

Abstract
Familial dysautonomia (FD) is an autosomal recessive neurodegenerative disease caused by a splicing mutation in the gene encoding Elongator complex protein 1 (ELP1, also known as IKBKAP). This mutation results in tissue-specific skipping of exon 20 with a corresponding reduction of ELP1 protein, predominantly in the central and peripheral nervous system. Although FD patients have a complex neurological phenotype caused by continuous depletion of sensory and autonomic neurons, progressive visual decline leading to blindness is one of the most problematic aspects of the disease, as it severely affects their quality of life. To better understand the disease mechanism as well as to test the in vivo efficacy of targeted therapies for FD, we have recently generated a novel phenotypic mouse model, TgFD9; IkbkapΔ20/flox. This mouse exhibits most of the clinical features of the disease and accurately recapitulates the tissue-specific splicing defect observed in FD patients. Driven by the dire need to develop therapies targeting retinal degeneration in FD, herein, we comprehensively characterized the progression of the retinal phenotype in this mouse, and we demonstrated that it is possible to correct ELP1 splicing defect in the retina using the splicing modulator compound (SMC) BPN-15477.
Introduction
Familial dysautonomia (FD, MIM223900), also known as Riley–Day syndrome or hereditary sensory and autonomic neuropathy type III (HSAN III), is a rare, recessive, early onset, sensory and autonomic neurodegenerative disorder caused by T to C nucleotide change in the 5′splice site of intron 20 of the Elongator complex protein 1 (ELP1 also known as IKBKAP). This mutation leads to tissue-specific skipping of exon 20 and to a corresponding reduction of ELP1 protein predominantly in the central and peripheral nervous system (CNS and PNS) (1–3). ELP1 is one of the scaffolding subunits of the Elongator complex (4–6), a highly conserved protein complex involved in transcriptional elongation, tRNA modification and cytoskeletal remodeling (6–9). Several studies have demonstrated the role of ELP1 in neurogenesis, neuronal survival and differentiation as well as peripheral tissue innervation (10–20).
The major clinical features of FD are all due to progressive depletion of sensory and autonomic neurons (2,21,22). Common symptoms include gastrointestinal dysfunction, gastroesophageal reflux, vomiting crises, recurrent pneumonia, seizures, gait abnormalities, kyphoscoliosis, postural hypotension, hypertension crises, absence of fungiform papillae on the tongue, decreased deep-tendon reflexes, defective lacrimation and impaired pain and temperature perception (3,23–29) Despite this complex neurological phenotype, FD patients also suffer from progressive visual dysfunction that severely affects their quality of life (30–32). Initially, it was reported that the loss of vision in FD patients resulted from corneal opacities, neovascularization and sensory defects such as corneal analgesia, severe dry eye, ulceration healing and incomplete closure of eye lids (33–37). However, recent detailed studies have shown that decreased visual acuity, loss of central vision and temporal optic nerve pallor occur in FD patients even without any corneal complications, suggesting a neuro-ophthalmic nature of the disease (31). In FD, visual impairment is usually early onset and often progresses to legal blindness in the third decade of life (30). Individuals with FD show a significant reduction in the thickness of the retinal nerve fiber layer (RNFL) due to death of retinal ganglion cells (RGCs) (30–32). FD shares several similarities with other optic neuropathies caused by mitochondrial gene mutations such as Leber’s hereditary optic neuropathy and dominant optic atrophy (31).
Many efforts have been undertaken in the field to develop therapies aimed to correct ELP1 splicing defects, including splicing modulator compounds (SMCs), antisense oligonucleotide (ASO) and modified exon-specific U1 small nuclear RNA (38–40). Previously, our team has identified the small-molecule kinetin (6-furfurylaminopurine) to be an orally active splicing modulator of ELP1 both in vitro and in vivo (41,42). As part of the NIH Blueprint Neurotherapeutics Network we have created a new class of highly potent SMCs, using kinetin as a starting molecule, and identified BPN-15477, a more potent and efficacious ELP1 splicing modulator (43). Despite this significant progress, the field has not yet developed a therapy to prevent retinal degeneration in FD. The creation and characterization of a mouse model able to recapitulate the retinal phenotype is the first step toward this effort. Two different Elp1 conditional knock-out (CKO) mice with retinal degeneration were previously described, the Tuba1a-cre; Elp1flox/flox and the Pax6-cre; Elp1flox/flox mice (44,45). In the Tuba1a-cre; Elp1flox/flox mouse, Elp1 is deleted in the nervous system and in the Pax6-cre; Elp1flox/flox mouse, Elp1 is deleted specifically in the retina (44,45). Both these mice exhibit loss of RGCs and they are useful models to investigate the retinal pathology associated with complete loss of Elp1. However, since FD is caused by a reduction, not loss, of ELP1, they do not precisely model the human molecular defect and therefore they cannot be used to evaluate the in vivo efficacy of splicing modulator therapies. Most recently, we have generated a new phenotypic mouse model for FD by introducing the human ELP1 transgene with the FD splice mutation (TgFD9) into a Elp1 hypomorphic mouse (Elp1Δ20/flox) (46,47). The TgFD9; Elp1Δ20/flox mouse recapitulates the same tissue-specific mis-splicing observed in FD patients and the major neurological symptoms of the disease (48). In the present study, we have characterized the progression of retinal disease in this FD mouse model and identified quantifiable retinal phenotypes which can be used to evaluate the efficacy of splicing targeted therapeutics in preventing retinal degeneration in FD.
Results
The FD phenotypic mouse recapitulates the optic neuropathy observed in FD patients
The RNFL is composed of non-myelinated axons of RGCs, which converge into the optic nerve. In several hereditary optic neuropathies, including glaucoma, the thinning of RNFL thickness results from damage of RGC axons which precedes the loss of RGCs (49–56). Many studies have demonstrated that patients with FD exhibit reduction in thickness of the RNFL layer due to the death of RGCs (30–32), and this loss is more profound near the temporal region of the optic nerve, specifically in the maculo-papillary region. Recently, we have generated a novel FD phenotypic mouse model, the TgFD9; Elp1Δ20/flox mouse, by introducing a transgene carrying the human ELP1 gene with the FD splice mutation (TgFD9) into an hypomorphic mouse that expresses low levels of endogenous Elp1 (Elp1Δ20/flox) (46–48). For simplicity, we will refer to TgFD9; Elp1Δ20/flox mouse as the FD phenotypic mouse throughout the manuscript. Seeking to develop therapies targeting retinal degeneration in FD, we comprehensively characterized the progression of the retinal phenotype in this FD mouse model. We measured the thickness of RNFL and ganglion cell-inner plexiform layer (GCIPL) in the superior, inferior, nasal and temporal hemispheres of the retina using high-definition spectral domain optical coherence tomography (SD-OCT) (Fig. 1A). At 3 months of age, we observed a significant reduction of RNFL and GCIPL layers in the temporal portion of the FD retina, as previously observed in FD patients. Starting from 6 months of age the thickness of RNFL and GCIPL layer becomes more uniformly reduced throughout the retina (Fig. 1B and C). The thinning of these layers further progresses at 12 and 18 months of age in FD mice compared to control mice (Fig. 1B and C) Even though, reduction of RNFL and GCIPL has been initiated in the temporal region of the retina, we observed uniform thinning of these layers throughout the retina after 3 months of age. These results indicate that our mouse model recapitulates the reduction of retinal thickness observed in FD patients.
Figure 1.

Measurement of RNFL and GCIPL thickness in FD mice using SD-OCT. (A) Representative SD-OCT b-scan images of FD and control retinae at 3, 6, 12 and 18 months. (B) Quantification of thickness measurements of RNFL in both central and peripheral regions of the retina in nasal and temporal hemispheres at 3 months (n = 8; central temporal: P < 0.0372), 6 months (n = 9; P < 0.0004), 12 months (n = 6; P < 0.0004) and 18 months (n = 3; central temporal: P < 0.0072; central nasal: P < 0.0148: peripheral temporal: P < 0.0012; peripheral nasal: P < 0.0088) of age. (C) Measurement of GCIPL thickness in both central and peripheral regions of the retina in nasal and temporal hemispheres at 3 months (n = 8; central temporal: P < 0.022), 6 months (n = 9; P < 0.0004), 12 months (n = 6; P < 0.0004) and 18 months (n = 3; central temporal: P < 0.0004; central nasal: P < 0.012: peripheral temporal: P < 0.0152; peripheral nasal: P < 0.0346) of age. Gray bars represent FD and white bars represent control mice. *P < 0.05, **P < 0.01 and ***P < 0.001. Error bars represent SEM. P value was calculated using students t-test with Bonferroni correction (4 tests, one for each retinal region).
