

Abstract
Many genes encoding nucleotide‐binding leucine‐rich repeat receptors (NLRs) are regulated and fine‐tuned by miR482 to balance the trade‐off between disease resistance and growth. Dicotyledonous plants, including cotton, usually have multiple miR482 isoforms. Each miR482 isoform can regulate several NLRs that in turn can be regulated by several different miR482 isoforms. Dissecting the functionality of individual miR482 isoforms in disease response and in balancing the disease resistance and growth trade‐off demands a collection of mutants mutated in individual miR482 members (single or multiple). In this study, we generated such a collection of cotton miR482 mutants using CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/Cas9) genome editing and transformation of pooled guide RNAs (gRNAs). In total, 84 T0 plants representing 40 independent transgenic events and harboring mutation in each of the 10 miR482 isoforms were generated. The average editing efficiency of the 18 transformed gRNAs is 75%, ranging from 0 (3 gRNAs) to 100% (8 gRNAs). Most miR482 isoforms have a diverse range of mutations, including small indels (1–44 bp) and substitutions, which are expected to impair biogenesis of miR482. All nine mutant populations used in Verticillium dahliae infection experiments showed a disease index lower than the control, with four being significantly lower. The disease assay also suggests a different role of different miR482 isoforms in disease response and a potential dosage effect of miR482l. The study demonstrates the feasibility of saturation mutagenesis of plant miRNA families with dozens of genetic loci using CRISPR/Cas9 and provides the cotton community a valuable resource for uncovering the miR482‐NLR module(s) underlying the interaction between cotton and different pathogens.
Keywords: cotton, disease response, gene editing, miRNA, mutagenesis
1. INTRODUCTION
Cotton production faces a wide range of challenges incurred by abiotic and biotic stresses, such as drought and diseases. Verticillium wilt (VW) caused by the soil‐born fungal pathogen Verticillium dahliae (Vd) is one of the most serious cotton diseases worldwide. Development of host plant resistance through genetic modification using traditional breeding and/or state‐of‐the‐art molecular strategies is the most effective measure for mitigating the detrimental impact of VW, which demands understanding the molecular mechanisms underlying cotton response to V. dahliae infection.
Nucleotide‐binding leucine‐rich repeat (NLR) proteins are intracellular immune receptors and can directly or indirectly detect pathogenic molecules or cognate effectors to activate plant immune system, particularly the effector‐triggered immunity (Ngou et al., 2022). Disease resistance conferred by NLRs is usually dominant and thus can be enhanced by increasing the expression level of NLRs. But the level of NLRs is intrinsically regulated and monitored at multiple layers, from transcription to post‐transcription to turnover (Ngou et al., 2022). miR482, a 22‐bp miRNA, is a negative post‐transcriptional regulator of NLRs by targeting their conserved P‐loop motif for transcript degradation or translational repression (Ouyang et al., 2014; Shivaprasad et al., 2012; Zhu et al., 2013). Repressing biogenesis of miR482 increases resistance to infection with the oomycete, bacterial or fungal pathogens in tomato due to enhanced expression of NLRs repressed by miR482 (Canto‐Pastor et al., 2019; Gao et al., 2021; Hong et al., 2021; Jiang et al., 2018).
miR482 is a conserved miRNA family mainly found in gymnospermous and dicotyledonous plants. Usually, multiple miR482 isoforms were found in the plant species, in which miR482 has been identified (Zhang et al., 2022). The tetraploid Upland cotton (Gossypium hisutum L.) genome has 36 MIR482 loci generating 11 different miR482 isoforms (Shen et al., 2020) that are predicted to regulate 105 (~17%; score ≥3) of the 624 NLRs annotated in the Upland cotton genome (Wang et al., 2019). Of these miR482‐targeted NLRs, 65 (~62%) are potentially regulated by ≥2 miR482 isoforms, suggesting an interwoven regulatory network among miR482 isoforms and their NLR targets. Deep understanding the functionality of individual miR482‐NLR modules in response to pathogen infection can provide genetic solution for breeding high yielding cotton cultivars resistant to diseases. But achieving the goal demands a collection of mutants mutated in single, double, and multiple MIR482 genes.
Traditionally, a mutant library can be created by mutagenesis using physical approach, for example, γ‐ray and neutron irradiation, or chemical approach, for example, ethyl methane sulfonate treatment. T‐DNA or transposon tagging has also been routinely applied to create plant mutant libraries for studies of functional genomics (Upadhyaya et al., 2011). However, saturation mutagenesis of a MIRNA family is difficult using the traditional approaches thanks to their nature of random mutagenisation and the small size of MIRNA genes. Owing to its versatility, efficiency, and accuracy at targeted genome modification, CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/Cas9)‐mediated gene editing has increasingly been adopted by the plant community to mutagenise single gene and gene family for functional genomics and to create novel germplasm of diverse crops for designed and precision crop breeding (Zhang et al., 2019). The functions of several miRNAs have also been investigated using CRISPR/Cas9‐based approaches (Deng et al., 2022). For example, functional specificity of all five Arabidopsis MIR172 genes in regulating shoot apex development and flowering time are elegantly distinguished using mutant alleles created by CRISPR/Cas9‐mediated gene editing (Lian et al., 2021; O'Maoileidigh et al., 2021).
