAFLD and obesity. The relationship between steatosis and insulin resistance is complex and incompletely understood. Although some studies suggest that systemic insulin resistance causes hepatic lipid accumulation via an Srebp1c-mediated pathway, others propose that causality proceeds in the opposite direction through the actions of bioactive lipid species such as ceramides and DAGs (77–80). The controversy centers on the idea that, during the course of obesity, the liver displays selective insulin resistance, where insulin fails to inhibit hepatic gluconeogenesis but continues to promote de novo lipogenesis (81). Here, we show that whole body deficiency in IRF3 protects against both HFD-induced steatosis and dysregulation in glucose homeostasis, but hepatocyte-specific ablation of IRF3 protects against only the latter. Thus, loss of hepatocyte IRF3 appears to uncouple hepatic insulin sensitivity and gluconeogenesis from steatosis. This begs the question, then, what cell type is IRF3 acting through to control steatosis? There is a small but burgeoning literature supporting the role of Kupffer cells (KCs) in promoting the development of steatosis during HFD feeding (36, 82–84). KCs are self-renewing liver-resident macrophages that originate from yolk sac–derived progenitor cells during embryonic development (82). Activation of KCs occurs during obesity and precedes the development of NAFLD, and depletion of KCs protects against steatosis (85, 86). How KCs exert their prosteatotic effects remains unclear. Initial work suggested a role for KC-mediated secretion of proinflammatory cytokines and chemokines such as TNFα, IL-1β, and MCP1 (84, 87, 88). However, anti-inflammatory strategies have demonstrated modest effects on obesity-induced dysglycemia and steatosis (15, 16, 89). A more recent study suggested that noninflammatory secretory products from KCs might be involved (83). It remains to be seen whether IRF3 activation in KCs plays a causal role in the development of HFD-induced steatosis and through which mediators.
It is noteworthy that LI3KO mice were not protected from steatosis, because the canonical function of AMPK is phosphorylation of ACC, which inhibits the production of malonyl-CoA, the first committed substrate for de novo lipogenesis and an inhibitor of fatty acid oxidation (90). Although numerous studies have shown that this results in decreased triglyceride accumulation in hepatocytes, the precise role of AMPK and ACC in steatosis remains controversial (91–94). One study showed that phosphorylation of ACC by AMPK is not required for regulation of fatty acid oxidation and another demonstrated that viral mediated overexpression of constitutively active AMPK in the liver actually lead to increased steatosis (46, 95). More recently, work from the Carling laboratory revealed that liver-specific AMPK activation had no effect on HFD-induced steatosis but protected against steatosis only when mice were fed a high-fructose diet (96). Their findings indicate that the predominant effect of AMPK activation on lipids in the liver is suppression of de novo lipogenesis, which is not a major driver of steatosis on HFD. Thus, the effect of AMPK activation on steatosis in experimental models may only be revealed under certain conditions, such as after a high-fructose diet. Our data are in agreement with this notion because the diet we used contains 60% fat without fructose. The reduced steatosis we observed in the HFD-fed IRF3KO mice was likely independent of AMPK signaling in hepatocytes.
Numerous studies have shown that activation of AMPK inhibits inflammation. AMPK signaling can inhibit the inflammatory response induced by NF-κB through several downstream targets such as SIRT1, FOXO, and PGC-1α (97–99). In particular, activation of AMPK in macrophages was demonstrated to decrease proinflammatory cytokine secretion and promote polarization to anti-inflammatory phenotype (100, 101). Furthermore, inactivation of macrophage AMPK has been shown to inhibit insulin signaling and compromise glucose metabolism, suggesting a role for AMPK-mediated suppression of inflammation in promoting insulin sensitivity (102, 103). In our studies, we found that HFD-fed LI3KO mice displayed increased liver AMPK activation and decreased macrophage infiltration. This suggests an additional mechanism by which IRF3-AMPK signaling might indirectly regulate hepatic glucose metabolism and insulin sensitivity in vivo through an inflammatory response. However, our studies in isolated hepatocytes clearly indicate that IRF3 affects glucose production in a cell-autonomous manner.