Selective loss of RGCs in the FD mouse retina
To evaluate if the reduction of RNFL and GCIPL thickness in the FD mice was due to loss of RGCs, we performed RGC counts in superior, inferior, nasal and temporal regions of the retina using retinal whole-mount analysis. We stained retinae from 3, 6- and 18-month-old mice using the RGC specific marker RNA-binding protein with multiple splicing (RPBMS) and we counted the number of RPBMS+ cells sited at 1 mm from the optic nerve head (ONH) (Fig. 2A and B). At 3 months of age, although we observed a trend toward reduction in RPBMS+ cells in the FD mice, the difference when compared with the control mice was not significant (Fig. 2C). The RGC loss further progresses and, at 6 months, the number of RPBMS+ cells became significantly lower in the FD mice in the temporal (P < 0.002), nasal (P < 0.001), superior (P < 0.005) and inferior (P < 0.006) regions of the retina. At 12 months of age, the number of RPBMS+ cells in the FD retinae, although significantly lower than in the control mice, is comparable with the 6-month-old counts, indicating that the major RGC loss occurs between 3 and 6 months of age (Fig. 2C). As expected, the control mice showed an age-dependent decline in the number of RGCs at 18 months. These results confirmed that the reduction in RNFL and GCIPL thickness results from progressive loss of RGCs.
Figure 2.

Progressive degeneration of RGCs in the phenotypic FD mouse model. Representative retinal whole-mount images from control and FD mice stained with RGC marker RBPMS. Scale bars, 100 μm. RPBMS+ cells in control and FD retinae were counted in each quadrant in superior (S), inferior (I), nasal (N) and temporal regions at 1 mm from the optic nerve head (ONH) at 3, 6 and 18 months of age. Scale bars, 350 μm. (B) Higher magnification representative RPBMS staining images of control and FD retinae. Scale bars, 100 μm. (C) Bar plots of RPBMS+ cell counts in 3 month- (n = 5), 6 month- (n = 6) and 18-month-old (n = 6) retinae from control and FD mice. Significant reduction in the number of RGCs was observed starting at 6 months of age and progresses at 18 months in FD mice compared to control mice. Gray bars represent FD and white bars represent control mice. *P < 0.05, **P < 0.01 and ***P < 0.001. Error bars represent SEM. P value was calculated using students t-test.
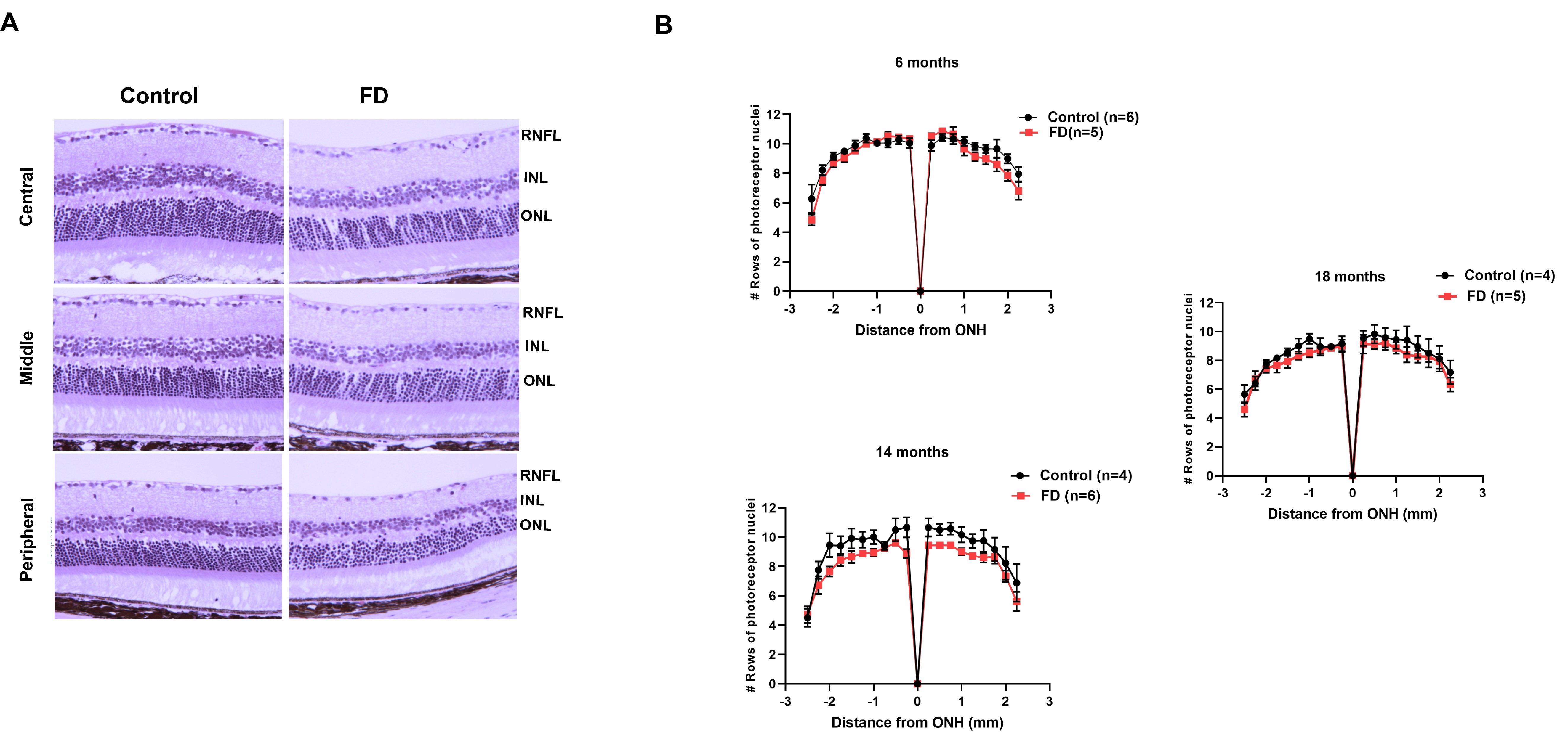
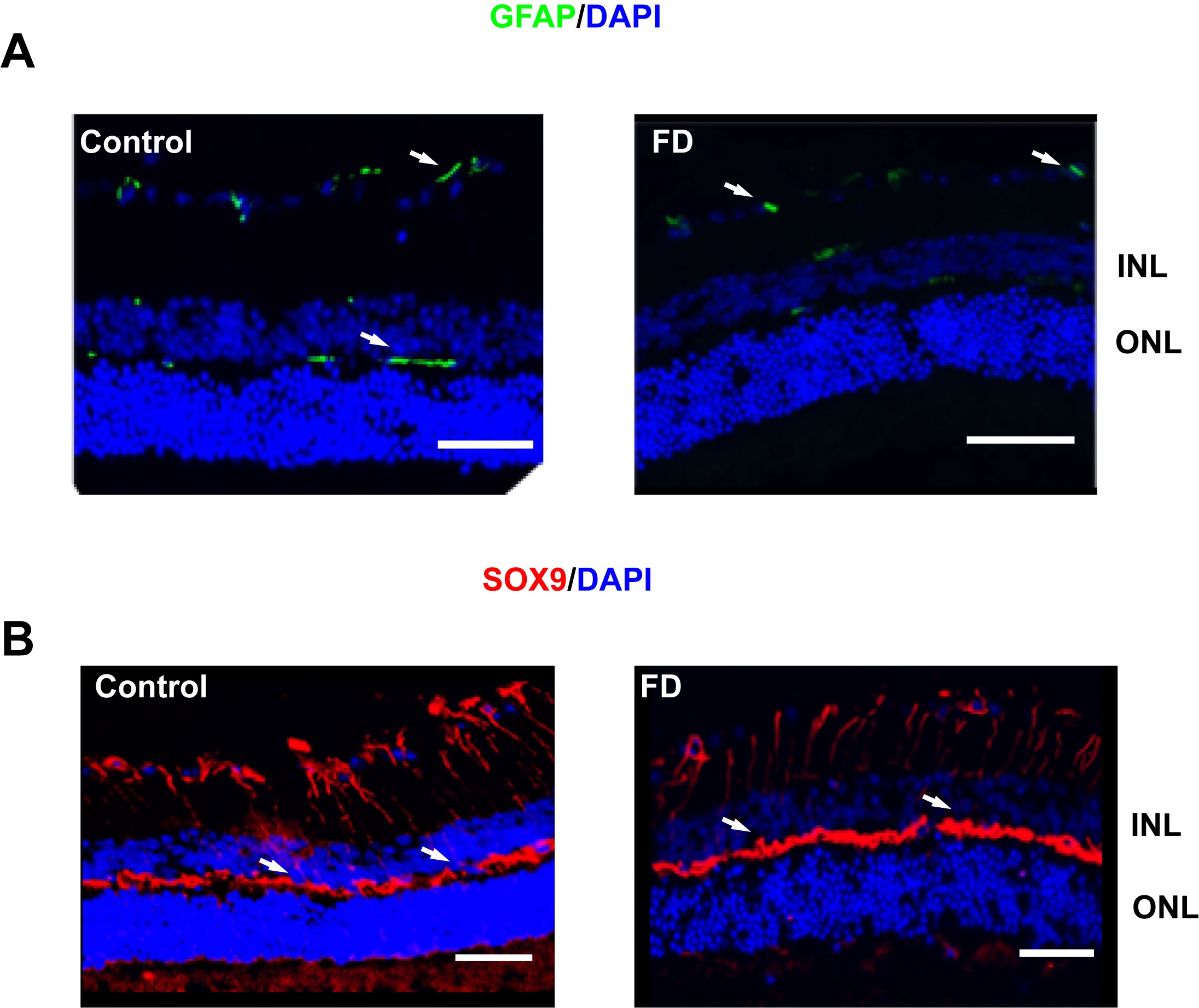
Patients with FD do not show any significant photoreceptor loss; however, degeneration of photoreceptors was observed in Tuba1a-cre; Elp1flox/flox mice (45). To investigate whether RGC loss led to photoreceptor degeneration, we analyzed the number of photoreceptors in our FD mice and we did not observe significant changes at any age points tested (Supplementary Material, Fig. S1). We also stained the retina with GFAP and Sox-9 antibodies to determine if there was any glial activation in the FD mouse as observed in the retina of Tuba1a-cre; Elp1flox/flox mice (45), and we did not observed any differences between control and FD mice (Supplementary Material, Fig. S2).
