

Abstract
Purpose:
Immune checkpoint inhibitors combined with anti-angiogenic agents produce benefits in the treatment of advanced hepatocellular carcinoma (HCC). We investigated the efficacy and immunomodulatory activity of cabozantinib alone and combined with anti-PD1 in experimental models of HCC, and explored the potential target population that might benefit from this combination.
Experimental Design:
C57BL/6J mice bearing subcutaneous Hepa1-6 or Hep53.4 tumours received cabozantinib, anti-PD1, their combination or placebo. Tumour and blood samples were analysed by flow cytometry, immunohistochemistry, transcriptome and cytokine profiling. Cabozantinib-related effects were validated in a colorectal cancer PDX model. Transcriptomic data from three human HCC cohorts (Cohort 1: n=167, Cohort 2: n=57, TCGA: n=319) were used to cluster patients according to neutrophil features, and assess their impact on survival.
Results:
The combination of cabozantinib and anti-PD1 showed increased anti-tumour efficacy compared to monotherapy and placebo (P<0.05). Cabozantinib alone significantly increased neutrophil infiltration and reduced intra-tumour CD8+PD1+T cell proportions, while the combination with anti-PD1 further stimulated both effects and significantly decreased Treg infiltration (all P<0.05). In blood, cabozantinib and especially combination increased the proportions of overall T cells (P<0.01) and memory/effector T cells (P<0.05), while lowering the neutrophil-to-lymphocyte ratio (P<0.001 for combination). Unsupervised clustering of human HCCs revealed that high tumour enrichment in neutrophil features observed with the treatment combination was linked to less aggressive tumours with more-differentiated, less-proliferative phenotypes.
Conclusions:
Cabozantinib in combination with anti-PD1 enhanced anti-tumour immunity by bringing together innate neutrophil-driven and adaptive immune responses, a mechanism of action which favours this approach for HCC treatment.
Keywords: Hepatocellular carcinoma, molecular therapies, immune therapies, precision oncology, checkpoint inhibitors
Statement of Translational Relevance
Hepatocellular carcinoma (HCC) is the most common type of liver cancer, which is the 3rd leading cause of cancer-related death worldwide. The combination of immune checkpoint inhibitors with anti-angiogenic agents produces survival benefits in advanced HCC, but only for around 30% of patients, so new strategies to overcome tumour-intrinsic resistance are urgently needed. Here we used preclinical models to explore the efficacy and immunological impact behind combining cabozantinib and anti-PD1 therapy for the treatment of HCC. We provide evidence that cabozantinib in combination with anti-PD1 enhances anti-tumour immunity by bringing together innate neutrophil-driven and adaptive immune responses, which is associated with greater anti-tumour responses than either monotherapy, thus providing a mechanistic rationale supporting this combination as a new treatment strategy for HCC. We also identify a subgroup of human HCC with reduced enrichment of active neutrophil phenotypes which displays significantly worse outcomes, and may therefore benefit the most from this combination.
Introduction
Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer, the fourth leading cause of cancer-related death and a major health problem globally(1,2). Around 50-60% of patients will be ultimately exposed to systemic therapies, where the combination of atezolizumab and bevacizumab has become the standard of care in first-line. In the Phase III IMbrave150 trial, this combination demonstrated significantly better overall and progression-free survival vs. sorafenib (median overall survival (OS) 19.2 vs. 13.4 months)(3,4), the multi-kinase inhibitor which had been the previous standard of care for advanced HCC for the past 12 years(5). This marks the first combination therapy and first treatment regimen involving an immunotherapy to induce a greater survival benefit for HCC patients compared to existing therapies, although objective responses were restricted to 30% of patients(3). Single-agent immune checkpoint inhibitors (ICIs) –i.e. nivolumab and pembrolizumab– had also shown promising results over the past decade, with median OS of 13-16 months reported in advanced HCC patients(6,7). However, no ICI monotherapy significantly extended overall survival in Phase III trials. In addition, recent data have revealed that non-viral HCC, particularly non-alcoholic steatohepatitis (NASH)-related HCC, might be less responsive to immunotherapy due to the intra-tumour accumulation of a population of dysfunctional activated CD8+PD1+ T cells(8). Thus, there is an unmet need to better understand the complex mechanisms underlying anti-tumour immunity and explaining response and resistance to immunotherapies, to favour the development of more efficacious therapeutic strategies with immunotherapies(9), including combination therapies.
Given that immunotherapeutic strategies show better results among tumours with an inflammatory microenvironment(10), efforts are being made to enhance the anti-tumour activity of ICIs by promoting the infiltration or re-activation of immune cells in tumours which lack an inflamed profile. One approach to this is via the combination of ICIs with anti-angiogenic agents, in light of the reported immunosuppressive role of pro-angiogenic molecules (mainly VEGF, angiopoietins, HGF and the PDGF family)(11,12). Encouraging results have been obtained for combinations of ICIs plus anti-angiogenic therapies in preclinical studies(13,14), as well as in clinical trials across different cancer types including HCC(3,15,16).
Cabozantinib, a tyrosine kinase inhibitor (TKI), proved its efficacy in a phase III trial with advanced HCC patients who had progressed on sorafenib, and extended median OS to 10.2 months from 8.0 months with placebo(17). Among other kinases, cabozantinib targets VEGFR, AXL and MET, key factors in pathways promoting angiogenesis, proliferation and epithelial-to-mesenchymal transition(18,19). Therefore, the combination of cabozantinib and ICIs may lead to enhanced clinical benefits over monotherapies in HCC, and in fact, the combination of nivolumab + cabozantinib conveyed a median progression-free survival (PFS) of 5.5 months in advanced HCC patients, with median OS not reached(20). The phase III study assessing the combination of atezolizumab + cabozantinib (COSMIC-312)(22) reported in the first interim analysis a significant improvement in PFS versus sorafenib (HR: 0.63, 99% CI: 0.44-0.91, P=0.0012), and a non-significant trend in terms of overall survival impact(23). Studies in experimental models of prostate cancer have also suggested that cabozantinib may have immunomodulatory activity(24), however this has not yet been explored in HCC.
Our study explored the anti-tumoural, mechanistic and immunomodulatory effects of cabozantinib alone and in combination with anti-PD1 in two preclinical HCC mouse models. The combination of cabozantinib with anti-PD1 was a) associated with greater efficacy than any single treatment alone, b) enhanced neutrophil recruitment and neutrophil activation profiles, and c) decreased CD8+PD1+ T and T regulatory cell infiltration in the tumour. Finally, human HCC data revealed that tumours with neutrophil-based enrichment are linked with better molecular and clinical features. Therefore, the combination of cabozantinib and anti-PD1 has the potential to bring together the activation of adaptive and innate immune responses against the tumour in patients with HCC.
Materials and Methods
Experimental mouse models
Immunocompetent murine models of HCC were generated by subcutaneously implanting 5x106 Hepa1-6 cells (Model 1) or 5x106 Hep 53.4 cells (Model 2) suspended in Matrigel (50% v/v) into the right flank of male 5-week old C57BL/6 mice (Model 1: n=80, Model 2: n=40). When tumours reached a volume of 200-300 mm3, animals were randomly assigned to receive: a) Placebo: Hamster IgG 10 mg/kg i.p. every 3 days for a total of 5 doses; b) Cabozantinib: 30 mg/kg p.o. daily until the end of the study; c) Anti-PD1: Hamster anti-murine PD1 mAb J43 (BioXCell) i.p. at 10 mg/kg every 3 days for a total of 5 doses; or d) Combination of cabozantinib with anti-PD1. Tumour volume measurements were taken every 2-3 days using bilateral callipers, and tumour response rates were calculated as described in the Supplementary Methods. In Model 1, six mice per group were randomly selected and culled on day 14 of treatment for pharmacodynamic analyses. The remaining 14 mice per group were monitored until their tumour volume surpassed 1500 mm3 or reached day 32 of treatment (Supplementary Figure 1). Model 2 was used as validation and all animals were culled at day 14. Animal experiments were approved by the corresponding review board. As a further independent model we also analysed the RNA sequencing data from paired human colorectal cancer patient-derived tumour explant (PDX) models treated with cabozantinib (30 mg/kg daily p.o.) or vehicle(25).
Flow cytometry
Peripheral blood samples were collected by cardiac puncture under deep terminal anaesthesia and subjected to red blood cell lysis. Tumour tissues were minced with forceps and scissors, followed by chemical and mechanical disaggregation. The resulting single-cell suspensions were incubated with antibody conjugates to identify specific populations of myeloid (dendritic cells, macrophages and neutrophils) and lymphoid (leukocytes, CD4+ T cells, CD8+ T cells, T regulatory cells, PD1-expressing CD8+ T cells, memory T cells and B cells) immune cells, and stained with a viability dye (Supplementary Tables 1–3) and in the presence of a Fc Receptor blocking reagent. Samples were analysed on a 5-laser Fortessa flow cytometer (BD Biosciences) using BD FACSDiva software v8.0.