Innate immunity provides nonspecific defense during the earliest stages of infection. However, it is now well appreciated that innate immune signaling also fuels the metabolic complications of obesity such as NAFLD and insulin resistance (104). Our work shows that obesity activates hepatocyte IRF3 in humans and mice. In the context of HFD feeding, IRF3 can be activated by multiple different damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) that are known to be elevated, including mitochondrial DNA, genomic DNA, free fatty acids, bacterial DNA, and lipopolysaccharide (LPS) (105–107). These ligands bind to specific PRRs such as cGAS, TLR4, and TLR9 triggering IRF3 phosphorylation. In the liver, TLR4, the PRR for LPS, has been shown to regulate obesity-induced inflammation, steatosis, and insulin resistance (106, 108). Similarly, hepatocyte mitochondrial DNA has been demonstrated to promote NAFLD progression and insulin resistance via activation of TLR9 (107, 109). Although there are potentially multiple HFD-induced DAMPs and PAMPs capable of activating IRF3 in hepatocytes, the exact contribution of each remains to be determined.
Multiple studies have investigated the role of the STING/TBK1-IKKε/IRF3 axis in metabolism. Mice deficient in STING demonstrated improved insulin sensitivity and glucose tolerance on HFD (110), with reduced hepatic steatosis, injury, and fibrosis (36). Similarly, TBK1 or IKKε ablation in mice protects against HFD-induced systemic glucose intolerance and insulin resistance, as well as hepatic steatosis (5, 111). In the case of IRF3, the story is more complex. IRF3 deficiency protects against alcohol-induced liver injury and steatosis, as well as HFD-induced insulin resistance (28, 112, 113). Our work with global IRF3-deficient mice corroborates these findings. However, some studies have linked IRF3 deficiency to increased liver injury, steatosis, and insulin resistance in response to HFD (37, 38). The precise rationale for these dichotomous results is unclear but might be related to the lack of appropriate littermate controls in many of the latter studies, as well as using HFDs of different composition. Furthermore, these investigations were all performed with global IRF3 knockout mice. In this work, we showed that IRF3 ablation in hepatocytes specifically protects against HFD-induced glucose mishandling and insulin resistance in the liver, without effects on steatosis or body weight.
Although IRF3 plays a clear role in metabolism, few studies have investigated which targets mediate its metabolic actions. As a transcription factor, IRF3 directly regulates the transcription of a set of inflammatory genes termed ISGs (33). Of these, IFN-α has been shown to promote HFD-induced hepatic steatosis and insulin resistance (114). Another ISG, viperin (RSAD2), was recently shown to regulate adipose tissue thermogenesis and fatty acid β-oxidation during HFD (115). In the context of coxsackievirus infection, ISG15 was demonstrated to reprogram hepatic metabolism by increasing oxidative capacity and gluconeogenesis in hepatocytes (116). However, outside of these IRF3-regulated inflammatory genes, there has never been a comprehensive analysis of all direct IRF3 targets in any cell type. Our transcriptome and cistrome profiling results show that IRF3 directly regulates numerous nonimmunological targets and metabolic pathways in hepatocytes. We identified Ppp2r1b as an IRF3 target gene and that it is responsible for IRF3’s metabolic actions on glucose homeostasis and insulin signaling. By driving the expression of Ppp2r1b, IRF3 increases PP2A activity, resulting in dephosphorylation of key metabolic kinases such as AMPKα and AKT. In the context on obesity, this leads to increased hepatic gluconeogenesis and insulin resistance. Thus, IRF3-PPP2R1B serves as a causal link between obesity-triggered inflammation and metabolic dysfunction. Furthermore, the dephosphorylation of multiple key kinases by PPP2R1B potentially explains how obesity-induced inflammation can fuel such a wide array of metabolic derangements.
There are several limitations to this study. Although we demonstrated that IRF3-mediated expression of PPP2R1B increased hepatic glucose production by dephosphorylating AMPK, it is undoubtedly not the only mechanism. The substrate specificity of PPP2R1B in hepatocytes is unknown. Thus, identifying the complete phosphoproteome in hepatocytes with and without PPP2R1B will be critical to better understanding the exact mechanisms by which the IRF3-PPP2R1B axis controls glucose production. Furthermore, although inhibiting IRF3 activation or PPP2R1B expression reduced hepatic glucose production, we did not observe any effect on HFD-induced steatosis. Further work will be needed to determine whether the present steatosis could trigger fibrosis and worsening of liver disease. Last, although data in humans show that liver PPP2R1B expression associates with obesity and diabetes, additional work is required to determine whether PPP2R1B modulation is a viable therapeutic modality in humans.