Optic nerve degeneration in the FD phenotypic mice
Histopathological analysis of the optic nerve from FD patients indicates diffuse degeneration of axonic bundles (30). To investigate if our FD mouse model also recapitulates this aspect of the disease, we collected optic nerves and retinae from FD mice and stained the axonal structures and the cell bodies of RGCs using the neurofilament (NF) antibody SMI-32. We performed this analysis at 8 months of age because our previous results have shown that the major RGC loss in FD mice occurs within the first 6 months of age. NF-staining of longitudinal optic nerve sections revealed that although the optic nerve fibers in the control mice were long, parallel and undistorted, the fibers in the FD optic nerve had significantly more retraction bulbs and holes (Fig. 3A). We therefore morphologically evaluated these alterations in the optic nerve sections using the SMI-32 score (57). This score assesses the structural integrity of the optic nerve fibers. It is usually calculated based on SMI-32 staining and it is graded as 0 for intact structure and no retraction bulbs, 1 for short axons and occasional retraction bulbs and 2 for numerous retraction bulbs and holes. The SMI-32 score of the FD optic nerves was 1.5, two times the score of the control optic nerve (P < 0.038; Fig. 3B). We then investigated whether these alterations led to thinning of the optic nerve by measuring the circumference of the DAPI-stained optic nerve cross sections (Fig. 3C). We found that the circumference of FD optic nerves was significantly smaller compared to the control optic nerves (P < 0.025; Fig. 3C and D). Finally, in order to analyze the number of axonal bundles entering the optic nerve, we performed NF-staining of whole-mount retinae (Fig. 3E and F). We counted the axonal nerve bundles entering the optic nerve in superior, inferior, nasal and temporal hemispheres. Interestingly, the number of axonal bundles in the FD retinae was significantly lower compared to the control retinae in every retinal region tested (Superior: P < 0.007, inferior: 0.0001, temporal: 0.0005, nasal: 0.005; Fig. 3G). Overall, our findings demonstrate that the FD mouse correctly models the optic nerve degeneration observed in FD patients.
Figure 3.

Loss of axonal bundles and thinning of optic nerve in the FD phenotypic mouse. (A) Representative optic nerve sections in 8-month-old FD and control retinae stained with NF (SMI-32) antibody. NF-staining indicates numerous retraction bulbs, shorter axons and holes in the optic nerve tissue of FD mice when compared with control mice. Scale bars, 100 μm. Arrows indicate holes and asterisks indicate retraction bulbs in the image. (B) Graph representing the scoring of SMI-32-stained optic nerve sections revealed significant degeneration of optic nerve evidenced by structural distortion of the optic nerve. One tailed Wilcoxon rank-sum test was used to analyze the statistical significance (P < 0.038, n = 3). (C) Staining of optic nerve cross sections at 8 months using DAPI. Scale bars, 50 μm. Optic nerve cross sections were obtained ~300 μm from the globe. (D) Bar plot representing the measurement of the optic nerve circumference using image J. Significant reduction in the circumference of the optic nerve was observed in FD mice at 8 months (P < 0.025: n = 3). (E) Representative picture of NF (SMI-32) staining of retinal whole-mounts from mice at 8 months of age. Scale bars, 200 μm. (F) Higher magnification image indicates axonic bundles surrounding the optic nerve. Some RGCs were also observed with NF staining. (G) Measurement of number of axonic bundles within the ONH. Axonic bundles were counted 0.5 mm from the ONH in 1 mm square at superior (P < 0.007), inferior (P < 0.0001), nasal (P < 0.0005) and temporal (P < 0.005) regions (n = 3). Gray bars represent FD and white bars represent control mice.
ELP1 exon 20 inclusion is lower in RGCs compared to other retinal cells
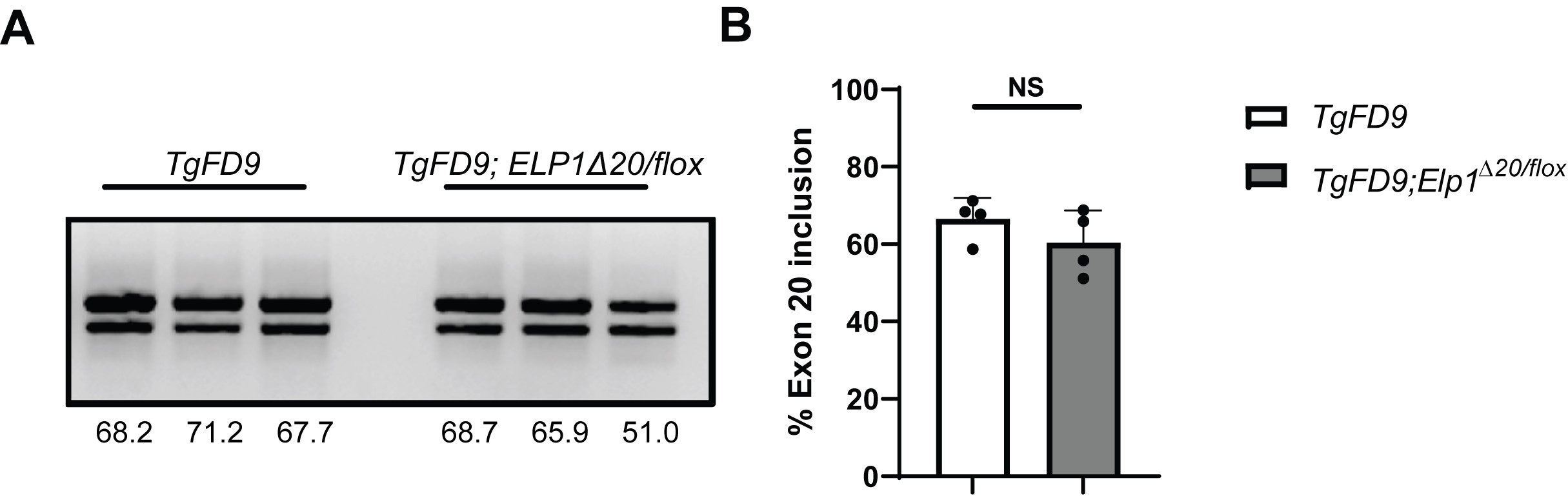
Although ELP1 is ubiquitously expressed, the major FD splicing mutation leads to a tissue-specific skipping of ELP1 exon 20 with the lowest amount of full-length ELP1 expression in the CNS and PNS (20,46,48,58). In order to assess the splicing of the human ELP1 transgene in different regions of the eye, we collected lens, cornea, retina, iris, optic nerve and posterior eye cup from the transgenic mice TgFD9 (46). For this analysis, we specifically used the TgFD9 mice because it is a suitable model to assess the FD splicing defect in vivo as the splicing pattern of the human ELP1 transgene is identical between the TgFD9 and the TgFD9; IkbkapΔ20/flox mice (46,48) (Supplementary Material, Fig. S4).