To have a set of MIR482 mutants for investigating the role of different miR482 isoforms in disease response and identifying the miR482‐NLR regulatory module(s) involved in resistance to V. dahliae and other cotton diseases, here we used CRISPR/Cas9 gene editing to mutate each isoform of the cotton MIR482 family and achieved saturated mutagenesis of the family. The results demonstrate the feasibility of using CRISPR/Cas9 and pooled guide RNAs (gRNAs) in mutagenising a miRNA family with dozens of genes in polyploid and a variable role of individual miR482 isoforms in response to V. dahliae infection.
2. RESULTS
2.1. Mutagenising the cotton MIR482 family by transformation of pooled gRNAs
The Upland cotton genome contains 36 MIR482 loci generating 11 different miR482 isoforms (miR482a to miR482h, miR482j to miR482l). Except miR482d, which is potentially generated from 19 loci, each of other miR482 isoforms is generated from a single (MIR482a, MIR482c, and MIR482j) or two homoeologous loci (Shen et al., 2020).
To mutate individual MIR482 genes, we designed 1–3 gRNAs for each MIR482 gene (except MIR482j, for which a suitable gRNA could not be designed) by targeting miR482 or miR482* to induce changes in miR482 sequence or to alter the hairpin structure of miR482 precursor (pre‐miR482) required for DICER‐LIKE1 processing, aiming for inactivating biogenesis of miR482 (Figure 1, Table 2). In total, 22 gRNAs were designed and individually sub‐cloned into the CRISPR/Cas9 binary vector linearized by BsaI digestion (Wang et al., 2018). Plasmids of the 22 constructs were pooled, transformed into Agrobacterium tumefaciens (LBA4404), and used in cotton transformation with leaf discs of Coker‐315 as explants. We obtained 84 T0 transgenics representing 40 independent transgenic events. Based on Sanger sequencing, 18 gRNAs were found to be successfully transformed, and no transgenic integrating gRNA33, gRNA43, gRNA47, or gRNA52 was detected (Figure 1, Table 3). If more transgenics were generated, it is likely these four gRNAs would have been integrated. However, since confirmation of the presence of each plasmid in the Agrobacterium pool was not attempted, the lack of integration for these gRNAs could also be due to absence of the plasmids containing those gRNAs in the Agrobacterium pool. While the majority transgenic events (82.5%) integrated a single gRNA, three (7.5%) and four (10%) transgenic events have two and three different gRNAs, respectively (Table S1).
FIGURE 1.
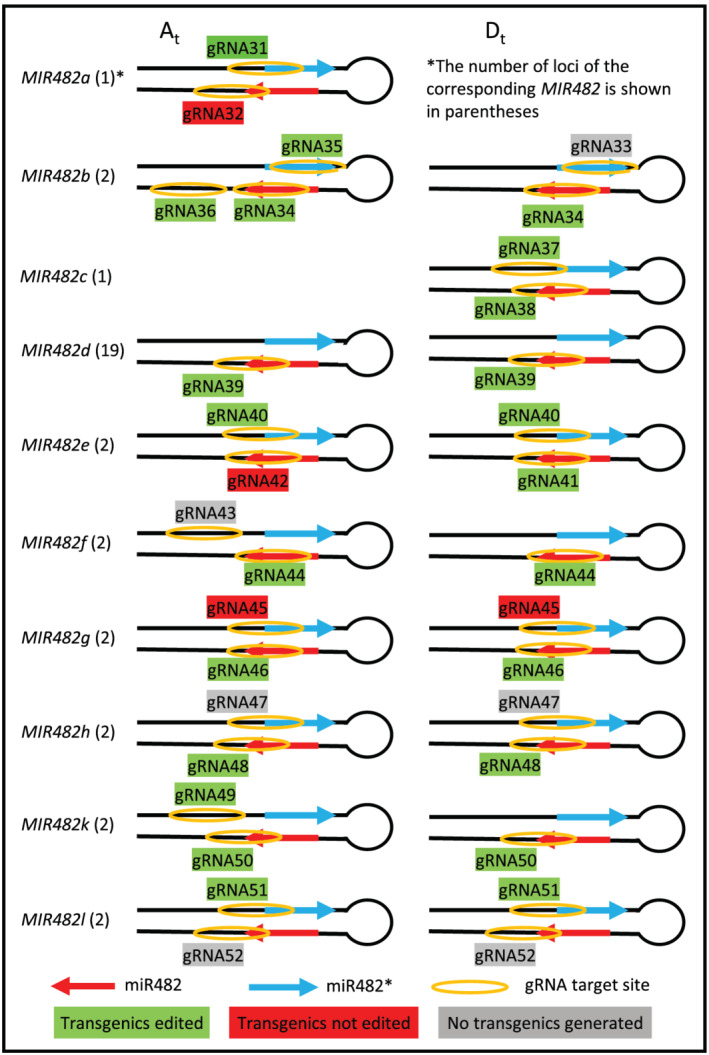
Schematic diagrams showing the hairpin structure of MIR482 genes and the target sites of gRNAs used in the study. miR482 is located at the 3′ arm of the hairpin
TABLE 2.