In summary, our data suggest an IRF3-centric model for how obesity-induced inflammation regulates hepatic glucose metabolism. At the center of this model lies Ppp2r1b, an IRF3-inducible target that functions as a critical component of the heterotrimeric phosphatase, PP2A. When overexpressed by IRF3 during pathological conditions, such as overnutrition, Ppp2r1b increases PP2A phosphatase activity, which, in turn, has pleiotropic effects on glucose homeostasis and insulin signaling. It dephosphorylates AMPKα, a key cellular energy sensor, and AKT, a critical signaling molecule in insulin signaling. Moreover, in obese individuals with NAFLD, hepatic Ppp2r1b expression tracks with HgbA1C concentrations and is elevated in patients with impaired fasting glucose. Future studies will be required to further elaborate the role of Ppp2r1b and to fully elucidate its target kinases in metabolic disease.
MATERIALS AND METHODS
Study design
The objective of this study was to investigate the contribution of hepatocyte IRF3 to the metabolic dysfunction associated with obesity and NAFLD in humans and mice. To evaluate IRF3 activation in humans, we measured gene expression of IRF3, gene expression of IRF3-regulated genes (ISGs), IRF3 transcriptional activity, and pIRF3 immunohistochemistry in liver tissue. To evaluate IRF3 activation in mice, we measured gene expression of Irf3, gene expression of IRF3-regulated genes (ISGs), protein expression of pIRF3, and nuclear accumulation of IRF3 in liver tissue. Hepatocyte-specific IRF3 loss-of-function and gain-of-function studies were conducted in mice placed on HFD to induce obesity and NAFLD. Metabolic dysfunction was analyzed by fed/fasting serum glucose, fed/fasting serum insulin, glucose tolerance test (GTT), insulin tolerance test (ITT), pyruvate tolerance test (PTT), liver TAG accumulation, in vitro glucose production, and liver expression of pAKT and pAMPKα. To analyze the molecular mechanism responsible for the metabolic actions of IRF3, we performed unbiased cistrome and transcriptome analysis by IRF3 CUT&RUN-seq and RNA-seq, respectively. PPP2R1B loss-of-function and gain-of-function studies were conducted using siRNA, plasmid-mediated overexpression, and viral AAV8-mediated expressing under the control of the hepatocyte-specific TBG promoter. For in vivo studies, the number of mice in each group is indicated in the figure legends. For in vitro studies, primary mouse hepatocytes were isolated, and the number of independent experiments performed is indicated in the figure legends. Power analyses were not used to calculate sample sizes; samples were not excluded, and investigators were not blinded during experiments.
HFD feeding
All animal experiments were performed with approval from the Institutional Animal Care and Use Committees of the Harvard Center for Comparative Medicine and Beth Israel Deaconess Medical Center. Mice were maintained at 12-hour/12-hour light/dark cycles, 23°C room temperature, and 30 to 70% humidity with ad libitum access to food and water in individually ventilated cages. At about 6 weeks of age, male mice were placed on either a SCD [8664 Harlan Teklad; 6.4% (w/w) fat] or a HFD consisting of 20% calories from protein, 60% from fat, and 20% from carbohydrate (Research Diets, D12492) for the indicated durations. Body weight was measured weekly. At the end of experiments, mice were euthanized, and tissues were collected, flash-frozen in liquid nitrogen, and stored at −80°C until use for downstream analysis.
In vivo AAV8 injections
Six-week-old IRF3LSL-2D mice were placed on either SCD or HFD for 6 weeks and then tail vein–injected with 1 × 1011 viral genomes (vg) of AAV8 expressing Cre recombinase or GFP under the control of hepatocyte-specific TBG promoter (AAV8-TBG-Cre and AAV8-TBG-GFP; Addgene). AAV8-infected HFD mice remained on HFD for two more weeks and were then analyzed by GTT. AAV8-infected SCD mice remained on SCD for two more weeks and were then euthanized for tissue harvesting or primary hepatocyte isolation.
Six-week-old LI3KO mice were placed on HFD for 12 weeks and then tail vein–injected with 1 × 1011 vg of AAV8 expressing Ppp2r1b or GFP under the control of hepatocyte-specific TBG promoter (AAV8-TBG-Ppp2r1b and AAV8-TBG-GFP; VectorBuilder, Addgene). AAV8-infected LI3KO mice remained on HFD and were then analyzed by GTT at week 14 and ITT at week 15. They were euthanized for tissue harvesting at week 16.