Gene expression profiling of mouse tumours
Gene expression profiling of mouse tumour samples was performed using a whole genome microarray platform (GeneChip Mouse Genome 430 2.0 Array, Affymetrix; GSE174770). Mouse expression data were retrieved, quality checked and processed through RMA using the R package affy (v.1.62.0). After quality filtering, probe-gene correlates were identified with the R package htmg430pmprobe (v.2.18.0). Subsequently, the mouse gene expression matrix was humanized using the mouse-human orthologs data available in ENSEMBL (retrieved using BioMart) and Mouse Genome Informatics (MGI, informatics.jax.org). Molecular Signature Database gene sets (MSigDB, broadinstitute.org/msigdb) and previously reported gene signatures representing different states of inflammation, HCC subclasses or distinct immune cell populations(26–29) were tested using enrichment tools implemented in GenePattern(30) (GSEA and ssGSEA). Additionally, the GenePattern module Comparative Marker Selection (CMS) was used to identify differential gene expression between treated and untreated tumours (FDR<0.05, FC≥1.5), and the Enrichr tool(31) was used to evaluate enrichment in specific pathways and biological functions among the differentially expressed genes (DEGs, Supplementary Tables 5–6). We considered as neutrophil-related genes those DEGs classified within biological functions linked to neutrophil activity. A signature reflecting the specific molecular effect of combination therapy (CAP-100) was retrieved by selecting the upregulated genes in the combination arm vs. placebo and deducting those genes commonly upregulated between the combination and the monotherapies.
Human samples and molecular profiling
We analysed the gene expression data of 167 paired HCC tumour and tumour-adjacent liver obtained by our group(26,32) from untreated, surgically-resected fresh-frozen samples (Cohort 1: Heptromic, GSE63898). Supplementary Table 7 depicts the clinical features of the subset of 167 patients, and a complete description of the full cohort can be found elsewhere(26,32). In addition, we used a second cohort of 57 untreated HCC patients with paired expression data from the tumour and tumour-adjacent liver (Cohort 2: Supplementary Table 7, GSE174570). Samples from both cohorts were collected through the International HCC Genomic Consortium under the corresponding IRB approval for each centre. We also used the survival and expression data of 319 HCC patients publicly available from The Cancer Genome Atlas (TCGA)(33).
RNA extractions from fresh-frozen samples were performed as previously reported(32), and RNA profiling was conducted using the Human Genome U219 Array Plate (Affymetrix). The processing of transcriptome data (normalization, background correction and filtering) was carried out as previously described(32). Prediction of mRNA-based signature positivity was performed with GenePattern’s Nearest Template Prediction (NTP) module, and gene expression signature enrichment as well as differential gene expression analysis were carried out as with the mouse tumour expression data. K-medoids clustering was used for the unsupervised classification of samples according to the enrichment in gene expression signatures (Supplementary Figure 9). The optimal number of clusters was determined as the k with the maximum silhouette width.
Statistical analysis
Statistical analyses were performed using R (version 3.6.1). Specifically, non-parametric tests were used for the comparison of distribution of continuous variables (Wilcoxon or Kruskal-Wallis tests). For the assessment of correlations between two continuous variables, we used Spearman’s rank correlation coefficient test. Univariate analysis of survival was conducted using the Log-Rank test, while a multivariate Cox Proportional Hazards model was conducted to identify independent predictors of survival. In the event of a multiple comparison testing, Benjamini-Hochberg’s correction was applied unless specified otherwise. In general, adjusted P-values <0.05 (two-sided) were considered to be statistically significant and are denoted as follows (unless stated otherwise in specific analyses): *P<0.05, **P<0.01, ***P<0.001. NS indicates not significant, P>0.05.
Data availability
Data generated or analysed by the authors is available in Gene Expression Omnibus (GEO) at GSE174770, GSE60939, GSE63898 and GSE174570.
Full details of the Materials and Methods are given in the Supplementary Data file.
Results
Cabozantinib plus anti-PD1 treatment displayed higher anti-tumour efficacy as compared to the monotherapies
We used subcutaneous murine models of HCC to assess the anti-tumour efficacy of cabozantinib, anti-PD1 and combination (Supplementary Figure 1), and evaluated tumour response rate according to RECIST criteria adapted for murine models. In the Hepa1-6 model, the combination of cabozantinib and anti-PD1 most rapidly induced an objective response (OR) out of all treatment arms, with statistically significant improvements vs. cabozantinib alone or anti-PD1 monotherapy, as well as placebo (median time to OR: 12.6 days for combination vs. 20.1 days for cabozantinib monotherapy, with time to OR not reached in the anti-PD1 and placebo arms having ORRs of 40% and 0% respectively, P<0.05, Figure 1A, Supplementary Figure 2A). Of note, cabozantinib and anti-PD1 monotherapies also significantly accelerated time to achieve an OR compared to placebo (P<0.05). In addition, the combination treatment induced the highest OR and complete response rates (70% ORR and 15% CRR), followed by cabozantinib (35% ORR and 5% CRR) and anti-PD1 (25% ORR and 15% CRR), and all treatment-induced ORRs were significantly higher as compared to placebo (0% ORR, P<0.05) (Figure 1B). All treatments also delayed tumour growth vs. placebo (P<0.05), and no adverse clinical signs or loss in starting body weight were detected throughout the study (Supplementary Figures 2B–C). These findings were also recapitulated in the Hep53.4 model (Supplementary Figures 2D–F).
Figure 1 |. Cabozantinib + anti-PD1 has a strong anti-tumour activity and promotes vascular normalisation in a murine HCC model.
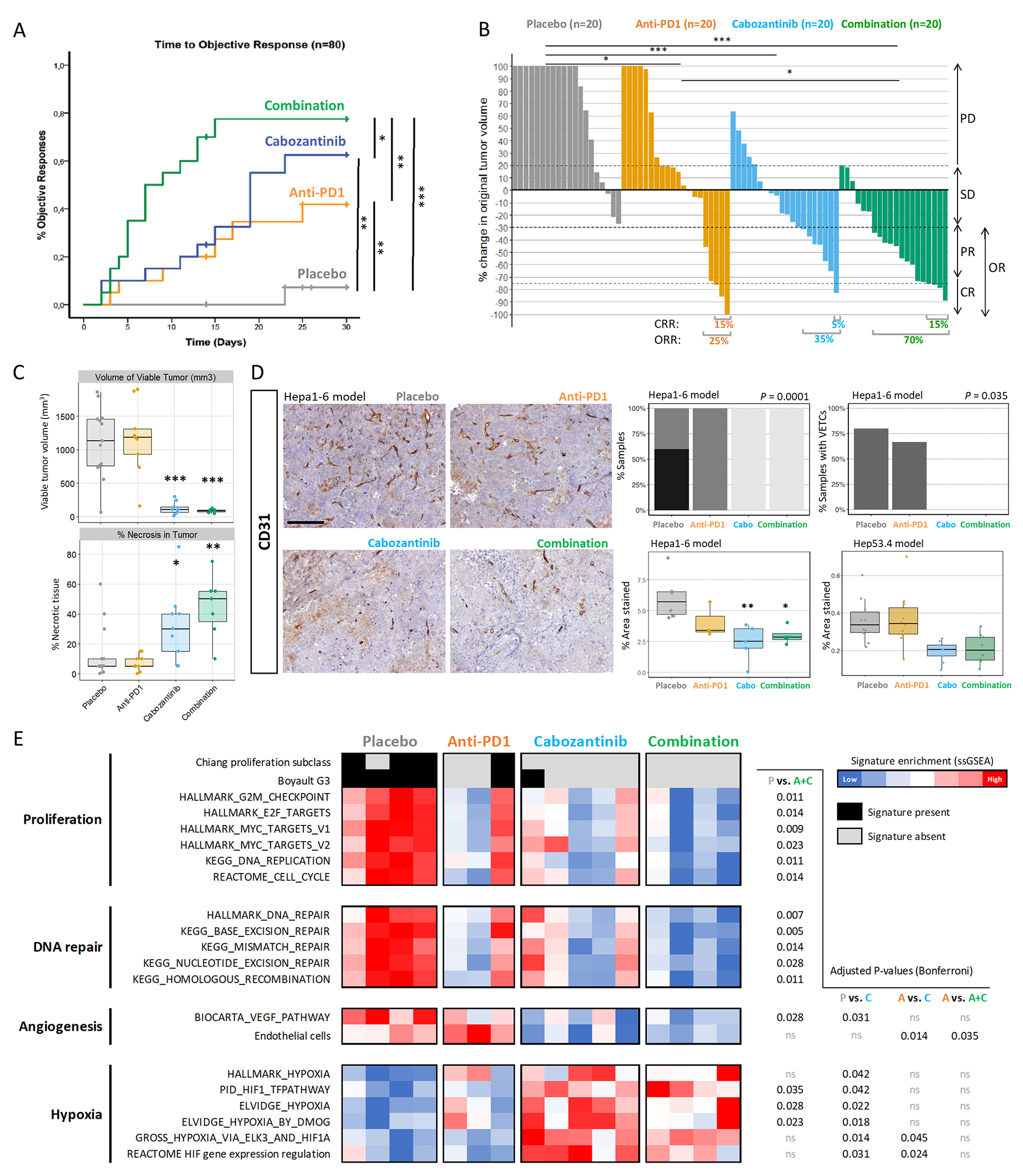
C57BL/6J mice bearing Hepa1-6 tumours were treated with anti-PD1 or placebo IgG ± oral cabozantinib (n=20 mice per arm). A. Time taken to achieve an objective response (Log-Rank test P-values depicted). B. Waterfall plot of tumour response at day 14 according to RECIST criteria (Fisher’s exact test P-values depicted). N.B. The Y-axis is capped at +100% and therefore only the first doubling in tumour volume is accurately depicted. PD = progressive disease, SD = stable disease, PR = partial response, CR = complete response, OR = objective response. C. Volume of viable tumour (mm3) and % of tumour necrosis from tumours at day 32 (compared vs. placebo). D. Representative images of CD31 staining across the four treatment groups, 100x magnification (left panel, scale bar = 200 μm). Plots on the right panel depict the proportion of samples with mild, moderate or marked CD31 staining, the quantification of tumour vascular endothelial cells stained with CD31 (compared vs. placebo, and validation in the Hep53.4 model), and the percentage of samples with vessels encapsulating tumour clusters (VETCs). Images taken at 100x magnification. Scale bar = 200 μm. E. Heatmap reflecting enrichment in signatures of proliferation, DNA repair, angiogenesis and hypoxia obtained through NTP and ssGSEA. * P<0.05, ** P<0.01, *** P<0.001, ns P>0.05.