gRNAs used in the study
| gRNA ID | gRNA + PAM | Target gene | Off‐target (mismatch) |
|---|---|---|---|
| gRNA31 | GGAGTTTATGGAAGTGGGATGGG | miR482a_A04 | miR482c_D04 (1 MM) |
| gRNA32 | AGAACTGGAAATAAGTGGTATGG | miR482a_A04 | miR482c_D04 (1 MM) |
| gRNA33 | TGGGTGAGGGGGTCAGACAATGG | miR482b_D13 | miR482b_scaf3700_A13 (1 MM) |
| gRNA34 | GCCTACTCCACCCATGCCACTGG | miR482b_A13/D13 | |
| gRNA35 | TGGGTGAGGGGGTAAGACAATGG | miR482b_scaf3700_A13 | miR482b_D13 (1 MM) |
| gRNA36 | TTAGGAGGAGGAGGAACACCAGG | miR482b_scaf3700_A13 | miR482b_D13 (1 MM) |
| gRNA37 | ATCCCACTTCCAAAAACTCCCGG | miR482c_D04 | miR482a_A04 (1 MM) |
| gRNA38 | ATAAGTGGAATGGGAGGAGTAGG | miR482c_D04 | miR482a_A04 (1 MM) |
| gRNA39 | ATCAGTGGAATGGGAGGAATTGG | miR482d (all 19 loci) | |
| gRNA40 | GTCTTCGGCTGTGGTTGATTCGG | miR482e_A05/D05 | |
| gRNA41 | ATCGTAGGCATGGGTGGAATCGG | miR482e_D05 | miR482e_A05 (1 MM) |
| gRNA42 | ATAGTAGGCATGGGTGGAATCGG | miR482e_A05 | miR482e_D05 (1 MM) |
| gRNA43 | GGAAACGGAGAGAGAGTTTCTGG | miR482f_A03 | miR482f_D02 (1 MM) |
| gRNA44 | TGGGGGGCATAGGAAAGATCTGG | miR482f_A03/D02 | |
| gRNA45 | AGTAATAGGTATGGGGGGTATGG | miR482g_A03/D02 | |
| gRNA46 | ATGAAGGGTATGGGAGGTGTTGG | miR482g_A03/D02 | |
| gRNA47 | GTTGGAGGTGTGGGAGAGTTGGG | miR482h_A05/D05 | |
| gRNA48 | ATCAAGGGTATGGGAGGGGTTGG | miR482h_A05/D05 | |
| gRNA49 | GAAAGGGAGATTGAAGTTTCTGG | miR482k_A07 | miR482k_D07 (1 MM) |
| gRNA50 | CTGGAAATCATTGGAATTGGAGG | miR482k_A07/D07 | |
| gRNA51 | ATTACAGGAATGGGGTCTTTGGG | miR482l_A12/A12 | |
| gRNA52 | ATAAATTGAAGTTCTTCCCGAGG | miR482l_A12/A12 |
TABLE 3.