Statistical analysis
Sample size, mean, and significance P values (P < 0.05) are indicated in the text, figure legends, or Materials and Methods. Error bars in the experiments represent SD from either independent experiments or independent samples. Statistical analyses were performed using GraphPad Prism.
Supplementary Material
Acknowledgments:
We thank the BNORC-supported Functional Genomics and Bioinformatics Core; the Vanderbilt MMPC (DK059637); and the surgery team, bariatric surgeons, and the biobank staff of the IUCPQ. We thank S. Henikoff for providing pA-MNase.
Funding:
This work was supported by NIH K08 DK125762, American Gastroenterological Association Research Scholar Award, MGH Tosteson Fellowship Award, and Gilead Liver Disease Research Scholar Award to S.J.P.; R01 DK085171, R01 DK102173, R01 DK102170, and R01 DK1113669 to E.D.R.; and R01 DK067536 to R.N.K.
Footnotes
Competing interests: A.T. receives funding from Johnson & Johnson, Medtronic, and GI Windows for studies related to bariatric surgery, as well as consulting fees from Bausch Health, Novo Nordisk, and Eli Lilly. The other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scitranslmed.abh3831
Data files S1 and S2
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All raw and processed RNA-seq and IRF3 CUT&RUN data have been deposited in the NCBI Gene Expression Omnibus (GEO) under GEO accession number GSE166369. A list of direct transcriptional targets of IRF3 in hepatocytes is given in data file S1. Raw data from figures are presented in data file S2.
REFERENCES AND NOTES
- 1.Han HS, Kang G, Kim JS, Choi BH, Koo SH, Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med 48, e218(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen MC, Vatner DF, Shulman GI, Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol 13, 572–587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okin D, Medzhitov R, The effect of sustained inflammation on hepatic mevalonate pathway results in hyperglycemia. Cell 165, 343–356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M, IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med 11, 191–198 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, Delproposto JB, Blackwell TS, Yull FE, Saltiel AR, The protein kinase IKKɛ regulates energy balance in obese mice. Cell 138, 961–975 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, Downes M, Yu RT, Liddle C, Evans RM, Oh D, Li P, Olefsky JM, Saltiel AR, An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat. Med 19, 313–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregor MF, Hotamisligil GS, Inflammatory mechanisms in obesity. Annu. Rev. Immunol 29, 415–445 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Osborn O, Olefsky JM, The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med 18, 363–374 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Hotamisligil GS, Shargill NS, Spiegelman BM, Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 259, 87–91 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Lumeng CN, Saltiel AR, Inflammatory links between obesity and metabolic disease. J. Clin. Invest 121, 2111–2117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asghar A, Sheikh N, Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell. Immunol 315, 18–26 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Hotamisligil GS, Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Jager J, Aparicio-Vergara M, Aouadi M, Liver innate immune cells and insulin resistance: The multiple facets of Kupffer cells. J. Intern. Med 280, 209–220 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Esser N, Paquot N, Scheen AJ, Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin. Investig. Drugs 24, 283–307 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, Glynn RJ, Libby P, Ridker PM, Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J. Am. Coll. Cardiol 71, 2392–2401 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R, Effects of an engineered human anti-TNF-α antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45, 881–885 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Wascher TC, Lindeman JHN, Sourij H, Kooistra T, Pacini G, Roden M, Chronic TNF-α neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol. Med 17, 189–193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G, NF-κB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology 129, 1663–1674 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Schreyer SA, Chua SC Jr., Leboeuf RC, Obesity and diabetes in TNF-alpha receptor-deficient mice. J. Clin. Invest 102, 402–411 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wunderlich FT, Ströhle P, Könner AC, Gruber S, Tovar S, Brönneke HS, Juntti-Berggren L, Li LS, Van Rooijen N, Libert C, Berggren PO, Brüning JC, Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 12, 237–249 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Baker RG, Hayden MS, Ghosh S, NF-κB, inflammation, and metabolic disease. Cell Metab. 