Excised HCC tumours were then evaluated at the molecular and histopathological level. Firstly, examination of the tumour sections revealed that cabozantinib and combination treatments were associated with significant reductions in viable tumour volume and increases in the percentage of tumour necrosis (Figure 1C). On the other hand, and confirming the anti-angiogenic activity of cabozantinib(18,19,25), immunohistochemical detection of endothelial cells by CD31 staining revealed that only placebo and anti-PD1 tumours contained marked or moderate levels of vascular endothelial cells, and cabozantinib-treated tumours (monotherapy and combination) were associated with significantly-reduced areas of CD31+ staining compared to placebo (P<0.05) (Figure 1D). Additionally, only cabozantinib and combination-treated tumours displayed an absence of vessels encapsulating tumour clusters (VETCs, Figure 1D).
Analysis of the tumour whole genome expression profile revealed the impact of therapies in reducing proliferation and angiogenesis. Regarding proliferation, using the NTP and ssGSEA modules of GenePattern we observed that placebo-treated tumours were associated with the Chiang proliferation(27) and the Boyault G3(29) subclasses of HCC, and also displayed increased expression of a wide array of proliferation and DNA repair signatures (Figure 1E). Furthermore, administration of cabozantinib (monotherapy or combination) significantly impacted angiogenesis by reducing VEGF and endothelial cell signalling and promoting enrichment in molecular signatures of hypoxia (Figure 1E). Combination was the only treatment associated with a significant reduction in proliferation and DNA repair signatures as compared to placebo, with a similar but non-significant trend also observed in cabozantinib monotherapy-treated tumours. Taken together, these data demonstrate that cabozantinib treatment induced anti-proliferative effects in the tumours and these were more marked when administered in combination with anti-PD1.
Combination treatment induced intra-tumoural neutrophil infiltration
Immune population analysis using flow cytometry, immunohistochemistry and transcriptomic data revealed that cabozantinib administration promoted the recruitment of neutrophils in the tumour tissue. Specifically, through flow cytometry we observed median 2.5 and 1.8-fold increases in the prevalence of tumour-infiltrating neutrophils in cabozantinib and combination treatment arms versus placebo (P<0.05 vs. placebo and anti-PD1, Figure 2A). Furthermore, in combination-treated animals the increased proportion of neutrophils coincided with high ESTIMATE immune cell infiltrate scores(34), suggesting that neutrophils were a prevalent immune subtype in these higher-infiltrated tumours (Supplementary Figure 2G). We further assessed neutrophil intra-tumour infiltration and activation by IHC using a panel of three markers: Ly6G, which is expressed by neutrophils and granulocytes; myeloperoxidase (MPO), which is expressed by active polymorphonuclear cells including neutrophils; and neutrophil elastase (NE), which is expressed in the lysosomal granules of active neutrophils. Overall, we observed a significantly elevated percentage of stained cells in tumours from the cabozantinib (median 4% for Ly6G, 12% for MPO, 8% for NE) and combination (8.5% for Ly6G, 15% for MPO, 4% for NE) arms vs. placebo (1.5% for Ly6G, 4.2% for MPO, 0.5% for NE; P<0.05, Figure 2B). These data suggest that active neutrophils are significantly increased in the intra-tumoural regions of mice treated with combination and cabozantinib, whereas no changes in peri-tumour neutrophil infiltration were observed across treatment groups (Supplementary Figure 4A–B). These findings were also recapitulated in the Hep53.4 model (Figure 2B), and when compared with Hep53.4 tumour samples treated with the anti-angiogenic lenvatinib ± anti-PD1 also generated by our lab(35), this effect was revealed to be cabozantinib specific (Supplementary Figure 2H).
Figure 2 |. Cabozantinib treatment induces intra-tumoural neutrophil infiltration and activation in preclinical models.
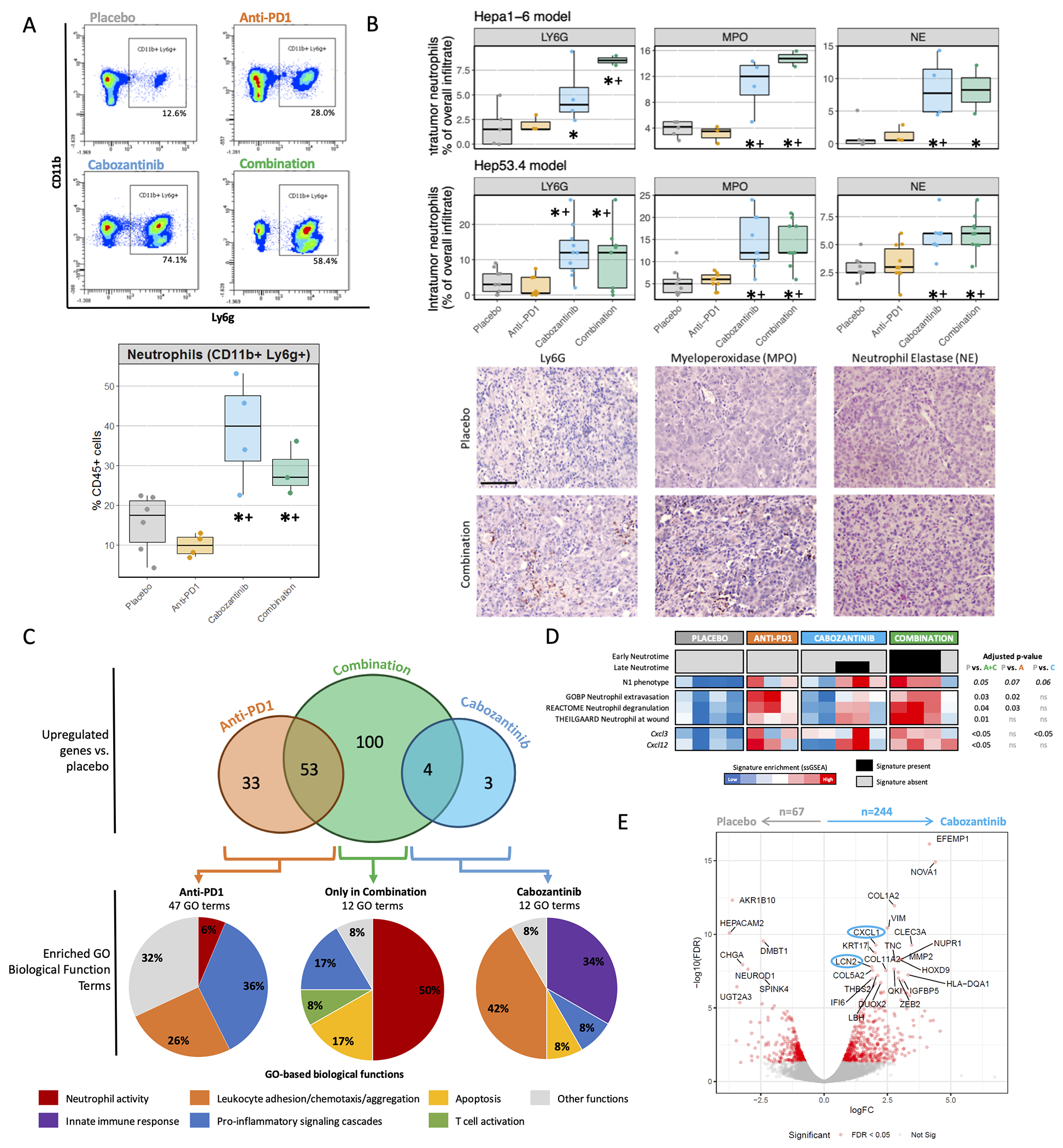
A. Representative flow cytometry results for the Ly6G gating among CD45+CD11b+ cells (upper panel), and resulting proportions of intra-tumoural neutrophils (CD11b+Ly6G+) across treatment arms (bottom panel). Data are depicted as the median percentage ± interquartile range of CD45+ cells. B. The mean percentage of the overall tumour immune infiltrate corresponding to intra-tumoural immune cells positive for the neutrophil markers Ly6G, myeloperoxidase (MPO) and neutrophil elastase (NE), as determined by IHC across treatment arms in Hepa1-6 and Hep53.4 tumours (upper panel). Representative images from Hepa1-6 tumours taken at 200x (bottom panel, scale bar = 100 μm). C. Venn diagram for the number of overexpressed genes among treatment groups as compared to placebo, false discovery rate (FDR) <0.05, fold change (FC) ≥ 1.5. Bottom pie charts depict the distribution of the GO-based biological function terms found to be significantly enriched (FDR < 0.05) in each group of genes using Enrichr analysis. GO = Gene ontology. D. Heatmap representing sample enrichment in neutrophil-related signatures and expression of differentially-expressed chemokines. A, C, D data obtained from the Hepa1-6 model. E. Volcano plot of differentially-expressed genes (DEGs) from the CRC PDX model by Song et al., as determined by RNAseq in paired tumours after 3 days of treatment, with significantly DEGs in red and top neutrophil-related genes circled in blue. * P<0.05 and FDR<0.1 compared to placebo; + P<0.05 and FDR<0.1 compared to anti-PD1.