Transgenic events, editing efficiency and events of the gRNAs used
| gRNA | MIR482 target a | Subgenome target b | Editing efficiency (%) c | Editing events d | |
|---|---|---|---|---|---|
| At | Dt | ||||
| gRNA31 | MIR482a (27/27) | At (13th) | 100 (2/2/4) | −8/−9 | WT |
| gRNA32 | MIR482a | At (2nd) | 0 | WT | WT |
| gRNA33 | MIR482b (24/23) | Dt (7th) | n/a | n/a | n/a |
| gRNA34 | MIR482b | Both | 66.7 (2/3/3) | −1/−2 | −2/−41 |
| gRNA35 | MIR482b | At (7th) | 85.7 (6/7/12) | −4/−5 | WT |
| gRNA36 | MIR482b | At (1st) | 75 (3/4/5) | −2/−7 | −3/−4 |
| gRNA37 | MIR482c (27/27) | Dt (8th) | 100 (4/4/8) | WT | −40 |
| gRNA38 | MIR482c | Dt (12th) | 100 (1/1/1) | −30 | −2/−8 |
| gRNA39 | MIR482d (35/27) | Both | 25.0 (1/4/11) | −1/−3/−4/−9/+1/WT | −1/−4/−9/+1/WT |
| gRNA40 | MIR482e (26/20) | Both | 100 (1/1/2) | WT | +1/WT |
| gRNA41 | MIR482e | Dt (18th) | 100 (3/3/10) | −3/−14 | −3/−15 |
| gRNA42 | MIR482e | At (18th) | 0 (0/1/2) | WT | WT |
| gRNA43 | MIR482f (13/7) | At (2nd) | n/a | n/a | n/a |
| gRNA44 | MIR482f | Both | 100 (2/2/4) | n/a | −1/−10 |
| gRNA45 | MIR482g (7/6) | Both | 0 (0/2/2) | WT | WT |
| gRNA46 | MIR482g | Both | 100 (1/1/1) | −1/−44 | −1a/−1b |
| gRNA47 | MIR482h (22/16) | Both | n/a | n/a | n/a |
| gRNA48 | MIR482h | Both | 50.0 (1/2/3) | −1/−11/sub | ‐1/−11/sub |
| gRNA49 | MIR482k (12/10) | At (18th) | 33.3 (1/3/3) | −38/WT | WT |
| gRNA50 | MIR482k | Both | 100 (1/1/13) | −1/−7/+1 | −1/+1 |
| gRNA51 | MIR482l (16/7) | Both | 85.7 (6/7/11) | −2/−3 | −9/+1 |
| gRNA52 | MIR482l | Both | n/a | n/a | n/a |
| Average | 75.0 (30/40/84) | ||||
The number in parentheses represent the total number of NLR targets of the corresponding miR482/the number of NLRs also targeted by at least one of other miR482 isoforms.
The mismatch position (counting from PAM) between the At and Dt subgenomes is given in parentheses.
The numbers in parentheses represent the number of: edited independent transgenic events/independent transgenic events/T0 transgenic plants.
Editing events from a representative T0 plant are shown. −1a/−1b (gRNA46) indicates two different −1 bp; sub = substitution (gRNA48).
2.2. Saturated mutagenesis of the cotton MIR482 family
The editing events in 71 of the 84 T0 plants were analyzed by sequencing of pooled amplicons amplified using barcoded primers (Table 1). After quality control, the reads were grouped based on barcode and target site of gRNA and subjected to editing event analysis. In the 40 independent transgenic events, editing events were found in 30, meaning an average editing efficiency of 75%. For individual gRNAs, the editing efficiency varied from 0 (gRNA32, gRNA42, and gRNA45) to 100% (gRNA31, gRNA37, gRNA38, gRNA40, gRNA41, gRNA44, gRNA46, and gRNA50) (Table 3). Approximately 52%, 31%, 10%, and 7% of the editing events are biallelic, chimeric, heterozygous, and homozygous, respectively. The major editing events are small indels, although insertion and substitution were also observed. The largest deletion is 44‐bp, observed in M482‐40‐1 (gRNA48) and M482‐7‐3 (gRNA51). Almost all gRNAs induced different editing events in the At and Dt subgenomes (Table 3, Table S1, Figures S1 and S2).
TABLE 1.
Primers used in the study
| Primer ID | Sequence (5′‐3′)* | Usage |
|---|---|---|
| Cas9_F | GCCTATTCTGTGCTGGTGGT | Presence/absence of transgene |
| Cas9_R | TCCAGGTAGTGCTTGTGCTG | |
| GhU6F1 | TGTGCCACTCCAAAGACATCA | Analyzing the integrated gRNA |
| gRNAR1 | ATGCACGCGCTAAAAACGGA | |
| GhU6F2 | TGGTCAGGACGTGGTAGCATA | Sequencing PCR product generated by GhU6F1/gRNAR1 |
| miR482ac_F | GTTCTCTCTCACTCTCACCCC | Editing events of gRNA31/37/38 |
| miR482ac_R | CCGATTTAACTTACCGAGTGC | |
| miR482b_F | GAAGAAGTATGGGTGTTAGCCG | Editing events of gRNA34/35/36 |
| miR482b_R | CATTTTCACCAAGGTTCCCTGC | |
| miR482d1_F | TACTTTCTCTTTCTCCTCACC | Editing events of gRNA39 |
| miR482d1_R | GGTTTACCGATTTCACTTAC | |
| miR482d2_F | TTTCTACTCATTTTCTGTGGC | Editing events of gRNA39 |
| miR482d2_R | ATATCTCCGCCGCTCATAACC | |
| miR482d3_F | TTAGACTGTTGTGAAGGAGGA | Editing events of gRNA39 |