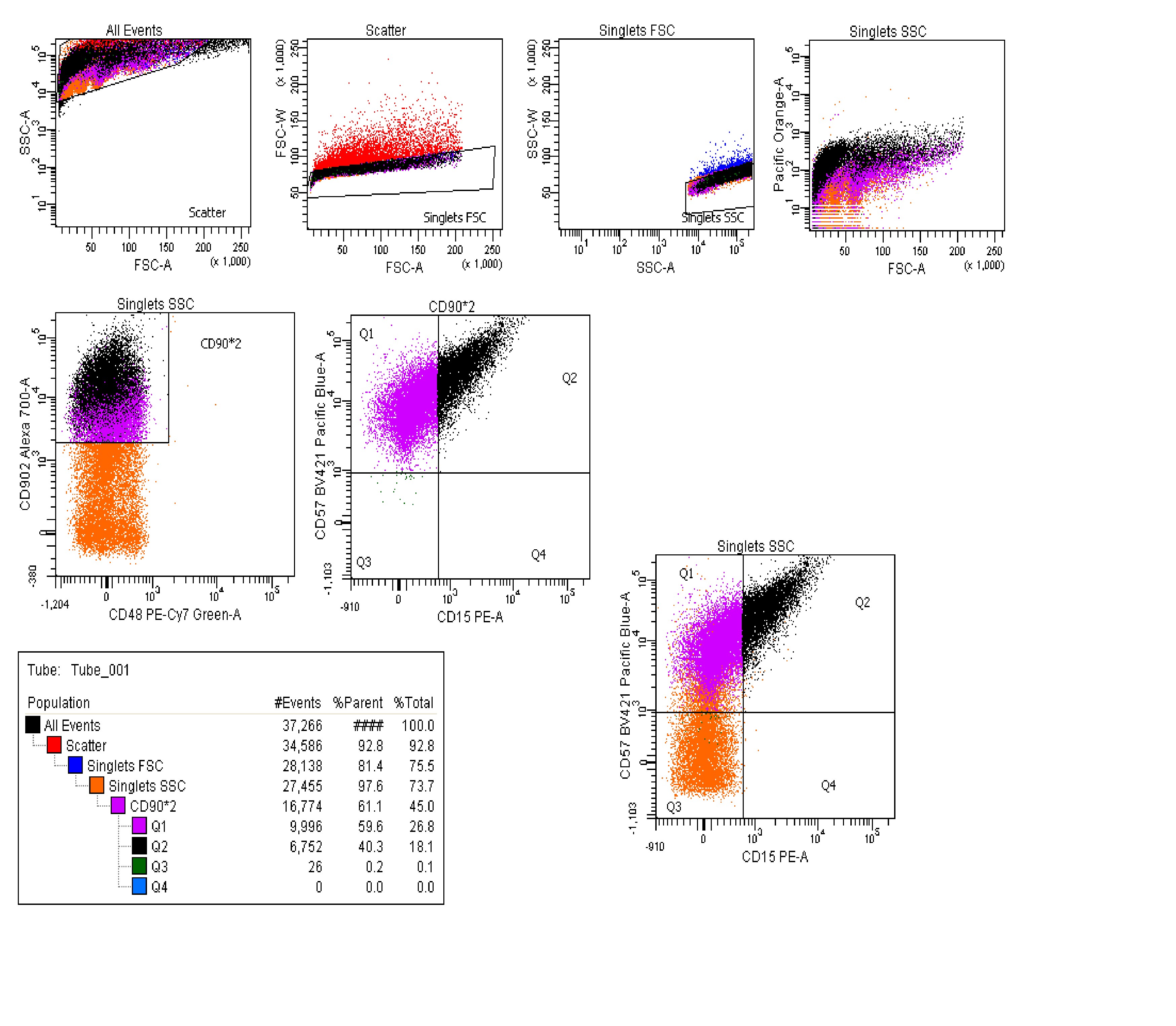
As shown in Figure 4, the amount of exon 20 inclusion was lower in the cornea and optic nerve when compared to other ocular tissues tested (Fig. 4A and B). Surprisingly, although the retina is part of the CNS, it was not the ocular tissues with the lowest amount of full-length ELP1 transcript expression. We therefore hypothesized that the mutant ELP1 transcript might splice differently in distinctive retinal cells and assessed ELP1 splicing in RGCs, because they are the most affected cell type in the FD (Fig. 4C). As RGCs comprise only 1% of the total retinal cells, the ELP1 splicing analysis of the entire retina might not necessarily represents the splicing efficacy of this specific subpopulation of neurons. RGCs were enriched from TgFD9 retinae with fluorescence activated cell sorting (FACS) using positive selection for the antithymocyte antigen (Thy1/CD90.2). Interestingly, exon 20 inclusion was significantly reduced in RGCs when compared to other retinal cells (Fig. 4C, D and Supplementary Material, Fig. S3) supporting the idea that the RGC loss observed in the FD retinae might result from ELP1 amounts falling below cell-specific threshold.
Figure 4.

Ocular- and cell-specific mis-splicing of the human FD ELP1 transgene. (A) Splicing analysis of human ELP1 transcripts in the retina, lens, optic nerve, iris, cornea and posterior eye cup of 3–4-month-old FD mice. (B) Quantification of the percentage of exon 20 inclusion in various ocular tissues (n = 6). (C) Splicing analysis of human ELP1 transgene in RGCs and non-RGC retinal cell population compared to whole retinal tissue. (D) Quantification of percentage of exon 20 inclusion in RGCs compared to other retinal cells (n = 3).
The small-molecule BPN-15477 corrects ELP1 splicing defect in the mouse retina
Our work has been focused on the development of small-molecule SMCs to correct ELP1 splicing defect in FD. We have shown that daily consumption of kinetin (6-furfuryl amino purine) rescues neuronal phenotype in our FD mouse model and, more recently, we have identified a novel SMC, BPN-15477, significantly more potent and efficacious than kinetin (40,60). However, we have never tested the ability of our SMCs to correct ELP1 splicing in the retina. Prior to initiating preclinical trials in our phenotypical FD mouse model, we sought to determine if BPN-15477 could modulate exon 20 inclusion in the retina of the TgFD9 mouse. This mouse is phenotypically normal, as it expresses normal amounts of endogenous Elp1 (46). However, it perfectly recapitulates the ELP1 splicing defect observed in patients and therefore is the ideal model to test the effect of SMCs on splicing in vivo (59). BPN-15477 was administered to the TgFD9 mice starting at birth through special formulated chow. The special chow was formulated so that each mouse received 70 mg/kg/day. At birth, pups were randomly assigned to vehicle- or BPN-15477-treated groups and were maintained in the same treatment regime for 5 weeks. The treatment was well tolerated, and no weight loss or adverse effects were observed in the treated group (60). To assess ELP1 mRNA splicing, RT-PCR assays were performed on total RNA extracted from brain and retina of the mice carrying the TgFD9 transgene. Remarkably, BPN-15477 treatment significantly improved ELP1 exon 20 inclusion in the brain and fully corrected ELP1 splicing in the retina in the BPN-15477-treated mice when compared to the vehicle-treated mice (Fig. 5). This result demonstrated that our splicing modulator BPN-15477 efficiently crosses the blood retinal barrier and therefore can be used to correct ELP1 splicing defect in the retina.
Figure 5.

BPN-15477 treatment improves exon 20 inclusion in the retina and brain of mice carrying the human FD transgene TgFD9. (A) Representative gel picture indicating splicing analysis of human ELP1 transcript in the brain from TgFD9 mice treated with −70 mg/Kg/day of BPN-15477. (B) Quantification of exon 20 inclusion in the brain (n = 6; P < 0.0001). (C) Representative gel picture of splicing analysis of human ELP1 transcript in the retina. Note the near complete correction of ELP1 splicing in TgFD9 mice after BPN-15477 treatment. (D) Quantification of exon 20 inclusion in the retina (n = 6; P < 0.0001). Gray bars represent FD and white bars represent control mice.
Discussion
FD is a devastating neurodegenerative disorder caused by a splicing mutation in the ELP1 gene. Despite the severe sensory and neurological manifestations, one of the most debilitating aspect of the disease is progressive visual loss. Previous studies using high-definition OCT showed that all FD patients suffer from an optic neuropathy featured by reductions of the RNFL which is due to loss of the macular RGCs (30,61). Although, pharmacological and surgical supportive treatments are available to alleviate some of the disease symptoms (62), to date there are no therapeutic interventions aimed at stopping vision loss and most of the patients become visually impaired or legally blind after their third decade of life. We have comprehensively characterized the retinal pathology of the FD phenotypic mouse model using a combination of in vivo imaging and histological analyses and show that this mouse accurately models the human phenotype both in terms of disease progression and severity (30). Using SD-OCT, we have shown that the reduction of RNFL and GCIPL layers in the FD mice starts as early as 3 months of age and continues to progress. These findings are consistent with previous studies in the Pax6-cre; Elp1flox/flox mouse showing that while Elp1 is required for RGC survival and maintenance, is not essential for their development (44). Our FD mouse also correctly recapitulates the severity of the retinal cell loss observed in FD patients (30–32). By 6 months of age the FD mice showed 50% reduction in the thickness of RNFL, similarly, the FD patients showed the same reduction in RNFL thickness by 27 years of age. Functional vision assessments in FD patients revealed that global reduction in RNFL thickness leads to a significant visual acuity loss (32), therefore, it is very likely that the reduction in RNFL thickness observed in our FD mice at 6 months of age would translate into a comparable functional deficit. However, we recognize that in the future, functional studies will be necessary to determine the extent of the visual loss caused by the structural defects observed in the FD mice. Although ELP1 is expressed in all major retinal neurons (44), we have shown that the retinal degeneration in FD is specific to RGCs and no significant differences in the photoreceptor number were observed even in old mice. Moreover, we have proven that our phenotypic mouse correctly models the optic nerve degeneration observed in patients. This extensive characterization allowed us to identify quantifiable endpoints that can be used for future preclinical trials aimed at testing the efficacy of existing or novel disease modifying therapies targeting the retina.