To further investigate this observation, we extracted differentially expressed genes (DEGs, FDR<0.05, fold-change>1.5) among the treatment groups through Comparative Marker Selection (CMS) analysis of tumour transcriptomic data. Combination treatment had the greatest impact reshaping the molecular profile of the tumour, since it was associated with the greatest number of DEGs compared to placebo at 157 genes, followed by anti-PD1 with 86 DEGs, while only 7 genes were significantly upregulated by cabozantinib. The 100 genes exclusively upregulated in the combination arm were used to generate the cabozantinib plus anti-PD1 100-gene signature (CAP-100), capturing the specific molecular effect of the combination (Supplementary Table 5). Gene Ontology enrichment analysis revealed that CAP-100 was significantly associated with 12 biological function GO-terms, of which 6 (50%) were linked to neutrophil activity (P<0.05, Figure 2C, Supplementary Table 6). The anti-tumour neutrophil phenotype in the combination arm was further supported by higher enrichments in gene expression signatures of the N1 neutrophil profile(36), neutrophil activity and maturation (late Neutrotime(37)) (Figure 2D). In parallel, the 7 genes upregulated by cabozantinib monotherapy were associated with the chemotaxis and migration of leukocytes, fundamentally neutrophils (5/12 GO-terms; 42%), and with innate immunity responses (4/12 GO-terms, 34%) (Figure 2C, Supplementary Table 6). On the other hand, the 86 genes upregulated by anti-PD1 monotherapy were linked to broader pro-inflammatory signalling cascades (17/47 GO-terms; 36%) and chemotaxis of a wide variety of leukocyte subpopulations (12/27 GO-terms, 26%).
We then validated the molecular effect of cabozantinib using RNAseq data from colorectal cancer (CRC) tumours in a patient-derived xenograft (PDX) model(25) (Supplementary Figure 3A). Gene Ontology enrichment analysis of the 244 genes up-regulated by cabozantinib versus placebo revealed a positive regulation of neutrophil chemotaxis (Supplementary Figure 3B, Figure 2E), and GSEA identified an enrichment in neutrophil, inflammation, hypoxia and apoptosis-related pathways (Supplementary Figure 3C).
Combination of cabozantinib and anti-PD1 induces a strong inflammatory phenotype
To further investigate the immunomodulatory roles of cabozantinib and its combination with anti-PD1, we assessed several infiltrating lymphoid and myeloid immune populations along with the tumour transcriptomic profiles. Regarding anti-tumour immunity features, anti-PD1 and combination induced a higher enrichment in stromal and immune microenvironment components based on expression data (Supplementary Figure 4D). Flow cytometry analysis revealed a specific reduction in the proportions of the exhausted CD8+PD1+ T cell subpopulation induced by cabozantinib and combination (mean 7% and 6% of CD45+ cells; P<0.05 vs. 25% in placebo and 35% in anti-PD1, Figure 3A), even though no significant differences in the IHC counts of infiltrating CD8+ and CD4+ T cells were observed (Supplementary Figure 4E–F). In addition, the location of CD8 positivity was mostly intra-tumoural in combination and anti-PD1 tumours (means of 73% and 60%, respectively; P<0.05 vs. 26% in placebo, Figure 3C), suggesting a higher capacity to infiltrate the tumour parenchyma to exert their effector function.
Figure 3 |. Cabozantinib + anti-PD1 therapy induces a strong pro-inflammatory effect.
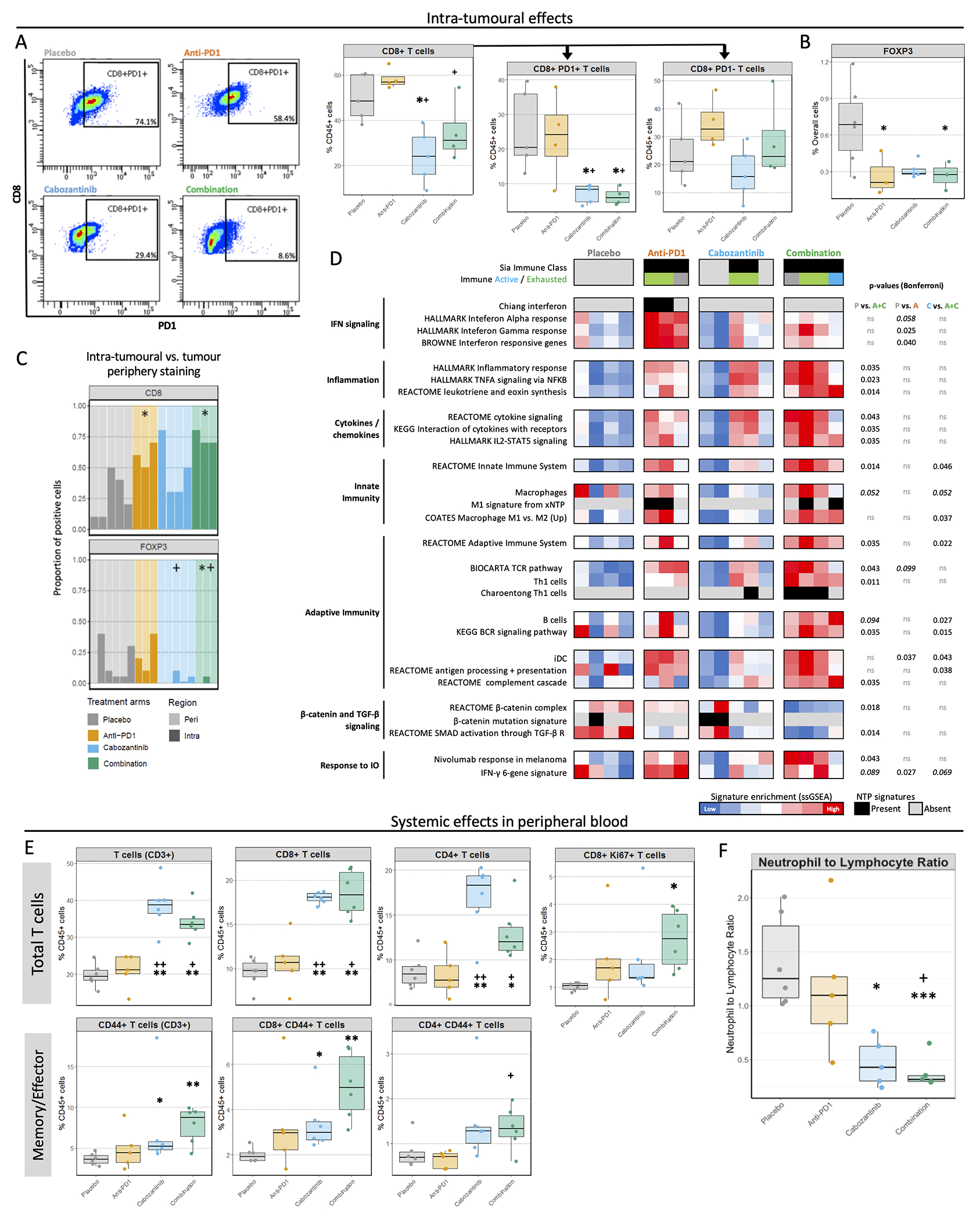
A. Representative dot plots showing PD1 gating among CD45+CD3+CD8+ cells (left), and proportions of intra-tumoural CD8+ T lymphocytes and their PD1+ and PD1− subsets as a % of CD45+ cells, as determined by flow cytometry (right). B. Mean percentage of FOXP3 positive cells in tumour parenchyma as determined by IHC using QuPath, relative to all cells. C. The proportion of total CD8 and FOXP3 staining in intra-tumoural or peripheral areas. Each bar represents a mouse, and the darker colour depicts which % of staining is intra-tumoural. D. Heatmap showing tumour immunity features. Gene signatures representing different states of inflammation, HCC subclasses or distinct immune cell populations were tested using NTP and ssGSEA. Bonferroni-corrected P-values from Dunn’s post-hoc test. E. Overall populations of T lymphocytes (CD3+), and specifically CD8+ T cells, CD4+ T cells and proliferating CD8+ T cells (defined as CD8+Ki67+) in peripheral blood after 14 days of treatment, and the proportions of the memory/effector subgroups within these populations (characterized by CD44 expression). F. The ratio of neutrophil to lymphocyte proportions in peripheral blood. * P<0.05 and FDR<0.1 compared to placebo; + P<0.05 and FDR<0.1 compared to anti-PD1. Data obtained from the Hepa1-6 model.