| miR482d3_R | CAAATATAGTGCTAACGTCCG | |
| miR482d4_F | GACGCATAATTCTCTGTGGC | Editing events of gRNA39 |
| miR482d4_R | GGTAAGGTTTACCGATTTCAC | |
| miR482e_F | AAGGTAAGGAAAGCCGCCAC | Editing events of gRNA40/41 |
| miR482e_R | CTTTCCAACTCCTCCATTCC | |
| miR482f_F | AGGCGATTGGTATGAGCACC | Editing events of gRNA44 |
| miR482f_R | GCGAAGGTGCAGGCATGAAA | |
| miR482g_F | ATATGCAATGAAGTTGATTTTGCC | Editing events of gRNA46 |
| miR482g_R | GAGTAAAAGGAAAAGCAAACC | |
| miR482h_F | CCTTCTCTTCGTTTGCCCAT | Editing events of gRNA48 |
| miR482h_R | GTTCTCTGAAAATCACAACCAG | |
| miR482k_F | CTTATGAAAGTTGGATGACC | Editing events of gRNA49/50 |
| miR482k_R | CATACCTTCTTCCATATACC | |
| miR482l_F | TTGAAACATAAAGGACCAGC | Editing events of gRNA51 |
| miR482l_R | CCTTACTTTCTTCCGTTTCC |
The mutations induced by CRISPR/Ca9 alter either the miR482 sequence per se and/or the secondary structure of pre‐miR482, which are expected to impair miR482 sequence and/or biogenesis of miR482. For instance, in the T0 plant M482c‐26‐1, gRNA38‐guided homozygous and biallelic editing were observed in the At (−30‐bp) and Dt (−2‐bp and −8‐bp) subgenome, respectively. The 30‐bp deletion in MIR482a_A t includes 14‐bp of miR482a, and the 2‐bp and 8‐bp deletions in MIR482c_D t are part of miR482c (Figure 2). These deletions also change the secondary structures of pre‐miR482a and pre‐miR482c, with a bigger bulge observed in the miR482/miR482* region or its flanking region that would affect processing of pre‐miR482a or pre‐miR482c by DICER‐LIKE1 (Figure S3). In another example (M482k‐27‐1), where gRNA50‐guided chimeric (−1‐bp, −8‐bp, and +1‐bp) and biallelic (−1‐bp and +1‐bp) editing were found in the At and Dt subgenome, respectively. All these changes occur within the sequence of miR482k (Figure 2) and result in more unpaired base‐pairs in the miR482k/miR482k* region (Figure S4).
FIGURE 2.
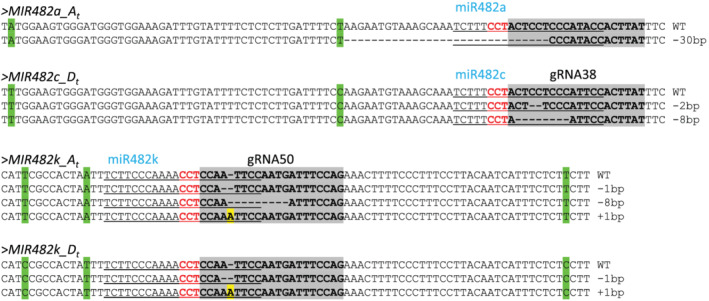
Sequence alignment showing gRNA38‐ and gRNA50‐mediated editing in miR482a/c and miR482k, respectively, in the At and Dt subgenomes. “‐” represents deletion. The inserted nucleotides are highlighted in yellow background. The miR482 sequences are underlined. The target sites of gRNA38 and gRNA50 are highlighted in gray background. PAM (protospacer adjacent motif) sequences are shown in red. The nucleotides used to distinguish the At and Dt subgenome are highlighted in green
Different individuals from the same transgenic event tend to have the same editing events, for example, the four T0 plants derived from the M482‐13 (gRNA35) transgenic event have the same biallelic editing in the At subgenome (−4‐bp and −5‐bp) and not edited in the Dt subgenome; however, different editing events were frequently observed in different individuals from the same transgenic event, for instance, in the five analyzed T0 plants from the M482‐11 (gRNA41) transgenic event (Table S1). Of the gRNAs used, seven gRNAs have a mismatch between the At and Dt subgenomes. Three (gRNA31, gRNA37, and gRNA49) of them seem to be specific for the designed target, as no editing was observed in the off‐target with a single base difference. But four (gRNA35, gRNA36, gRNA38, and gRNA41) of them showed editing activity at the off‐target with one mismatch (Table 3). For both gRNA41 and gRNA49, the mismatch in the At and Dt subgenome targets is at the 18th position, gRNA49 edits only the fully matched target (At subgenome) and gRNA41 edits both the fully matched target (Dt subgenome) and the target with one mismatch (At subgenome). While the mismatch of gRNA36 in the two subgenomes is located at the first position, gRNA36‐guided editing is not specific (Table 3). These results suggest that a single mismatch, regardless of its position relative to protospacer adjacent motif (PAM), may or may not be enough to define targeting specific.
Despite no transgenic for four gRNAs (gRNA33, gRNA43, gRNA47, and gRNA52) and no editing for three gRNAs (gRNA32, gRNA42, and gRNA45), saturation mutagenesis is achieved for the cotton MIR482 family as each MIR482 gene is edited by at least one gRNA (Table 3, Table S1).