Many efforts have been undertaken in the field to better understand the pathophysiological mechanisms underlying retinal degeneration and to develop disease modifying therapies for FD. The majority of these approaches aim to correct the ELP1 splicing defect and include SMCs, ASO and modified exon-specific U1 small nuclear RNA (38–40). Gene replacement strategies have been hindered by the large size of the ELP1 gene. The creation and characterization of a mouse model able to recapitulate the retinal phenotype now provides a crucial resource for testing all of these potential therapies. The previously described Elp1 CKO mice, Tuba1a-cre; Elp1flox/flox and Pax6-cre; Elp1flox/flox, are useful models to investigate the retinal pathology associated with complete loss of Elp1, however, they do not accurately model the molecular cause of the disease and therefore they cannot be used to evaluate the in vivo efficacy of splicing modulator therapies (44,45). Conversely, the retinal phenotype of the TgFD9; Elp1Δ20/flox mouse recapitulates the pathogenic mechanism of disease and therefore is the ideal model system to assess the in vivo efficacy of splicing correction in the retina (48). We have previously shown that daily consumption of kinetin rescues neuronal phenotypes in this mouse model (40), however, prior to initiation of a long-term efficacy study to evaluate BPN-15477 on retinal degeneration, we utilized the TgFD9 humanized transgenic mouse to confirm that oral administration of the drug would reach the retina and modulate exon 20 inclusion. This mouse, although phenotypically normal, perfectly recapitulates the tissue-specific ELP1 splicing defect observed in FD patients and has been used to evaluate the effect of drugs in vivo (38,39,48,59). It is worth noting that during the generation of these transgenic mouse models insertional mutations might be occurred. Over the past 14 years, these mice have been extensively used to test in vivo splicing correction mediated by different therapeutic strategies including SMCs (48,59), ASOs (38) and modified exon-specific U1 small nuclear RNA (39). Splicing analysis of the human FD ELP1 transgene in different eye regions of the TgFD9 mouse showed that the highest amount of exon 20 inclusion was observed in the lens, whereas cornea and optic nerve were among the ocular tissues with lowest amount of full-length ELP1 transcript. During embryonic development, lens and cornea are derived from the surface ectoderm, whereas retina and optic nerve extends from the diencephalon (63). We have previously shown that the amount of full-length ELP1 expression is significantly lower in the CNS and PNS when compared to other tissues both in FD patients and in mouse models (20,46,48). Therefore, we were surprised to find that the retina, although part of the CNS and largely affected by the disease, expressed 63% of ELP1 full-length transcript. We then hypothesized that the mutant ELP1 transcript could splice differently in different retinal cells and assessed ELP1 splicing in RGCs because they are the most affected retinal neuronal subtype in FD. As anticipated, we found that ELP1 exon 20 inclusion was significantly lower in this specific subpopulation of neurons when compared to the rest of the retina, demonstrating that the RGC loss in FD might result from ELP1 amounts falling below a cell-specific threshold. Publicly available single-cell RNA-seq data in neonatal mice RGCs showed that, Elp1 is uniformly expressed in different RGC subtypes (64). However, it is very likely that in FD mice the splicing of the mutant transcript might vary in different RGCs rendering specific RGC subtype more vulnerable to the disease than others. In the future it would be interesting to evaluate ELP1 splicing in different RGC subtypes to determine if the effect of the FD mutation is cell-type specific (64).
Our work to develop SMCs that can correct ELP1 splicing led to the generation of BPN-15477, a potent and efficacious splice modifier (60). Transcriptome analysis of BPN-15477-treated fibroblasts showed that this compound modulates splicing selectively (60). To test the ability of our newly identified SMC to correct ELP1 splicing in the retina, we administered BPN-15477 to the TgFD9 mice starting at birth through special formulated chow. BPN-15477 fully corrects ELP1 splicing defect in the retina, proving for the first time the therapeutic potential of an oral treatment to prevent retinal degeneration in FD. Taken together, the identification of quantifiable retinal phenotypes as well as the demonstration that BPN-15477 can correct ELP1 splicing in the retina sets the stage for future preclinical testing that will move targeted treatments for blindness in FD to the clinic.
Materials and Methods
Animals: The generation of the human ELP1 transgene carrying the IVS20 + 6 T > C mutation and the creation of the TgFD9 mouse line can be found in Hims et al. (46). Descriptions of the original targeting vector used to generate the Elp1flox allele as well as the strategy to generate the Elp1Δ20 allele have been previously published (18,47). The generation of the mice carrying the Elp1flox allele as well as the mice carrying the Elp1 Δ20 allele can be found in Dietrich et al. (47). To generate the experimental TgFD9; Elp1Δ20/flox mouse, we crossed the TgFD9 transgenic mouse (TgFD9+/−) to mice heterozygous for the Elp1flox allele (Elp1flox/+). Pups were genotyped to identify those carrying both the TgFD9 transgene and the Elp1flox allele (F1: TgFD9; Elp1flox/+). These animals were then crossed with the mouse line heterozygous for the Elp1Δ20 allele (E1p1Δ20/+) to generate the FD mouse TgFD9; Elp1Δ20/flox (F2). Controls are littermates of the FD mice that carry the transgene but are phenotypically normal because they express the endogenous Elp1 gene (TgFD9; Elp1+/+, TgFD9; Elp1 flox/+ or TgFD9; Elp1 Δ20/+). Genetic background analysis using MiniMUGA v0009 panel (Transnetyx) of control and FD mice revealed that our mice are on a mixed background that includes C57BL/6 J and C57BL/6 N (65). Analysis of 156 potential diagnostic SNPs unique for C57BL/6 J indicated that ~56% of these SNPs are found in both our control and FD mice. These data are available upon request. Both sexes were included in this study. Mice were maintained according to the Association for Research in Vison and Ophthalmology statement for the use of animals in ophthalmic and vision research and with protocols approved by Institutional Animal Care and Use Committee of the Massachusetts General Hospital. The mice used for this study were housed in the animal facility of Massachusetts General Hospital (Boston, MA, USA), provided with constant access to a standard diet of food and water, and maintained on a 12-hour light/dark cycle. For BPN-15477 treatment, assigned mice were fed on BPN-15477 containing chow. Mice were genotyped by PCR amplification of the genomic DNA obtained from tail biopsies and using the following primers: Elp1 1F (5′-TGATTGACACAGACTCTGGCCA-3′) and Elp1 4R (5′-CTTTCACTCTGAAATTACAGGAAG-3′) to discriminate the Elp1 alleles and Tg Probe1 F (5′-GCCATTGTACTGTTTGCGACT-3′) and Tg Probe 1R(5′TGAGTGTCACGATTCTTTCTGC-3′) to discriminate the TgFD9 transgene.
Spectral domain optical coherence tomography
For in vivo imaging of the retina, mice were anesthetized by placing them in a mobile isoflurane induction chamber and the vaporizer was set to an isoflurane concentration of 2% at 2 L/min O2. Pupils of the mice were dilated using 2.5% phenylephrine, 1% tropicamide. About 0.5% proparacaine is used as a topical anesthetic during the procedure. SD-OCT imaging was performed using a Leica EnvisuR2210 OCT machine. Measurements were made 100 μm from the optic nerve for central retina and 1.5 mm from the optic nerve for peripheral retina. Control and FD mice at 3, 6, 12 and 18 months of age were analyzed. Linear B-scans of central and peripheral retina were performed, and the thickness of the RNFL and GCIPL layers were manually measured using Bioptigen InVivoView Clinc software. Each OCT image comprises of 100 B-scans, with each B-scan consisting of 1000 A-scans. We then analyzed four representative images per mouse, two for each eye and measurements from both eyes were included in the analysis.
RT-PCR analysis of ELP1 transcripts
Following euthanasia of the mice, eyes were enucleated along with the optic nerve and placed in phosphate buffered saline (PBS) on ice. Dissection was performed to separate cornea, lens, iris, optic nerve, retina and posterior eye cup from each eye and snap frozen using liquid nitrogen. The tissues were homogenized in ice-cold QIAzol (Qiagen) lysis reagent using a Tissue Lyser (Qiagen). Total RNA was extracted using the standard chloroform extraction procedure. Concentration of the total RNA for each sample was determined with a Nanodrop ND-1000 spectrophotometer. cDNA was synthesized by reverse transcription with ~0.5 μg of total RNA, Random Primers (Promega), and Superscript III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. To perform RT-PCR and detect ELP1 exon 20 inclusion, 100 ng of starting RNA equivalent cDNA was subjected to PCR reaction with the GoTaq green master mix (Promega) and 30 amplification cycles (94°C for 30 s, 58°C for 30 s and 72°C for 30 s). The human-specific forward and reverse primers spanning exon 19–21 of the ELP1 gene 5′-CCTGAGCAG CAATCATGTG−3′ and 5′-TACATGGTCTTCGTGACATC-3′ were used for amplification both wild type and mutant isoforms of human ELP1 transcript. The PCR products were resolved on 1.5% agarose gels containing ethidium bromide for detection. The relative amounts of normal and mutant ELP1 isoforms were quantified using ImageJ software, and the integrated density value for each band was determined and expressed as percentage as described previously (48).