Aligning with these results, transcriptomic analysis revealed that all anti-PD1 and combination-treated tumours recapitulated the HCC Immune Class(26), compared to none in the placebo arm (Figure 3D). Combination further contributed to the pro-inflammatory and anti-tumour immunity effect of anti-PD1 with the highest enrichments in pathways of cytokine and chemokine signalling, T cell activation and Th1-related responses, B cell signalling, classically activated M1 macrophages, dendritic cells, antigen presentation and features of response to nivolumab in melanoma(38) (P<0.05 vs. placebo, Figure 3D, Supplementary Figure 4D). Flow cytometry did not reveal significant changes in the immune infiltrate composition for B cells or dendritic cells, but showed a cabozantinib-related reduction in macrophage percentages (Supplementary Figure 4G).
In terms of immune suppression features, IHC analysis revealed decreased infiltration of T regulatory cells (FOXP3+) in combination and anti-PD1 arms versus placebo (P<0.05), with a clear trend for cabozantinib (Figure 3B). Additionally, combination and cabozantinib-treated tumours excluded T regulatory cells from intra-tumoural areas, with mean 1.7% and 3.2% of the total FOXP3+ positivity in the IHC sections being intra-tumoural (P<0.05 vs. 15% in placebo, Figure 3C). In parallel, gene expression data revealed that combination treatment also caused a significantly reduced enrichment in TGFβ and β-catenin signalling pathways, associated with immune exclusion features in HCC and resistance to immunotherapies(26,39–41) (P<0.05 vs. placebo, Figure 3D). Furthermore, cabozantinib and combination arms were also linked to the strongest PD-L1 expression in tumour cells and increased CTLA4 staining (Supplementary Figure 4H), which could potentially be associated with the activation of feedback loops to negatively regulate T cell activation. Overall, tumour molecular data suggest that combination enhanced anti-tumour immunity by stimulating both innate and adaptive components.
Combination treatment with cabozantinib promotes adaptive immune cell populations in blood
We then investigated treatment-related effects on systemic immunity in our Hepa1-6 model through flow cytometry and chemokine profiling in blood. In contrast to what was observed in tumour tissue, the proportions of circulating overall T lymphocytes (CD45+CD3+) as well as CD4+ and CD8+ subpopulations were significantly increased by cabozantinib and combination (mean 38% and 34% of CD45+ cells) as compared to placebo and anti-PD1 (20% and 21%, P<0.05, Figure 3D). In addition, combination induced a significant increase in the proportions of proliferating CD8+ T cells in blood (P<0.05 vs. placebo, Figure 3E), and the greatest enrichment in the subset of memory/effector T cells (CD45+CD3+CD44+), particularly memory/effector CD8+ T cells (P<0.01 vs. placebo), which were seen increased to a lesser extent by cabozantinib monotherapy (P<0.05 vs. placebo, Figure 3D). We also assessed the neutrophil to lymphocyte ratio (NLR), associated with poor prognosis in HCC patients(42), as the rate between CD3+ T cell and CD11b+Ly6G+ cell percentages. Cabozantinib and combination induced a significant reduction of the NLR as compared to placebo (mean 0.47 and 0.38 vs. 1.4, P<0.05, Figure 3F). Of note, no significant changes were observed for circulating T regulatory cells, B cells, macrophages or dendritic cells (Supplementary Figure 5A). Finally, cytokine and chemokine profiling of peripheral blood revealed significantly increased levels of two chemoattractants in the combination arm: CTACK/CCL27 (chemotactic agent for T lymphocytes) and IL-16 (leukocyte chemoattractant and modulator of T cell activation) (P<0.05 vs. placebo, Supplementary Figure 5B). Overall, cabozantinib and especially combination impacted systemic immunity by promoting an enrichment in adaptive immune cell subsets.
Human HCCs with combination-like neutrophil features have favourable clinical outcomes and molecular profiles
We then elucidated whether these active neutrophil profiles were linked to any specific molecular or clinical features in human HCC, using two cohorts of patients (Cohort 1: n=167, and Cohort 2: n=57 including paired tissue and blood samples), and the MCPCounter(43) neutrophil signature which correlated strongly with the neutrophil infiltration and activation profile in combination-treated mice (Supplementary Figure 6). Unsupervised clustering of human HCC based on enrichment of the neutrophil signature revealed three distinct clusters of samples Neutrophil Enriched, Neutrophil Depleted, and Tumour-only Neutrophil Depleted (Supplementary Figure 7A–B).
The Neutrophil Enriched cluster, accounting for 25% (42/167) of patients, was characterized by high neutrophil enrichment both in the tumour and in the adjacent tissue. These samples exhibited significant enrichment in the S3 HCC molecular class(28) and liver metabolic pathways - capturing more differentiated tumours- and suppression of TGF-β signalling(44), but a reduced activation of proliferation and DNA repair pathways (Figure 4A). In terms of immunity, Neutrophil Enriched HCCs were neither associated with adaptive immunity pathways, the HCC Inflamed(45) or Immune Classes(26) nor their Active or Exhausted subclasses, and they lacked HCC immune exclusion features derived from β-catenin pathway activation(40).
Figure 4 |. Unsupervised clustering reveals distinct neutrophil profiles in human HCC.