2.3. Diversity of the cotton MIR482 gene editing library
To investigate the transmission and diversity of the editing events, 116 T1 progeny from 11 T0 plants (with eight gRNAs targeting six miR482 isoforms generated from 31 MIR482 loci) were analyzed by amplicon sequencing. The major editing events observed in T0 plants were found to be faithfully passed onto next generation, in which different combinations of the At and Dt editing events were observed in different individuals. Based on distinct combination of the editing events in the At and Dt subgenomes, in the 8–12 T1 progeny per T0 plant investigated, four to 10 genotypes were observed in individual T1 populations. All 10 T1 individuals from M482d‐29‐3 have different editing genotypes due to a total of 19 MIR482d loci being the targets of gRNA39 (Figure 3a). In total, 71 different editing genotypes were found among the 116 T1 progeny, indicating a high diversity of the gene editing mutant library. The editing‐genotype diversity of T1 population could depend on the nature of the editing events (biallelic and chimeric) and the number of integrated transgenes in T0 plants, the presence/absence of CRISPR/Cas9 in T1 individuals, and the number of targets of a gRNA. For instance, five and eight editing‐genotypes were observed among the 12 and 11 T1 progeny of M482c‐26‐1 and M482k‐27‐1, respectively (Figure 3a–c).
FIGURE 3.
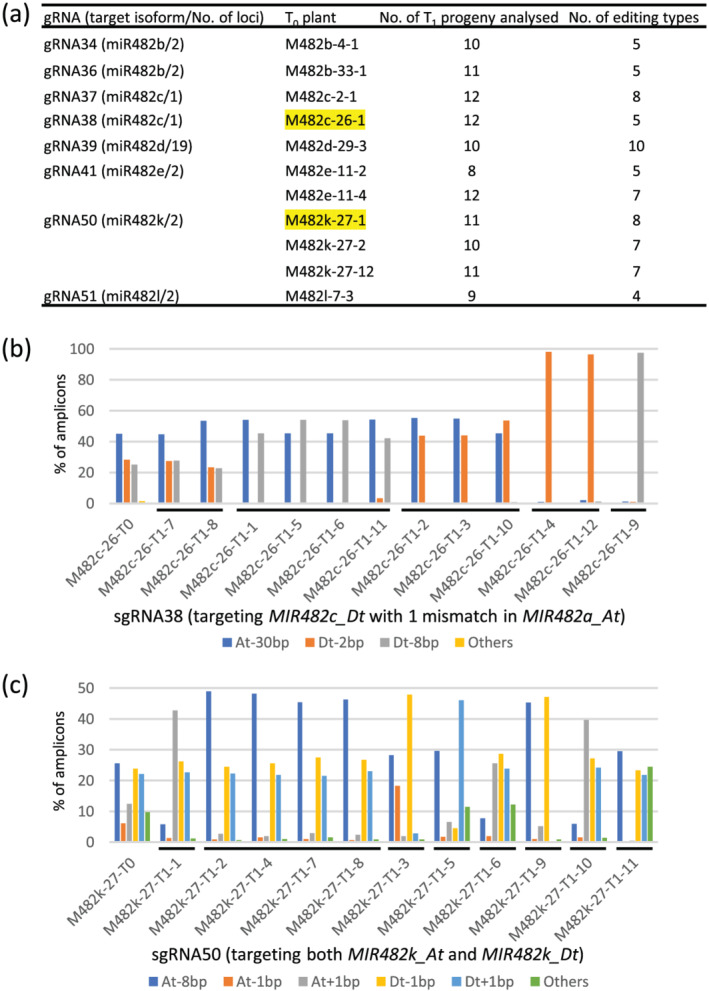
Characterization of representative MIR482 mutants. (a) The number of editing types observed in T1 progeny from eight independent transgenic events. The editing details of the two T0 plants highlighted in yellow and their T1 progeny are shown in (b) and (c). (b,c) The editing types of M482b‐33‐1 (M482b‐33‐T0) and M482k‐27‐1 (M482k‐27‐T0) and their progeny, respectively. T1 plants with the same editing events are indicated by a black bar underneath the X‐axis
2.4. Response of miR482 mutants to V. dahliae infection
To investigate the function of different miR482 isoforms in disease response, we generated T2 progeny from T1 plants mutated in MIR482b, MIR482c, MIR482k, or MIR482l and analyzed their editing events by amplicon sequencing. T3 populations derived from nine T2 plants were selected based on their mutation zygosity in the two subgenomes were inoculated with a nondefoliating V. dahliae (Vd‐ND) isolate using root‐dipping. Upon Vd‐ND infection, all nine T3 populations had a lower disease index (DI) than the control Coker‐315, with a significant lower DI observed in the populations mutated in MIR482c or MIR482l (Figure 4a,e), suggesting a variable role of different miR482 isoforms in response to Vd‐ND infection. Of the three MIR482l populations, the two with homozygous (M482l‐7‐T3_At/Dt_1, −44‐bp) or biallelic (M482l‐7‐T3_At/Dt_2, −2‐bp and −8‐bp) mutations in both subgenomes (Figure 4b,c) performed better than the one with heterozygous mutations in both subgenomes (M482l‐7‐T3_At‐H/Dt‐H) (Figure 4a,d), suggesting a potential dosage effect of miR482l.