RGC enrichment
RGCs were enriched from total retinal cells from a humanized transgenic mouse TgFD9. We followed previously published protocol with slight modifications for RGC enrichment (66). Briefly, a total of 10, 2–3-month-old TgFD9 mice were sacrificed, and eyes were enucleated. Subsequently, the retinae were pooled for FACS analysis. Retinal cell suspension was prepared in a solution containing PBS with 1% fetal bovine serum by gentle maceration and filtration by passing through a 70 μm nylon strainer. The retinal cells were immunolabeled using CD90.2AF-700, (1 μg) CD48 PE-Cyanine 7(0.4 μg), CD15 PE (0.02 μg) and untagged CD57 (0.4 μg). After 30 min incubation and washing cell suspension was incubated with BV421 (0.1 μg) secondary antibody. After washing twice, labeled retinal cell suspension was subjected to FACS sorting to enrich CD90.2+, CD48−, CD15−, CD57− phenotype. One eye from each mouse was used.
Immunohistochemistry
Eyes were enucleated and fixed in 4% paraformaldehyde overnight at 4°C. After a single PBS wash, eyes were cryoprotected in 30% sucrose overnight at 4°C and embedded in optimal cutting temperature compound (Sakura Finetek, Torrance, CA, USA) and cryosectioned into 15-μm thin sections. Sections were fixed in dry ice-cold acetone for 15 min and washed with PBS three times. Antigen retrieval was then performed by incubating the sections using the buffer containing 0.1mtris.HCL ph 8, 50 mM EDTA ph 8.0 and 20 μg/ml proteinase K for 10 min at room temperature. After washing two times in PBS, retinal sections were blocked with animal-free blocker (Vector Laboratories, Burlingame, CA, USA) containing 0.5% Triton X-100 for 1 h at room temperature, then primary antibodies anti-GFAP (Glial fibrillary acidic protein) (NeuroMab, Davis, CA, USA), anti-Sox9 (SRY-Box Transcription Factor 9) (EMD Millipore, Billerica, MA, USA) were applied and incubated at 4°C overnight in a moist chamber. Sections were washed three times with PBS with 0.1% triton X 100 and incubated with secondary antibodies (Invitrogen; Jackson Immuno Research, West Grove, PA, USA) for 1 h at room temperature. Sections were mounted with hard-set mounting media containing DAPI (Vector labs) and fluorescent microscopy was performed. One eye from each mouse was included in this analysis.
Retinal whole mounting and RGC counting
Staining and RGC counting of retinal whole-mounts was performed according to the method previously described by Ueki et al. (44,45). Briefly, fixation of the eyes was performed at room temperature for 1 h in 4% PFA and eyes were marked with a yellow tissue marking dye on the temporal surface. After fixation, retinae were isolated, with each temporal retina marked with a small cut. Relaxing cuts in the spherical retina were made on all four corners. Nonspecific binding was blocked by incubating with animal-free blocker containing 0.5% Triton X-100 overnight at 4°C, and anti-RPBMS antibody was applied overnight at 4°C. Retinae were incubated with secondary antibodies for 1 h at room temperature and mounted on slides. Images were acquired using a LeicaDMi8 epifluorescent microscope and MetaMorph 4.2 acquisition software (Molecular Devices, San Jose, CA, USA). Whole scans of complete flat mount samples were attained at 20X magnification using scan stage and autofocus. The total retinal area in the whole scan was ~14 mm2. With ImageJ software, the number of RPBMS+ cells were measured at an area of 1 × 1 mm square at 1 mm from the ONH at superior, inferior, temporal and nasal hemispheres as shown in the Supplementary Material, Figure S5 (45). If a specific square area was damaged, due to rips or folds in the retina, we have counted the RGCs in an adjacent undamaged area. Moreover, we have intentionally avoided to count RGCs in the edges of the retina because these areas usually have higher cell counts due to the downward pressure caused by the flattening cuts to the retina. We analyzed ~30% of the retina from one eye of each mouse. The investigator conducting the analysis was masked to the genotype.
Histological examination
Following euthanasia of the mice with CO2, eyes were marked with a yellow tissue marking dye on the temporal surface. Eyes were then enucleated and fixed overnight in 4% and embedded in paraffin for sectioning. Approximately 5-μm sections were cut through the center of the eye based on the position of the optic nerve and subjected to hematoxylin and eosin (H&E) staining. Images were acquired by light microscopy using MetaMorph 4.2 software. Whole retina images were digitally developed using scan stage function for automated image patching. Morphology of the retina of both FD and control mice (n = 5 each group) was analyzed with ImageJ. The number of rows of photoreceptor nuclei in a single column in the outer nuclear layer was counted as described previously (44). Nuclei counting was performed at intervals of 0.25 mm starting at the ONH and toward both the temporal and nasal retinal hemispheres. Each sample was imaged and measured in triplicate and the investigator conducting the experiment was masked to the genotype. One eye from each mouse was included in the analysis.
Oral administration of BPN-15477 using specially formulated diet
Transgenic mice carrying human ELP1 transgene with FD mutation, TgFD9; Elp1+/+, were fed special diet containing 0.08% of BPN-15477 (LabDiet 5P00 with 800 ppm BPN-15477), corresponding to a daily consumption of 70 mg/Kg/day from birth until 5 weeks of age. These mice are phenotypically normal as they express normal levels of endogenous Elp1. However, they perfectly recapitulate the ELP1 splicing defect observed in patients and therefore are the ideal model to test the effect of SMCs on splicing in vivo (48,59). In the control group, TgFD9 transgenic mice were fed with vehicle diet (LabDiet 5P00). The mice were given water ad libitum.
Statistical analysis
The data were statistically analyzed using graph pad prism software (La jolla CA, USA). Comparison between the two groups was performed using two-tailed independent Student’s t-tests. When one group was compared to more than one other group, we corrected for multiple comparisons by applying Bonferroni correction, and adjusted P values were reported. The data are presented as the mean—standard error. P values <0.05, <0.01 and <0.001 were considered as significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Dr Horacio Kaufmann at the Dysautonomia Treatment and Evaluation Center at New York University Medical School, Dr Frances Lefcort at Montana State University and Dr Carlos Mendoza at the Bascom Palmer Eye Institute for their long-standing collaboration and helpful discussions. We also thank Nate Nowak at Transnetyx for his help in interpreting the genetic background analysis of our mice.
Conflict of interest statement: The authors declare competing financial interests.
Susan A. Slaugenhaupt is a paid consultant to PTC Therapeutics and is an inventor on several U.S. and foreign patents and patent applications assigned to the Massachusetts General Hospital, including U.S Patents 8 729 025 and 9 265 766, both entitled ‘Methods for altering mRNA splicing and treating familial dysautonomia by administering benzyladenine,’ filed on August 31, 2012 and May 19, 2014 and related to use of kinetin; and U.S. Patent 10 675 475 entitled, ‘Compounds for improving mRNA splicing’ filed on July 14, 2017 and related to use of BPN-15477. Elisabetta Morini and Susan A. Slaugenhaupt are inventors on an International Patent Application Number PCT/US2021/012103, assigned to Massachusetts General Hospital and entitled ‘ RNA Splicing Modulation’ related to use of BPN-15477 in modulating splicing. Luk H. Vandenberghe (LHV) holds equity in Affinia Therapeutics and Akouos and serves on the Board of Directors of Affinia Therapeutics, Addgene and Odylia. LHV is compensated for his scientific advisory position with Affinia and Akouos. LHV is an SAB member to Akouos, consultant to Affinia and Novartis and receives research support from Novartis. LHV’s interests were reviewed and are managed by Mass Eye and Ear and Mass General Brigham in accordance with their conflict-of-interest policies.
Contributor Information
Anil Chekuri, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA; Department of Neurology, Massachusetts General Hospital Research Institute and Harvard Medical School, Boston, MA, USA; Grousbeck Gene Therapy Center, Schepens Eye Research Institute and Massachusetts Eye and Ear Infirmary, Boston, MA, USA.
Emily M Logan, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA.
Aram J Krauson, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA.
Monica Salani, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA.
Sophie Ackerman, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA.
Emily G Kirchner, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA.
Jessica M Bolduc, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA.
Xia Wang, Grousbeck Gene Therapy Center, Schepens Eye Research Institute and Massachusetts Eye and Ear Infirmary, Boston, MA, USA.
Paula Dietrich, Department of Physiology, The University of Tennessee, Health Science Center, Memphis, TN, USA.
Ioannis Dragatsis, Department of Physiology, The University of Tennessee, Health Science Center, Memphis, TN, USA.
Luk H Vandenberghe, Grousbeck Gene Therapy Center, Schepens Eye Research Institute and Massachusetts Eye and Ear Infirmary, Boston, MA, USA.
Susan A Slaugenhaupt, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA; Department of Neurology, Massachusetts General Hospital Research Institute and Harvard Medical School, Boston, MA, USA.
Elisabetta Morini, Center for Genomic Medicine, Massachusetts General Hospital Research Institute, Boston, MA, USA; Department of Neurology, Massachusetts General Hospital Research Institute and Harvard Medical School, Boston, MA, USA.