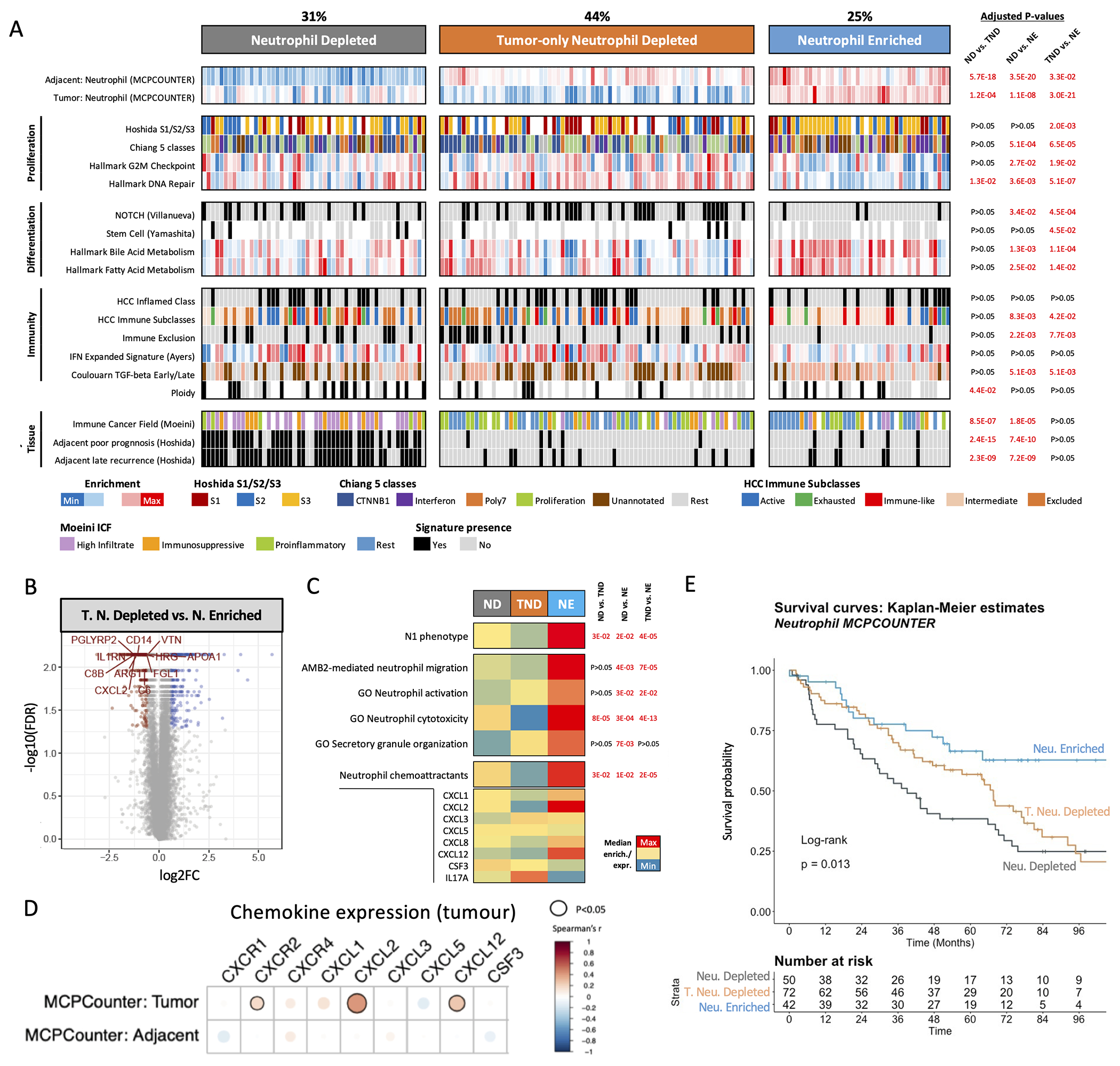
A. Heatmap displaying molecular features linked to the three neutrophil-related profiles: Neutrophil Depleted (ND), Tumour-only Neutrophil Depleted (TND), and Neutrophil Enriched (NE), with adjusted P-values comparing between groups. B. Volcano plot displaying the top differentially-expressed genes between the Tumour-only Neutrophil Depleted and Neutrophil Enriched group. C. Heatmap representation of the median enrichment in neutrophil-related gene expression signatures across neutrophil-related clusters. Genes included in the Neutrophil chemoattractants signature are depicted below. D. Correlation between the tumour expression of a set of neutrophil-attracting chemokines/chemokine receptors, and the enrichment in MCPCounter neutrophil signature in the tumour and adjacent tissues. E. Kaplan-Meier survival analysis (with log-rank P-value). All data presented are from Cohort 1 (n=167).
The Neutrophil Depleted cluster captured 31% of cases (52/167) which presented low neutrophil enrichment in both the tumour and adjacent non-tumour tissue. Finally, the Tumour-only Neutrophil Depleted (TND) cluster encompassed 44% of cases (73/167) displaying low neutrophil enrichment in the tumour but high enrichment in the adjacent tissue. Both the Neutrophil Depleted and TND groups presented pro-proliferative traits, with a higher prevalence of stem cell features and an enrichment in oncogenic TGF-β signalling (late TGF-β)(44) (Figure 4A). In addition, Neutrophil Depleted HCCs were more polyploid (FDR<0.05 vs. TND) and displayed a marked enrichment in poor prognosis signatures in the adjacent tissue (FDR<0.05 vs. TND and Neutrophil Enriched). Further, we observed a reduced proportion of hepatitis B and C virus infections in Neutrophil Enriched cases (Supplementary Figure 8A) and a higher proportion of cirrhotic cases in the Neutrophil Depleted subgroup (FDR <0.05). We also observed an overall lack of correlation between tumour and adjacent neutrophil enrichment values (Supplementary Figure 7C–D).
To gain insight into the neutrophil phenotypes and potential molecular mechanisms driving neutrophil recruitment in the tumours, we analysed the differentially expressed genes in tumour tissue among clusters (FDR<0.05, fold-change>1.5). We identified 74 genes commonly upregulated in the Neutrophil Enriched group when compared to TND, some of which were linked to innate immune cells like the macrophage-related CD14 and the neutrophil chemoattractant CXCL2 (Figure 4B, Supplementary Figure 9B). In parallel, we confirmed that Neutrophil Enriched HCCs were significantly associated with transcriptomic features of anti-tumour activity like the N1 phenotype, neutrophil migration, activation and cytotoxicity (Figure 4C, Supplementary Figure 9C). We also detected an enrichment in neutrophil chemokines mostly driven by CXCL2 and CXCL12 expression, which were more strongly correlated with MCPCounter tumour enrichment (P<0.05, Figure 4D, Supplementary Figure 9D). In addition, following up on the absence of β-catenin-driven immune exclusion among Neutrophil Enriched tumours, we observed that β-catenin activation (assessed through the presence of the Chiang CTNNB1 subclass) was linked to lack of active neutrophil features and a reduced expression of neutrophil chemoattractants (P<0.05 for CXCL3 and CXCL12 in the CTNNB1 subclass vs. Rest, Supplementary Figure 9E).
Importantly, and consistent with the favourable molecular features of Neutrophil Enriched tumours, patients of this cluster presented significantly longer survival as compared to the rest of the cohort (Figure 4E, HR=0.49 [95% CI 0.28–0.85]), log-rank P=0.011, which remained significant in a multivariate analysis (Cox proportional hazards model, P=0.025, Table 1). On the other hand, the Neutrophil Depleted subgroup presented reduced survival in the univariate analysis (Figure 4E, Table 1, log-rank P=0.02, HR=1.62 [95% CI 1.07–2.45]). Furthermore, we observed that tumours with the highest MCPCounter signature enrichment were associated with significantly better outcomes as compared to the rest (n=164, HR=0.45 [95% CI 0.25–0.81], multivariate Cox P=0.007) and these results were recapitulated in TCGA (n=319, HR=0.37 [95% CI 0.21-0.66], multivariate Cox P<0.001) (Supplementary Figure 10, Supplementary Table 9). Additionally, we confirmed all the above molecular findings in an independent cohort (Cohort 2: n=57, Supplementary Figures 8B–9A).
Table 1 |. Univariate and multivariate analysis of the 3 neutrophil clusters and key clinical features associated with survival in Cohort 1 (n=167).
Statistically significant values (P<0.05) are in bold.
| Variable | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
|
| ||||
| Hazard Ratio (95% CI) | Log-Rank P-value | Hazard Ratio (95% CI) | Cox PH P-value | |
| MCPCounter clusters | ||||
| Neutrophil Depleted | 1.62 (1.07 - 2.45) | 0.02 | (Reference) | |
| Tumor-only Neutrophil Depleted | 1.04 (0.70 - 1.57) | 0.8 | 0.79 (0.50 - 1.26) | 0.329 |
| Neutrophil Enriched | 0.49 (0.28 - 0.85) | 0.011 | 0.50 (0.27 - 0.91) | 0.025 |
|
| ||||
| Age (≥ 65) | 0.70 (0.47 - 1.05) | 0.079 | ||
|
| ||||
| Cirrhosis | 1.20 (0.73 - 1.99) | 0.5 | ||
|
| ||||
| Poor Differentiation | 1.23 (0.74 - 2.03) | 0.4 | ||
|
| ||||
| Gender (male) | 0.60 (0.39 - 0.93) | 0.022 | 0.55 (0.35 - 0.88) | 0.012 |
|
| ||||
| High AFP (≥400 ng/mL) | 1.47 (0.82 - 2.62) | 0.19 | ||
|
| ||||
| Multinodularity (>1 nodule) | 2.22 (1.46 - 3.36) | <0.001 | 2.34 (1.50 - 3.65) | <0.001 |
|
| ||||
| Satellites (yes/no) | 1.69 (1.10 - 2.60) | 0.015 | 1.52 (0.99 - 2.35) | 0.056 |
|
| ||||
| Size (≥3.5 cm) | 1.60 (1.05 - 2.44) | 0.027 | 1.04 (0.99 - 1.10) | 0.145 |
|
| ||||
| Vascular Invasion (micro/macro) | 2.10 (1.40 - 3.15) | <0.001 | 1.49 (0.97 - 2.30) | 0.07 |
Overall, we identified that a subset of human HCC recapitulates the specific neutrophil-related molecular effect of cabozantinib and anti-PD1 combination from our preclinical model, and these tumours of the Neutrophil Enriched cluster were linked to favourable molecular and clinical features.
Discussion
The recent approval of atezolizumab + bevacizumab has been a pivotal milestone in the treatment of advanced HCC and suggests that combination therapies with immune checkpoint inhibitors are the future for HCC treatment. However, responses are typically observed in a subset of patients, and there is currently a surge of research into broadening the spectrum of responders by overcoming potential tumour-intrinsic resistance to immune checkpoint blockade. The fact that recent reports suggest that immune therapies might be more effective in viral than non-viral related HCCs(8,46) also further highlights the need to understand the benefits of combining distinct agents with ICIs, since not all TKIs lead to equal immunomodulatory effects. In this study, we determined using preclinical models that the experimental combination of cabozantinib with anti-PD1 was associated with a greater proportion of anti-tumour responses in a shorter time, and enhanced molecular features of anti-tumour immunity including higher neutrophil recruitment and activation. To our knowledge this is the first study reporting a dominant role of neutrophils, particularly of the N1 phenotype, in the immune effects associated with a TKI combined with anti-PD1 therapy in HCC. We also identified distinct subgroups of human HCCs based on neutrophil features with potential clinical relevance (Figure 5).