FIGURE 4.
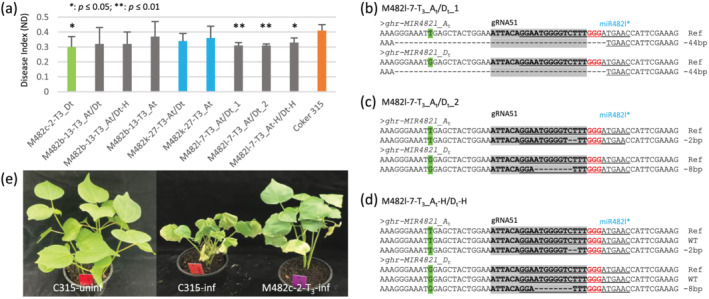
Response of miR482 edited mutants to infection of V. dahliae. (a) Disease index of T3 seedlings infected by the non‐defoliating V. dahliae (Vd‐ND) isolate. T3 progeny with different editing events in MIR482c (M482c), MIR482b (M482b), MIR482k (M482k), or MIR482l (M482l) were used. At, Dt, and At/Dt represent homozygous or biallelic editing in the subgenome At, Dt, and both, respectively. At‐H and Dt‐H represent heterozygous editing in the subgenome At and Dt, respectively. (b–d) editing genotypes of the parental plants of the three M482l T3 populations used in the disease assay. M482l‐7‐T3_At/Dt‐1: Homozygous for −44‐bp in both At and Dt subgenomes; M482l‐7‐T3_At/Dt‐2: Homozygous for −2‐bp and −8‐bp in At and Dt subgenome, respectively; M482l‐7‐T3_At‐H/Dt‐H: Heterozygous for both At and Dt subgenomes. The miR482l* sequence is underlined. The target sites of gRNA51 are highlighted in gray background. PAM (protospacer adjacent motif) sequences are shown in red. The nucleotides used to distinguish the At and Dt subgenome are highlighted in green. (e) Comparison of T3 plants infected by Vd‐ND with infected and uninfected control Coker‐315
3. DISCUSSION
CRISPR/Cas9‐mediated genome editing has revolutionized plant biology studies since its first application in targeted gene modification in plants in 2013 (Li et al., 2013; Nekrasov et al., 2013; Shan et al., 2013). While editing a single or few genes of interest may still be the major use of the technology, large‐scale mutagenesis of a gene family or thousands of genes, aiming for generation of knockout mutant collections for functional genomics and identification of genes important for crop performance, has been reported in several plant species (Bai et al., 2020; Liu et al., 2020; Lu et al., 2017; Meng et al., 2017). Regarding MIRNA genes, however, CRISPR/Cas9‐mediated editing of all members of a family has been documented only for the Arabidopsis MIR172 family with five members (Lian et al., 2021; O'Maoileidigh et al., 2021). Here, we mutated each of the 10 isoforms of the cotton MIR482 family, significantly advancing CRISPR/Cas9‐mediated mutagenesis of plant MIRNA family.
The MIR482 mutant library was generated by transformation of pooled gRNAs. Of the 22 gRNAs, 18 were successfully transformed, a coverage rate of ~82%. The coverage could be higher if the presence of all 22 gRNAs in the Agrobacterium culture were ensured and/or more independent transgenic plants were generated. For the transformed gRNAs, the average editing efficiency was 75%, slightly lower than the results reported in cotton (Ramadan et al., 2021) and maize (Liu et al., 2020) but higher than that in tomato (Jacobs et al., 2017) and soybean (Bai et al., 2020).
One of the key questions to be addressed when performing transformation of pooled gRNAs is how many T0 plants or independent transgenic events should be generated in order to have a complete coverage of the gRNAs used and the target genes. This is particularly important for the plant species that are recalcitrant to transformation, such as cotton. The required T0 population size depends on several factors, such as the number of target genes, mutagenic efficiency, the number of independent lines required to associate mutant genotypes with phenotypes, and concentration variation of the pooled gRNAs (Gaillochet et al., 2021). According to the published results, in rice, 2326 loci were found to be edited in the 5541 transgenic T0 plants analyzed (Lu et al., 2017), suggesting that the minimum number of T0 plants needed is ~2 times of the number of targets; in maize, the minimum number of T0 plants needed was suggested to be ~4 times of the number of vectors based on simulation analysis (Liu et al., 2020). In our case, the 18 transformed gRNAs or vectors were found in 40 independent transgenic events, indicating an ~2:1 ratio of independent transgenic event and gRNA/vector. While cotton transformation is restricted by few transformable genotypes and time‐consuming and labor‐intensive tissue culture procedure, to have a good coverage of the targets, the minimum number of independent T0 plants should be at least two times of the number of gRNAs to be transformed.