Web resources
Analysis of Elp1 expression in different RGC subtypes was performed using publicly available single-cell RNA-seq dataset (https://health.uconn.edu/neuroregeneration-lab/rgc-subtypes-gene-browser).
Funding
The National Eye Institute (NEI) grant (1R01EY029544-01 to S.A.S. and L.H.V.) and by Grousbeck Family Foundation (to L.H.V).
References
- 1. Axelrod, F.B., Nachtigal, R. and Dancis, J. (1974) Familial dysautonomia: diagnosis, pathogenesis and management. Adv. Pediatr. Infect. Dis., 21, 75–96. [PubMed] [Google Scholar]
- 2. Pearson, J. and Pytel, B.A. (1978) Quantitative studies of sympathetic ganglia and spinal cord intermedio-lateral gray columns in familial dysautonomia. J. Neurol. Sci., 39, 47–59. [DOI] [PubMed] [Google Scholar]
- 3. Pearson, J. (1979) Familial dysautonomia (a brief review). J. Auton. Nerv. Syst., 1, 119–126. [DOI] [PubMed] [Google Scholar]
- 4. Otero, G., Fellows, J., Li, Y., de Bizemont, T., Dirac, A.M., Gustafsson, C.M., Erdjument-Bromage, H., Tempst, P. and Svejstrup, J.Q. (1999) Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell, 3, 109–118. [DOI] [PubMed] [Google Scholar]
- 5. Hawkes, N.A., Otero, G., Winkler, G.S., Marshall, N., Dahmus, M.E., Krappmann, D., Scheidereit, C., Thomas, C.L., Schiavo, G., Erdjument-Bromage, H. et al. (2002) Purification and characterization of the human elongator complex. J. Biol. Chem., 277, 3047–3052. [DOI] [PubMed] [Google Scholar]
- 6. Kim, J.H., Lane, W.S. and Reinberg, D. (2002) Human Elongator facilitates RNA polymerase II transcription through chromatin. Proc. Natl. Acad. Sci. U.S.A., 99, 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Creppe, C., Malinouskaya, L., Volvert, M.L., Gillard, M., Close, P., Malaise, O., Laguesse, S., Cornez, I., Rahmouni, S., Ormenese, S. et al. (2009) Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell, 136, 551–564. [DOI] [PubMed] [Google Scholar]
- 8. Close, P., Hawkes, N., Cornez, I., Creppe, C., Lambert, C.A., Rogister, B., Siebenlist, U., Merville, M.P., Slaugenhaupt, S.A., Bours, V. et al. (2006) Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol. Cell, 22, 521–531. [DOI] [PubMed] [Google Scholar]
- 9. Huang, B., Johansson, M.J. and Bystrom, A.S. (2005) An early step in wobble uridine tRNA modification requires the Elongator complex. RNA, 11, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackson, M.Z., Gruner, K.A., Qin, C. and Tourtellotte, W.G. (2014) A neuron autonomous role for the familial dysautonomia gene ELP1 in sympathetic and sensory target tissue innervation. Development, 141, 2452–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zeltner, N., Fattahi, F., Dubois, N.C., Saurat, N., Lafaille, F., Shang, L., Zimmer, B., Tchieu, J., Soliman, M.A., Lee, G. et al. (2016) Capturing the biology of disease severity in a PSC-based model of familial dysautonomia. Nat. Med., 22, 1421–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abashidze, A., Gold, V., Anavi, Y., Greenspan, H. and Weil, M. (2014) Involvement of IKAP in peripheral target innervation and in specific JNK and NGF signaling in developing PNS neurons. PLoS One, 9, e113428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunnicutt, B.J., Chaverra, M., George, L. and Lefcort, F. (2012) IKAP/Elp1 is required in vivo for neurogenesis and neuronal survival, but not for neural crest migration. PLoS One, 7, e32050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee, G. and Studer, L. (2011) Modelling familial dysautonomia in human induced pluripotent stem cells. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 366, 2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee, G., Papapetrou, E.P., Kim, H., Chambers, S.M., Tomishima, M.J., Fasano, C.A., Ganat, Y.M., Menon, J., Shimizu, F., Viale, A. et al. (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature, 461, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen, Y.T., Hims, M.M., Shetty, R.S., Mull, J., Liu, L., Leyne, M. and Slaugenhaupt, S.A. (2009) Loss of mouse Ikbkap, a subunit of elongator, leads to transcriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol. Cell. Biol., 29, 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Solinger, J.A., Paolinelli, R., Kloss, H., Scorza, F.B., Marchesi, S., Sauder, U., Mitsushima, D., Capuani, F., Sturzenbaum, S.R. and Cassata, G. (2010) The Caenorhabditis elegans Elongator complex regulates neuronal alpha-tubulin acetylation. PLoS Genet., 6, e1000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dietrich, P., Yue, J. and E, S. and Dragatsis, I. (2011) Deletion of exon 20 of the familial Dysautonomia gene Ikbkap in mice causes developmental delay, cardiovascular defects, and early embryonic lethality. PLoS One, 6, e27015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson, S.L., Coli, R., Daly, I.W., Kichula, E.A., Rork, M.J., Volpi, S.A., Ekstein, J. and Rubin, B.Y. (2001) Familial dysautonomia is caused by mutations of the IKAP gene. Am. J. Hum. Genet., 68, 753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Slaugenhaupt, S.A., Blumenfeld, A., Gill, S.P., Leyne, M., Mull, J., Cuajungco, M.P., Liebert, C.B., Chadwick, B., Idelson, M., Reznik, L. et al. (2001) Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet., 68, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pearson, J., Pytel, B.A., Grover-Johnson, N., Axelrod, F. and Dancis, J. (1978) Quantitative studies of dorsal root ganglia and neuropathologic observations on spinal cords in familial dysautonomia. J. Neurol. Sci., 35, 77–92. [DOI] [PubMed] [Google Scholar]
- 22. Pearson, J. and Pytel, B. (1978) Quantitative studies of ciliary and sphenopalatine ganglia in familial dysautonomia. J. Neurol. Sci., 39, 123–130. [DOI] [PubMed] [Google Scholar]
- 23. Macefield, V.G., Norcliffe-Kaufmann, L., Gutierrez, J., Axelrod, F.B. and Kaufmann, H. (2011) Can loss of muscle spindle afferents explain the ataxic gait in Riley-Day syndrome? Brain, 134, 3198–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ford, D.M., Bagnall, K.M., Clements, C.A. and McFadden, K.D. (1988) Muscle spindles in the paraspinal musculature of patients with adolescent idiopathic scoliosis. Spine (Phila Pa 1976), 13, 461–465. [DOI] [PubMed] [Google Scholar]
- 25. Mahloudji, M., Brunt, P.W. and McKusick, V.A. (1970) Clinical neurological aspects of familial dysautonomia. J. Neurol. Sci., 11, 383–395. [DOI] [PubMed] [Google Scholar]
- 26. Axelrod, F.B. (2004) Familial dysautonomia. Muscle Nerve, 29, 352–363. [DOI] [PubMed] [Google Scholar]
- 27. Hilz, M.J., Kolodny, E.H., Neuner, I., Stemper, B. and Axelrod, F.B. (1998) Highly abnormal thermotests in familial dysautonomia suggest increased cardiac autonomic risk. J. Neurol. Neurosurg. Psychiatry, 65, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunt, P.W. (1967) Unusual cause of Charcot joints in early adolescence (Riley-Day syndrome). Br. Med. J., 4, 277–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaplan, L., Margulies, J.Y., Kadari, A., Floman, Y. and Robin, G.C. (1997) Aspects of spinal deformity in familial dysautonomia (Riley-Day syndrome). Eur. Spine J., 6, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mendoza-Santiesteban, C.E., Palma, J.A., Hedges, T.R., 3rd, Laver, N.V., Farhat, N., Norcliffe-Kaufmann, L. and Kaufmann, H. (2017) Pathological confirmation of optic neuropathy in familial Dysautonomia. J. Neuropathol. Exp. Neurol., 76, 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mendoza-Santiesteban, C.E., Hedges, T.R., 3rd, Norcliffe-Kaufmann, L., Warren, F., Reddy, S., Axelrod, F.B. and Kaufmann, H. (2012) Clinical neuro-ophthalmic findings in familial dysautonomia. J. Neuroophthalmol., 32, 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mendoza-Santiesteban, C.E., Hedges Iii, T.R., Norcliffe-Kaufmann, L., Axelrod, F. and Kaufmann, H. (2014) Selective retinal ganglion cell loss in familial dysautonomia. J. Neurol., 261, 702–709. [DOI] [PubMed] [Google Scholar]
- 33. Liebman, S.D. (1956) Ocular manifestations of Riley-Day syndrome; familial autonomic dysfunction. A.M.A. Arch. Ophthalmol., 56, 719–725. [DOI] [PubMed] [Google Scholar]