Figure 5 |.
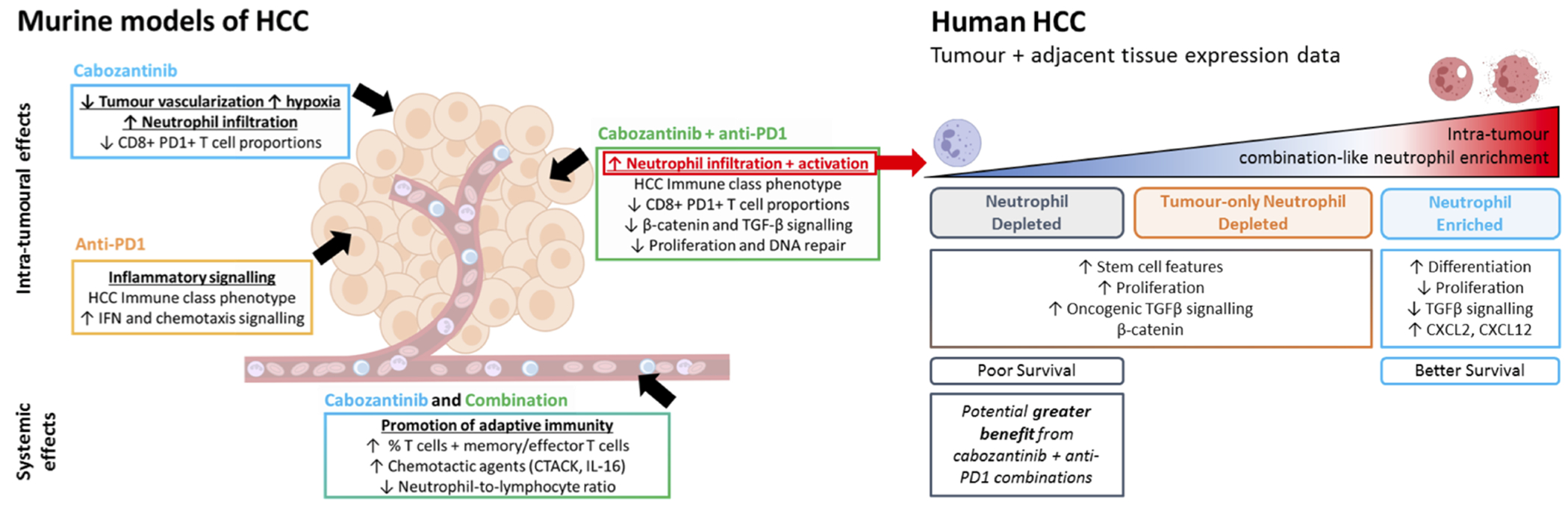
Schematic summary of the study results.
The observed increase in neutrophil infiltration after cabozantinib administration in our murine models had only been previously reported in a prostate cancer model, where neutrophil recruitment proved to be essential for the anti-tumour effect of the drug(24). These results were also supported by the transcriptomic data from a human colorectal cancer PDX model(25), contributing to the notion that this effect of cabozantinib may not be limited to a specific cancer type. Our transcriptomic analysis revealed that cabozantinib could be driving neutrophil infiltration through the upregulation of ligands for the neutrophil receptor CXCR2 (CXCL3 in our subcutaneous tumours, CXCL1 in the CRC PDX model), and that the upregulation of CXCL12 could promote their retention in the tumour, as previously reported in prostate cancer(24). In light of the phenotypic plasticity attributed to neutrophils in cancer biology(47), we observed that cabozantinib in combination with anti-PD1 could further enhance a molecular phenotype of neutrophil activation, possibly due to a contributing pro-inflammatory effect of the immunotherapy. In fact, the combination had the greatest impact upregulating genes linked to N1 phenotype, neutrophil maturity and degranulation, as well as immunoglobulin-related genes, which could also contribute to an anti-tumour neutrophil phenotype through antibody-dependent cellular cytotoxicity(48). Moreover, combination treatment was linked to reduced β-catenin signalling, which is associated with immune exclusion in HCC(39,40), as well as lower enrichment of TGF-β signalling, which has been demonstrated to polarize neutrophils towards a pro-tumour N2 phenotype(36,47). Overall, this demonstrates the potential of cabozantinib to trigger a neutrophil-mediated anti-tumour innate immune response, which is enhanced when combined with anti-PD1 therapy.
Additionally, cabozantinib significantly reduced infiltrating CD8+PD1+ T cells, a clinically interesting effect given that the accumulation of a dysfunctional CD8+PD1+ T cell subpopulation could be responsible for the reduced efficacy of ICIs in non-viral HCC, particularly NASH-HCC(8). Further studies are required to confirm whether cabozantinib and anti-PD1 combinations could improve NASH-HCC patient response by depleting this CD8+PD1+ T cell subset while increasing neutrophil infiltration. Of note, the evaluation of this combination in preclinical NASH-HCC models (e.g. high-fat diet) and a subgroup analysis of the COSMIC-312 trial(22) could shed light on this question.
In parallel to these effects, the combination of cabozantinib and anti-PD1 had the most favourable impact on the tumour immune microenvironment out of the treatment arms investigated, such as a significant reduction of T regulatory cells, enriched Th1 and M1 macrophage phenotypes, and enriched innate/adaptive immune pathways. The increased proportions of memory/effector T cells found in blood suggest that adaptive immunity was also activated with this treatment, a feature associated with a significant lowering of the neutrophil-to-lymphocyte ratio (P<0.001 for combination). HCC progression is closely linked with inflammation, and the importance of this is underscored by the wealth of evidence linking inflammation-based scores such as the NLR with HCC prognosis (49). Finally, consistent with the inhibitory activity of cabozantinib against tyrosine kinases including VEGFR2(19), our data indicate that at least part of the anti-tumour mechanism of action of cabozantinib is derived from its anti-angiogenic properties (reduced endothelial cells and VEGF signalling, and absence of VETCs, which have been proposed as a predictor of aggressive HCC(49)).
In HCC patients, using MCPCounter(43) as a transcriptomic tool to evaluate active neutrophil infiltration in tumour and non-tumour adjacent tissue, we confirmed that neutrophil enrichment in the adjacent tissue or in blood is not always linked to intra-tumour enrichment. This suggests that neutrophil recruitment mechanisms may be independent in liver adjacent and tumour tissues, and the circulating NLR associated with poor prognosis(42) may not reflect intra-tumoural neutrophil levels nor their phenotype. This finding has significance given we found that a high enrichment of active neutrophils in the tumour was linked to better outcomes in two independent cohorts, along with less proliferative and more differentiated molecular features, and no enrichment in inflammation profiles (HCC Immune Class(26)). In addition, intra-tumour neutrophil enrichment was linked to the expression of the chemokines CXCL2 (a CXCR2 ligand like CXCL1 and CXCL3, upregulated by cabozantinib in mice) and CXCL12 (upregulated in our model and previously described as a cabozantinib effect in prostate cancer(24)), suggesting that relevant mechanisms of neutrophil chemoattraction in human HCC were recapitulated in the preclinical models. Moreover, β-catenin pathway activation(40) may negatively influence the presence of this neutrophil phenotype in HCC as we observed it was linked to reduced enrichment in neutrophil signatures and expression of neutrophil chemokines; this may represent an additional immune exclusion effect of β-catenin activation that is worth further exploration.
Taken together, these observations suggest that cabozantinib, particularly in combination with anti-PD1 treatment, contributes to inducing neutrophil infiltration, decreasing an immune suppressive environment and enhancing anti-tumour activity compared to the monotherapies in HCC. Those patients with a reduced enrichment of active neutrophil phenotypes in the tumour (~30% of cases) could potentially gain the greatest benefits from this combination (Figure 5), although the molecular assessment of tumour samples from HCC patients exposed to cabozantinib in combination with PD1/PDL1 inhibitors will be necessary to test these hypotheses.
Overall, the combination of cabozantinib and anti-PD1 has the potential to bring together the activation of adaptive and innate immune responses against the tumour, and this provides a mechanistic rationale for combining cabozantinib and anti-PD1 therapy to render enhanced anti-tumour immune responses among HCC patients.