13, 11–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Catrysse L, van Loo G, Inflammation and the metabolic syndrome: The tissue-specific functions of NF-κB. Trends Cell Biol. 27, 417–429 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE, Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med 11, 183–190 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polkinghorne E, Lau Q, Cooney GJ, Kraegen EW, Cleasby ME, Local activation of the IκK-NF-κB pathway in muscle does not cause insulin resistance. Am. J. Physiol. Endocrinol. Metab 294, E316–E325 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Ke B, Zhao Z, Ye X, Gao Z, Manganiello V, Wu B, Ye J, Inactivation of NF-κB p65 (RelA) in liver improves insulin sensitivity and inhibits cAMP/PKA pathway. Diabetes 64, 3355–3362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Z, Zhang J, Henagan TM, Lee JH, Ye X, Wang H, Ye J, P65 inactivation in adipocytes and macrophages attenuates adipose inflammatory response in lean but not in obese mice. Am. J. Physiol. Endocrinol. Metab 308, E496–E505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Honda K, Taniguchi T, IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol 6, 644–658 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, Tenen D, Roh HC, Kong X, Kazak L, Ahmad R, Rosen ED, IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Invest 126, 2839–2854 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P, Crosstalk between LXR and Toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 12, 805–816 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell RM, Cheng G, Saez E, Miller JF, Tontonoz P, LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119, 299–309 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ, Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, eaaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Lucchini FC, Wueest S, Challa TD, Item F, Modica S, Borsigova M, Haim Y, Wolfrum C, Rudich A, Konrad D, ASK1 inhibits browning of white adipose tissue in obesity. Nat. Commun 11, 1642(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J, Transcriptional profiling of interferon regulatory factor 3 target genes: Direct involvement in the regulation of interferon-stimulated genes. J. Virol 76, 5532–5539 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR, TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell 172, 731–743.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, Liu FQ, Shen YH, Hou XG, Chen L, Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 81, 13–24 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X, STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest 129, 546–555 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanz-Garcia C, McMullen MR, Chattopadhyay S, Roychowdhury S, Sen G, Nagy LE, Nontranscriptional activity of interferon regulatory factor 3 protects mice from high-fat diet-induced liver injury. Hepatol. Commun 3, 1626–1641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T, Gao L, Deng W, Zhang SM, Zhu LH, Guo S, Chen K, Zhang XD, Liu DP, Li H, Interferon regulatory factor 3 constrains IKKβ/NF-κB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 59, 870–885 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Roh HC, Tsai LTY, Lyubetskaya A, Tenen D, Kumari M, Rosen ED, Simultaneous transcriptional and epigenomic profiling from specific cell types within heterogeneous tissues in vivo. Cell Rep. 18, 1048–1061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahrens M, Ammerpohl O, Von Schönfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, Erhart W, Egberts J, Sipos B, Schreiber S, Häsler R, Stickel F, Becker T, Krawczak M, Röcken C, Siebert R, Schafmayer C, Hampe J, DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18, 296–302 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Baselli GA, Dongiovanni P, Rametta R, Meroni M, Pelusi S, Maggioni M, Badiali S, Pingitore P, Maurotti S, Montalcini T, Taliento AE, Prati D, Rossi G, Fracanzani AL, Mancina RM, Romeo S, Valenti L, Liver transcriptomics highlights interleukin-32 as novel NAFLD-related cytokine and candidate biomarker. Gut 69, 1855–1866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Hilda Ye B, Califano A, Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet 48, 838–847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marbach D, Lamparter D, Quon G, Kellis M, Kutalik Z, Bergmann S, Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 13, 366–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samuel VT, Shulman GI, Nonalcoholic fatty liver disease, insulin resistance, and ceramides. N. Engl. J. Med 381, 1866–1869 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B, Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147, 2432–2441 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B, Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54, 1331–1339 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Zhou B, Zhang Y, Li S, Wu L, Fejes-Toth G, Naray-Fejes-Toth A, Soukas AA, Serum- and glucocorticoid-induced kinase drives hepatic insulin resistance by directly inhibiting AMP-activated protein kinase. Cell Rep. 37, 109785(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong YH, Varanasi US, Yang W, Leff T, AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem 278, 27495–27501 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Jing Y, Liu W, Cao H, Zhang D, Yao X, Zhang S, Xia H, Li D, Wang YC, Yan J, Hui L, Ying H, Hepatic p38α regulates gluconeogenesis by suppressing AMPK. J. Hepatol 62, 1319–1327 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Chen X, Chen S, Shen T, Yang W, Chen Q, Zhang P, You Y, Sun X, Xu H, Tang Y, Mi J, Yang Y, Ling W, Adropin regulates hepatic glucose production via PP2A/AMPK pathway in insulin-resistant hepatocytes. FASEB J. 34, 10056–10072 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, Zhang D, Graham MJ, Unterman TG, Shulman GI, Sztalryd C, Bennett MJ, Ahima RS, Birnbaum MJ, Lazar MA, Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med 18, 934–942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corton JM, Gillespie JG, Hawley SA, Hardie DG, 5-Aminoimidazole-4-carboxamide ribonucleoside: A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem 229, 558–565 (1995). [DOI] [PubMed] [Google Scholar]