- 34. Kroop, I.G. (1956) The production of tears in familial dysautonomia; preliminary report. J. Pediatr., 48, 328–329. [DOI] [PubMed] [Google Scholar]
- 35. Josaitis, C.A. and Matisoff, M. (2002) Familial dysautonomia in review: diagnosis and treatment of ocular manifestations. Adv. Exp. Med. Biol., 506, 71–80. [DOI] [PubMed] [Google Scholar]
- 36. Liebman, S.D. (1968) Riley-Day syndrome: long-term ophthalmologic observations. Trans. Am. Ophthalmol. Soc., 66, 95–116. [PMC free article] [PubMed] [Google Scholar]
- 37. Goldberg, M.F., Payne, J.W. and Brunt, P.W. (1968) Ophthalmologic studies of familial dysautonomia. The Riley-Day syndrome. Arch. Ophthalmol., 80, 732–743. [DOI] [PubMed] [Google Scholar]
- 38. Sinha, R., Kim, Y.J., Nomakuchi, T., Sahashi, K., Hua, Y., Rigo, F., Bennett, C.F. and Krainer, A.R. (2018) Antisense oligonucleotides correct the familial dysautonomia splicing defect in IKBKAP transgenic mice. Nucleic Acids Res., 46, 4833–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donadon, I., Pinotti, M., Rajkowska, K., Pianigiani, G., Barbon, E., Morini, E., Motaln, H., Rogelj, B., Mingozzi, F., Slaugenhaupt, S.A. et al. (2018) Exon-specific U1 snRNAs improve ELP1 exon 20 definition and rescue ELP1 protein expression in a familial dysautonomia mouse model. Hum. Mol. Genet., 27, 2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morini, E., Gao, D., Montgomery, C.M., Salani, M., Mazzasette, C., Krussig, T.A., Swain, B., Dietrich, P., Narasimhan, J., Gabbeta, V. et al. (2019) ELP1 splicing correction reverses proprioceptive sensory loss in familial Dysautonomia. Am. J. Hum. Genet., 104, 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Slaugenhaupt, S.A., Mull, J., Leyne, M., Cuajungco, M.P., Gill, S.P., Hims, M.M., Quintero, F., Axelrod, F.B. and Gusella, J.F. (2004) Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum. Mol. Genet., 13, 429–436. [DOI] [PubMed] [Google Scholar]
- 42. Axelrod, F.B., Liebes, L., Gold-Von Simson, G., Mendoza, S., Mull, J., Leyne, M., Norcliffe-Kaufmann, L., Kaufmann, H. and Slaugenhaupt, S.A. (2011) Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr. Res., 70, 480–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salani, M., Urbina, F., Brenner, A., Morini, E., Shetty, R., Gallagher, C.S., Law, E.A., Sunshine, S., Finneran, D.J., Johnson, G. et al. (2019) Development of a screening platform to identify small molecules that modify ELP1 pre-mRNA splicing in familial Dysautonomia. SLAS Discov, 24, 57–67. [DOI] [PubMed] [Google Scholar]
- 44. Ueki, Y., Shchepetkina, V. and Lefcort, F. (2018) Retina-specific loss of Ikbkap/Elp1 causes mitochondrial dysfunction that leads to selective retinal ganglion cell degeneration in a mouse model of familial dysautonomia. Dis. Model. Mech., 11, dmm033746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ueki, Y., Ramirez, G., Salcedo, E., Stabio, M.E. and Lefcort, F. (2016) Loss of Ikbkap causes slow, progressive retinal degeneration in a mouse model of familial Dysautonomia. eNeuro, 3. doi: 10.1523/ENEURO.0143-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hims, M.M., Shetty, R.S., Pickel, J., Mull, J., Leyne, M., Liu, L., Gusella, J.F. and Slaugenhaupt, S.A. (2007) A humanized IKBKAP transgenic mouse models a tissue-specific human splicing defect. Genomics, 90, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dietrich, P., Alli, S., Shanmugasundaram, R. and Dragatsis, I. (2012) IKAP expression levels modulate disease severity in a mouse model of familial dysautonomia. Hum. Mol. Genet., 21, 5078–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morini, E., Dietrich, P., Salani, M., Downs, H.M., Wojtkiewicz, G.R., Alli, S., Brenner, A., Nilbratt, M., LeClair, J.W., Oaklander, A.L. et al. (2016) Sensory and autonomic deficits in a new humanized mouse model of familial dysautonomia. Hum. Mol. Genet., 25, 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Howell, G.R., Libby, R.T., Jakobs, T.C., Smith, R.S., Phalan, F.C., Barter, J.W., Barbay, J.M., Marchant, J.K., Mahesh, N., Porciatti, V. et al. (2007) Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol., 179, 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hoyt, C.S. (1980) Autosomal dominant optic atrophy. A spectrum of disability. Ophthalmology, 87, 245–251. [DOI] [PubMed] [Google Scholar]
- 51. Votruba, M. (2004) Molecular genetic basis of primary inherited optic neuropathies. Eye (Lond.), 18, 1126–1132. [DOI] [PubMed] [Google Scholar]
- 52. Kerrison, J.B. (2005) Latent, acute, and chronic Leber's hereditary optic neuropathy. Ophthalmology, 112, 1–2. [DOI] [PubMed] [Google Scholar]
- 53. Ajax, E.T. and Kardon, R. (1998) Late-onset Leber's hereditary optic neuropathy. J. Neuroophthalmol., 18, 30–31. [PubMed] [Google Scholar]
- 54. Barboni, P., Savini, G., Valentino, M.L., Montagna, P., Cortelli, P., De Negri, A.M., Sadun, F., Bianchi, S., Longanesi, L., Zanini, M. et al. (2005) Retinal nerve fiber layer evaluation by optical coherence tomography in Leber's hereditary optic neuropathy. Ophthalmology, 112, 120–126. [DOI] [PubMed] [Google Scholar]
- 55. Francois, J. (1976) Hereditary optic atrophies. J. Genet. Hum., 24, 183–200. [PubMed] [Google Scholar]
- 56. Alasil, T., Wang, K., Yu, F., Field, M.G., Lee, H., Baniasadi, N., de Boer, J.F., Coleman, A.L. and Chen, T.C. (2014) Correlation of retinal nerve fiber layer thickness and visual fields in glaucoma: a broken stick model. Am J. Ophthalmol., 157, 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Noristani, R., Kuehn, S., Stute, G., Reinehr, S., Stellbogen, M., Dick, H.B. and Joachim, S.C. (2016) Retinal and optic nerve damage is associated with early glial responses in an experimental autoimmune glaucoma model. J. Mol. Neurosci., 58, 470–482. [DOI] [PubMed] [Google Scholar]
- 58. Cuajungco, M.P., Leyne, M., Mull, J., Gill, S.P., Lu, W., Zagzag, D., Axelrod, F.B., Maayan, C., Gusella, J.F. and Slaugenhaupt, S.A. (2003) Tissue-specific reduction in splicing efficiency of IKBKAP due to the major mutation associated with familial dysautonomia. Am. J. Hum. Genet., 72, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shetty, R.S., Gallagher, C.S., Chen, Y.T., Hims, M.M., Mull, J., Leyne, M., Pickel, J., Kwok, D. and Slaugenhaupt, S.A. (2011) Specific correction of a splice defect in brain by nutritional supplementation. Hum. Mol. Genet., 20, 4093–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gao, D., Morini, E., Salani, M., Krauson, A.J., Chekuri, A., Sharma, N., Ragavendran, A., Erdin, S., Logan, E.M., Li, W. et al. (2021) A deep learning approach to identify gene targets of a therapeutic for human splicing disorders. Nat. Commun., 12, 3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Norcliffe-Kaufmann, L., Slaugenhaupt, S.A. and Kaufmann, H. (2017) Familial dysautonomia: history, genotype, phenotype and translational research. Prog. Neurobiol., 152, 131–148. [DOI] [PubMed] [Google Scholar]
- 63. Graw, J. (2010) Eye development. Curr. Top. Dev. Biol., 90, 343–386. [DOI] [PubMed] [Google Scholar]
- 64. Rheaume, B.A., Jereen, A., Bolisetty, M., Sajid, M.S., Yang, Y., Renna, K., Sun, L., Robson, P. and Trakhtenberg, E.F. (2018) Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun., 9, 2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sigmon, J.S., Blanchard, M.W., Baric, R.S., Bell, T.A., Brennan, J., Brockmann, G.A., Burks, A.W., Calabrese, J.M., Caron, K.M., Cheney, R.E. et al. (2020) Content and performance of the MiniMUGA genotyping array: a new tool to improve rigor and reproducibility in mouse research. Genetics, 216, 905–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chintalapudi, S.R., Patel, N.N., Goldsmith, Z.K., Djenderedjian, L., Wang, X.D., Marion, T.N., Jablonski, M.M. and Morales-Tirado, V.M. (2017) Isolation of primary murine retinal ganglion cells (RGCs) by flow cytometry. J. Vis. Exp., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mead, B., Thompson, A., Scheven, B.A., Logan, A., Berry, M. and Leadbeater, W. (2014) Comparative evaluation of methods for estimating retinal ganglion cell loss in retinal sections and wholemounts. PLoS One, 9, e110612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.