Supplementary Material
Acknowledgements
This study was sponsored by Ipsen Pharmaceuticals. R. Esteban-Fabró is supported by a doctoral training grant (BES-2017-081286) from MCIN/AEI/10.13039/501100011033 and the European Social Fund (ESF) and a mobility grant from Fundació Universitària Agustí Pedro i Pons. C.E. Willoughby is supported by a Sara Borrell fellowship (CD19/00109) from the Instituto de Salud Carlos III (ISCIII) and ESF. M. Piqué-Gili is supported by a doctoral training grant (PRE2020-094716) from MCIN/AEI/10.13039/501100011033 and ESF, and a mobility grant from Fundació Universitària Agustí Pedro i Pons. C. Montironi is supported by a Rio Hortega fellowship (CM19/00039) from ISCIII and ESF. J. Abril-Fornaguera is supported by a doctoral training grant from the University of Barcelona (PREDOCS-UB 2020) and a mobility grant from Fundació Universitària Agustí Pedro i Pons. J. Peix is supported by a PERIS ICT-Suport grant from the Departament de Salut de la Generalitat de Catalunya (SLT017/20/000206). A. Mesropian is supported by is supported by a FI-SDUR pre-doctoral support grant (BDNS 550325) from the Agency for Management of University and Research Grants (AGAUR) and the Generalitat de Catalunya. U. Balaseviciute is supported by an EILF-EASL Juan Rodés PhD Studentship (EASL_JR_12_20) from the European Association for the Study of the Liver (EASL) and the EASL International Liver Foundation (EILF). J.M. Llovet is supported by grants from the European Commission (EC) Horizon 2020 Program (HEPCAR, proposal number 667273-2), the NIH (RO1DK56621 and RO1DK128289), the Samuel Waxman Cancer Research Foundation, the Spanish National Health Institute (MICINN, SAF-2016-76390 and PID2019-105378RB-I00), through an Accelerator award in partnership between Cancer Research UK, Fondazione AIRC and Fundación Científica de la Asociación Española Contra el Cáncer (HUNTER, ref. C9380/A26813), and by the Generalitat de Catalunya (AGAUR, SGR-1358). We are indebted to the Cytometry and Cell Sorting Core Facility of the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), particularly Dr. Isabel Crespo, for excellent flow cytometry technical assistance.
Conflicts of interest
This study was supported by a research grant from Ipsen. J.M.L. is receiving research support from Bayer HealthCare Pharmaceuticals, Eisai Inc, Bristol-Myers Squibb, Boehringer-Ingelheim and Ipsen, and consulting fees from Eli Lilly, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Eisai Inc, Celsion Corporation, Exelixis, Merck, Ipsen, Genentech, Roche, Glycotest, Nucleix, Sirtex, Mina Alpha Ltd and AstraZeneca. All other authors report no conflicts of interest.
List of abbreviations
- CRR
complete response rate
- DEG
differentially-expressed genes
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- ICI
immune checkpoint inhibitor
- MPO
myeloperoxidase
- NASH
non-alcoholic steatohepatitis
- NE
Neutrophil Elastase
- ORR
objective response rate
- OS
overall survival
- PD1
programmed cell death protein 1
- PDGF
platelet-derived growth factor
- PFS
progression free survival
- TKI
tyrosine kinase inhibitor
- VEGF
vascular endothelial growth factor
- VETCs
vessels encapsulating tumour clusters
References
- 1.International Agency for Research / World Health Organization (IARC/WHO). Global Cancer Observatory (GLOBOCAN). Int Agency Res World Heal Organ [Internet]. 2021;419:2–3. Available from: https://gco.iarc.fr/today/fact-sheets-cancers [Google Scholar]
- 2.Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2021;7:6. [DOI] [PubMed] [Google Scholar]
- 3.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. United States; 2020;382:1894–905. [DOI] [PubMed] [Google Scholar]
- 4.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J Clin Oncol. Wolters Kluwer; 2021;39:267–267. [Google Scholar]
- 5.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib for advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. [DOI] [PubMed] [Google Scholar]
- 6.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–52. [DOI] [PubMed] [Google Scholar]
- 8.Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol 2018. page 599–616. [DOI] [PubMed] [Google Scholar]
- 10.Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22:1865–74. [DOI] [PubMed] [Google Scholar]
- 11.Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol Nature Publishing Group; 2018. page 310–24. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8++ T cells in tumors. J Exp Med. The Rockefeller University Press; 2015;212:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. 2017;9:eaak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol Res. 2014;2:632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abou-Alfa GK, Meyer T, Cheng A-L, El-Khoueiry AB, Rimassa L, Ryoo B-Y, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou L, Liu XD, Sun M, Zhang X, German P, Bai S, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. 2016;35:2687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang Q, Chen W, Ren M, Wang J, Zhang H, Deng DYB, et al. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin Cancer Res. 2014;20:2959–70. [DOI] [PubMed] [Google Scholar]
- 20.Yau T, Zagonel V, Santoro A, Acosta-Rivera M, Choo SP, Matilla A, et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J Clin Oncol. American Society of Clinical Oncology; 2020;38:478–478. [Google Scholar]
- 21.Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. Massachusetts Medical Society; 2021;384:829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rimassa L, Cheng A-L, Braiteh F, Chaudhry A, Benzaghou F, Thuluvath P, et al. Phase III (COSMIC-312) study of cabozantinib (C) in combination with atezolizumab (A) vs sorafenib (S) in patients (pts) with advanced hepatocellular carcinoma (aHCC) who have not received previous systemic anticancer therapy. Ann Oncol. 2019;30:v320. [Google Scholar]
- 23.Kelley RK, Yau T, Cheng A-L, Kaseb A, Qin S, Zhu AX, et al. VP10-2021: Cabozantinib (C) plus atezolizumab (A) versus sorafenib (S) as first-line systemic treatment for advanced hepatocellular carcinoma (aHCC): Results from the randomized phase III COSMIC-312 trial. Ann Oncol. 2022;33:114–6. [Google Scholar]
- 24.Patnaik A, Swanson KD, Csizmadia E, Solanki A, Landon-Brace N, Gehring MP, et al. Cabozantinib eradicates advanced murine prostate cancer by activating antitumor innate immunity. Cancer Discov. 2017;7:750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song EK, Tai WM, Messersmith WA, Bagby S, Purkey A, Quackenbush KS, et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int J Cancer. 2015;136:1967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology. 2017;153:812–26. [DOI] [PubMed] [Google Scholar]
- 27.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009/09/03. 2009;69:7385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyault S, Rickman DS, De Reyniès A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2006/12/26. 2007;45:42–52. [DOI] [PubMed] [Google Scholar]
- 30.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0 [2]. Nat. Genet 2006. page 500–1. [DOI] [PubMed] [Google Scholar]
- 31.Kuleshov M V, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Méndez-González J, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. 2015;61:1945–56. [DOI] [PubMed] [Google Scholar]
- 33.Ally A, Balasundaram M, Carlsen R, Chuah E, Clarke A, Dhalla N, et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torrens L, Montironi C, Puigvehí M, Mesropian A, Leslie J, Haber PK, et al. Immunomodulatory Effects of Lenvatinib Plus Anti–Programmed Cell Death Protein 1 in Mice and Rationale for Patient Enrichment in Hepatocellular Carcinoma. Hepatology. 2021;74:2652–69. [DOI] [PubMed] [Google Scholar]
- 36.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. NIH Public Access; 2009;16:183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grieshaber-Bouyer R, Radtke FA, Cunin P, Stifano G, Levescot A, Vijaykumar B, et al. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat Commun. 2021;12:2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Galarreta MR, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-catenin activation promotes immune escape and resistance to anti–PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9:1124–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinyol R, Sia D, Llovet JM. Immune exclusion-WNT/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin Cancer Res. 2019;25:2021–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res. 2019;25:2116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao WK, Chen D, Li SQ, Fu SJ, Peng BG, Liang LJ. Prognostic significance of neutrophil-lymphocyte ratio in hepatocellular carcinoma: A meta-analysis. BMC Cancer. 2014;14:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-β gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47:2059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montironi C, Castet F, Haber PK, Pinyol R, Torres‐Martín M, Mesropian A, et al. Immune classification of hepatocellular carcinoma based on new molecular features: the inflamed class. J Hepatol. 2021;75:S241–2. [Google Scholar]
- 46.Haber PK, Puigvehí M, Castet F, Lourdusamy V, Montal R, Tabrizian P, et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology. 2021;161:879–98. [DOI] [PubMed] [Google Scholar]
- 47.Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. Elsevier B.V; 2015;10:562–73. [DOI] [PubMed] [Google Scholar]
- 48.Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. Elsevier B.V; 2018;23:3946–3959.e6. [DOI] [PubMed] [Google Scholar]
- 49.Renne SL, Woo HY, Allegra S, Rudini N, Yano H, Donadon M, et al. Vessels Encapsulating Tumor Clusters (VETC) Is a Powerful Predictor of Aggressive Hepatocellular Carcinoma. Hepatology. 2020;71:183–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated or analysed by the authors is available in Gene Expression Omnibus (GEO) at GSE174770, GSE60939, GSE63898 and GSE174570.
Full details of the Materials and Methods are given in the Supplementary Data file.