- 53.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B, Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 120, 2355–2369 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hasenour CM, Ridley DE, Hughey CC, James FD, Donahue EP, Shearer J, Viollet B, Foretz M, Wasserman DH, 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J. Biol. Chem 289, 5950–5959 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mu J, Brozinick JT, Valladares O, Bucan M, Birnbaum MJ, A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7, 1085–1094 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC, Ubiquitination of the transcription factor IRF-3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 44, 1151–1161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanz-Garcia C, Poulsen KL, Bellos D, Wang H, McMullen MR, Li X, Chattopadhyay S, Sen G, Nagy LE, The non-transcriptional activity of IRF3 modulates hepatic immune cell populations in acute-on-chronic ethanol administration in mice. J. Hepatol 70, 974–984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu M, Fang T, Li S, Meng K, Guo D, Bipartite nuclear localization signal controls nuclear import and DNA-binding activity of IFN regulatory factor 3. J. Immunol 195, 289–297 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Skene PJ, Henikoff S, An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, Nakamura Y, Yuan GC, Bauer DE, Xu J, Bulyk ML, Orkin SH, Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173, 430–442.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM, Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol 24, 8778–8789 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yadav H, Devalaraja S, Chung ST, Rane SG, TGF-β1/Smad3 pathway targets PP2A-AMPK-FoxO1 signaling to regulate hepatic gluconeogenesis. J. Biol. Chem 292, 3420–3432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho US, Xu W, Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445, 53–57 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Seshacharyulu P, Pandey P, Datta K, Batra SK, Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 335, 9–18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ning S, Pagano JS, Barber GN, IRF7: Activation, regulation, modification and function. Genes Immun. 12, 399–414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ekberg K, Landau BR, Wajngot A, Chandramouli V, Efendic S, Brunengraber H, Wahren J, Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes 48, 292–298 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Millward TA, Zolnierowicz S, Hemmings BA, Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci 24, 186–191 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Cazzolli R, Carpenter L, Biden TJ, Schmitz-Peiffer C, A role for protein phosphatase 2A-like activity, but not atypical protein kinase Cζ, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes 50, 2210–2218 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Xian L, Hou S, Huang Z, Tang A, Shi P, Wang Q, Song A, Jiang S, Lin Z, Guo S, Gao X, Liver-specific deletion of Ppp2cα enhances glucose metabolism and insulin sensitivity. Aging (Albany NY) 7, 223–232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng YS, Seibert O, Klöting N, Dietrich A, Straßburger K, Fernández-Veledo S, Vendrell JJ, Zorzano A, Blüher M, Herzig S, Berriel Diaz M, Teleman AA, PPP2R5C couples hepatic glucose and lipid homeostasis. PLOS Genet. 11, e1005561(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chavez JA, Summers SA, A ceramide-centric view of insulin resistance. Cell Metab. 15, 585–594 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz-Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, MacArthur D, Kellis M, Thomson A, Young T, Gelfand E, Donovan M, Meng Y, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Im HK, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis ET, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Little AR, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF, The genotype-tissue expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kemp BE, Stapleton D, Campbell DJ, Chen ZP, Murthy S, Walter M, Gupta A, Adams JJ, Katsis F, Van Denderen B, Jennings IG, Iseli T, Michell BJ, Witters LA, in Biochemical Society Transactions (Portland Press Ltd., 2003), vol. 31, pp. 162–168. [DOI] [PubMed] [Google Scholar]
- 74.Nair S, Chacko VP, Arnold C, Diehl AM, Hepatic ATP reserve and efficiency of replenishing: Comparison between obese and nonobese normal individuals. Am. J. Gastroenterol 98, 466–470 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Zhao P, Sun X, Chaggan C, Liao Z, Wong KI, He F, Singh S, Loomba R, Karin M, Witztum JL, Saltiel AR, An AMPK–caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 367, 652–660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steinberg GR, Michell BJ, van Denderen BJW, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Görgün CZ, Carling D, Hotamisligil GS, Febbraio MA, Kay TW, Kemp BE, Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 4, 465–474 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Könner AC, Brüning JC, Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 16, 144–152 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Jornayvaz FR, Shulman GI, Diacylglycerol activation of protein kinase Cε and hepatic insulin resistance. Cell Metab. 15, 574–584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GKK, Birnbaum MJ, Summers SA, Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 5, 167–179 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell 6, 77–86 (2000). [PubMed] [Google Scholar]
- 81.Brown MS, Goldstein JL, Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 7, 95–96 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, Grønbæk H, The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol 16, 145–159 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Morgantini C, Jager J, Li X, Levi L, Azzimato V, Sulen A, Barreby E, Xu C, Tencerova M, Näslund E, Kumar C, Verdeguer F, Straniero S, Hultenby K, Björkström NK, Ellis E, Rydén M, Kutter C, Hurrell T, Lauschke VM, Boucher J, Tomčala A, Krejčová G, Bajgar A, Aouadi M, Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat. Metab 1, 445–459 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JEM, Van Rooijen N, Staels B, Kersten S, Müller M, Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 51, 511–522 (2010). [DOI] [PubMed] [Google Scholar]
- 85.Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, Scott DK, O’Doherty RM, Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 59, 347–357 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Song M, Schuschke DA, Zhou Z, Zhong W, Zhang J, Zhang X, Wang Y, McClain CJ, Kupffer cell depletion protects against the steatosis, but not the liver damage, induced by marginal-copper, high-fructose diet in male rats. Am. J. Physiol. Gastrointest. Liver Physiol 308, G934–G945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS, Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem 287, 40161–40172 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E, Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol 302, G1310–G1321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pollack RM, Donath MY, LeRoith D, Leibowitz G, Anti-inflammatory agents in the treatment of diabetes and its vascular complications. Diabetes Care 39, S244–S252 (2016). [DOI] [PubMed] [Google Scholar]
- 90.Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR, Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab 311, E730–E740 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Garcia D, Hellberg K, Chaix A, Wallace M, Herzig S, Badur MG, Lin T, Shokhirev MN, Pinto AFM, Ross DS, Saghatelian A, Panda S, Dow LE, Metallo CM, Shaw RJ, Genetic liver-specific AMPK activation protects against diet-induced obesity and NAFLD. Cell Rep. 26, 192–208.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, Anagnostopoulos AE, Wang A, Broadfield LA, Ford RJ, Foster RA, Bates J, Sun H, Wang T, Liu H, Ray AS, Saha AK, Greenwood J, Bhat S, Harriman G, Miao W, Rocnik JL, Westlin WF, Muti P, Tsakiridis T, Harwood HJ, Kapeller R, Hoshida Y, Tanabe KK, Steinberg GR, Fuchs BC, Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 29, 174–182.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M, AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28, 194–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Foretz M, Even PC, Viollet B, AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int. J. Mol. Sci 19, 2826(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zordoky BNM, Nagendran J, Pulinilkunnil T, Kienesberger PC, Masson G, Waller TJ, Kemp BE, Steinberg GR, Dyck JRB, AMPK-dependent inhibitory phosphorylation of ACC is not essential for maintaining myocardial fatty acid oxidation. Circ. Res 115, 518–524 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly-Y M, Carling D, Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 18, 3043–3051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J, AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A, The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem 282, 30107–30119 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Álvarez-Guardia D, Palomer X, Coll T, Davidson MM, Chan TO, Feldman AM, Laguna JC, Vázquez-Carrera M, The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res 87, 449–458 (2010). [DOI] [PubMed] [Google Scholar]
- 100.Sag D, Carling D, Stout RD, Suttles J, Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol 181, 8633–8641 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mounier R, Théret M, Arnold L, Cuvellier S, Bultot L, Göransson O, Sanz N, Ferry A, Sakamoto K, Foretz M, Viollet B, Chazaud B, AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 18, 251–264 (2013). [DOI] [PubMed] [Google Scholar]
- 102.Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, Izon D, Honeyman J, Chen ZP, Van Denderen BJ, Kemp BE, Steinberg GR, Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Invest 121, 4903–4915 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang Z, Kahn BB, Shi H, Xue BZ, Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem 285, 19051–19059 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cai J, Zhang XJ, Li H, Role of innate immune signaling in non-alcoholic fatty liver disease. Trends Endocrinol. Metab 29, 712–722 (2018). [DOI] [PubMed] [Google Scholar]
- 105.Bai J, Liu F, The cGAS-cGAMP-STING pathway: A molecular link between immunity and metabolism. Diabetes 68, 1099–1108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jia L, Chang X, Qian S, Liu C, Lord CC, Ahmed N, Lee CE, Lee S, Gautron L, Mitchell MC, Horton JD, Scherer PE, Elmquist JK, Hepatocyte Toll-like receptor 4 deficiency protects against alcohol-induced fatty liver disease. Mol. Metab 14, 121–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E, Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 139, 323–334.e7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, Lee S, Scherer PE, Elmquist JK, Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat. Commun 5, 3878(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ, Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Invest 126, 859–864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, Song J, Fujiwara K, Abe JI, Lemaire SA, Wang XL, Shen YH, STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler. Thromb. Vasc. Biol 37, 920–929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cruz VH, Arner EN, Wynne KW, Scherer PE, Brekken RA, Loss of Tbk1 kinase activity protects mice from diet-induced metabolic dysfunction. Mol. Metab 16, 139–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, Szabo G, STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. U.S.A 110, 16544–16549 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luther J, Khan S, Gala MK, Kedrin D, Sridharan G, Goodman RP, Garber JJ, Masia R, Diagacomo E, Adams D, King KR, Piaker S, Reinecker HC, Yarmush ML, Argemi J, Bataller R, Dienstag JL, Chung RT, Patel SJ, Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl. Acad. Sci. U.S.A 117, 11667–11673 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, Shen L, D’Angelo JA, Horton P, Chapman WC, Brockmeier D, Woo M, Engleman EG, Adeyi O, Hirano N, Jin T, Gehring AJ, Winer S, Winer DA, Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol 2, eaai7616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eom J, Kim JJ, Yoon SG, Jeong H, Son S, Lee JB, Yoo J, Seo HJ, Cho Y, Kim KS, Choi KM, Kim IY, Lee HY, Nam KT, Cresswell P, Seong JK, Seo JY, Intrinsic expression of viperin regulates thermogenesis in adipose tissues. Proc. Natl. Acad. Sci. U.S.A 116, 17419–17428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kespohl M, Bredow C, Klingel K, Voß M, Paeschke A, Zickler M, Poller W, Kaya Z, Eckstein J, Fechner H, Spranger J, Fähling M, Wirth EK, Radoshevich L, Thery F, Impens F, Berndt N, Knobeloch KP, Beling A, Protein modification with ISG15 blocks coxsackievirus pathology by antiviral and metabolic reprogramming. Sci. Adv 6, eaay1109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gautier L, Cope L, Bolstad BM, Irizarry RA, Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 118.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Kähäri AK, Keenan S, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Overduin B, Parker A, Patricio M, Perry E, Pignatelli M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Aken BL, Birney E, Harrow J, Kinsella R, Muffato M, Ruffier M, Searle SMJ, Spudich G, Trevanion SJ, Yates A, Zerbino DR, Flicek P, Ensembl 2015. Nucleic Acids Res. 43, D662–D669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li B, Dewey CN, RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kang S, Kong X, Rosen ED, in Methods in Enzymology (Academic Press Inc., 2014), vol. 537, pp. 1–16. [DOI] [PubMed] [Google Scholar]
- 122.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T, Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13, 539–548 (2000). [DOI] [PubMed] [Google Scholar]
- 123.Silva RC, Castilho BA, Sattlegger E, A rapid extraction method for mammalian cell cultures, suitable for quantitative immunoblotting analysis of proteins, including phosphorylated GCN2 and eIF2α. MethodsX 5, 75–82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Folch J, Lees M, Sloane Stanley GH, A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem 226, 497–509 (1957). [PubMed] [Google Scholar]
- 125.Morrison WR, Smith LM, Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J. Lipid Res 5, 600–608 (1964). [PubMed] [Google Scholar]
- 126.De Jesus DF, Zhang Z, Kahraman S, Brown NK, Chen M, Hu J, Gupta MK, He C, Kulkarni RN, m 6 A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat. Metab 1, 765–774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim D, Langmead B, Salzberg SL, HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liao Y, Smyth GK, Shi W, FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 129.Robinson MD, McCarthy DJ, Smyth GK, edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fresno C, Fernández EA, RDAVIDWebService: A versatile R interface to DAVID. Bioinformatics 29, 2810–2811 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All raw and processed RNA-seq and IRF3 CUT&RUN data have been deposited in the NCBI Gene Expression Omnibus (GEO) under GEO accession number GSE166369. A list of direct transcriptional targets of IRF3 in hepatocytes is given in data file S1. Raw data from figures are presented in data file S2.