CRISPR/Cas9 gene editing has been used to edit three of the tomato MIR482 genes. In one study, CRISPR/Cas9‐induced small deletions (2‐bp to 9‐bp) at immediately down‐stream of the mature slymiR482f sequence significantly reduces the expression level of slymiR482f, and the mutated tomato plants are more resistance to Fusarium oxysporum f. sp. Lycopersici, the causative fungal pathogen of tomato wilt disease (Gao et al., 2021). In another study, a CRISPR/Cas9 vector containing three gRNAs targeting slymiR482b and three gRNAs targeting slymiR482b was used in transformation, and transgenic tomato plants simultaneously edited in both slymiR482b and slymiR482c or edited in slymiR482b alone were generated. The mutants showed enhanced resistance to Phytophthora infestans, an oomycete pathogen responsible for late blight disease, with a stronger resistance observed in the double mutants (Hong et al., 2021). In line with these results, we found that four of the nine tested cotton MIR482 mutants were more resistant to Vd‐ND and that the MIR482l mutants mutated in both subgenomes seemed to have a better resistance than the MIR482l mutant heterozygous in both subgenomes (Figure 4). We expect that that is a result of enhanced expression level of the NLRs targeted by the miR482 that is mutated, which is what we are going to verify.
We anticipate that the regulatory relationship between miR482 and NLRs in cotton is quite complicated, as ~62% of the predicted NLR targets of miR482 are potentially regulated by ≥2 miR482 isoforms (Shen et al., 2020). For instance, all the 27 predicted NLR targets of miR482c are regulated by at least one of other miR482 isoforms, and for miR482l, seven of its 16 NLR targets are regulated by at least one of other miR482 isoforms (Table 3). The miR482 mutants reported here provide essential materials for untangling the interwoven network composed of miR482 isoforms and their NLR targets to dissect the redundant function of individual miR482‐NLR modules responding to infection of V. dahliae and other cotton pathogens. But given the possibly complicated redundant functionality of different miR482 isoforms, it also demands high‐order mutants of different MIR482 genes, which are now being generated by crossing plants mutated in different miR482 isoforms, and requires simultaneous investigation of the pathogen‐induced expression changes of all miR482 isoforms and their NLR targets.
While many NLRs, typically with a major effect in disease resistance, have been functional characterized in plants (Deng et al., 2020), only few have been reported in cotton (Li et al., 2019), possibly that many NLRs each with a small contribution are required to confer disease resistance because of balancing disease response and growth to avoid autoimmunity caused by high expression of single NLRs. Given the complex regulatory relationship between miR482 isoforms and NLRs, miR482 may play a critical role in fine‐tuning the expression level of individual NLRs to limit the fitness costs resulted from overactive resistance caused by high expression of NLRs; that is, miR482 may be the key in balancing the growth‐defense trade‐off in cotton. The quantitative nature of disease resistance conferred by NLRs, as proposed in tomato (Canto‐Pastor et al., 2019), makes it unrealistic to enhance cotton disease resistance by manipulating individual NLRs, but it could be achieved by modulating the abundance of miR482 isoforms that regulate the NLRs involved.
To conclude, by combining CRISPR/Cas9 gene editing with transformation of pooled gRNAs, we accomplished saturated mutagenesis of the cotton MIR482 family with dozens of members in one go, demonstrating the feasibility of generating a mutant library by transformation of pooled gRNAs in polyploid. We showed that different miR482 isoforms function differently in response to Vd‐ND infection. The MIR482 mutant collection reported here provides not only the essential materials for dissecting the role of individual MIR482 genes and identifying the miR482‐NLR module(s) responding to V. dahliae and other pathogens, but also novel germplasm for breeding new cotton cultivars resistant to diseases.
4. METHODS
4.1. Plant materials and growth conditions
The Upland cotton (Gossypium hirsutum L.) accession Coker‐315 was used in cotton transformation as the recipient of gRNAs. All plants were grown in a greenhouse in Canberra, Australia, at 28°C ± 2°C with natural lighting.
4.2. gRNA design and generation of CRISPR/Cas9‐gRNA constructs
The published precursor sequences of all Upland cotton MIR482 genes were retrieved from our previous study (Shen et al., 2020) and uploaded onto the targetDesign webserver (http://skl.scau.edu.cn/targetdesign/) to get all potential gRNAs using the default settings based on the reference genome of TM‐1 (Zhang et al., 2015). The candidate gRNAs were manually checked to select a final list of gRNAs for each MIR482 based on the criteria of (1) preferring targeting miR482 to miR482* and to other region of the stem‐loop structure, and (2) without additional off‐target except the one located on the homoeologous chromosome.Forward and reverse strands of each gRNA with an additional 4‐bp (“GGCA” for the forward oligo and “AAAC” for the reverse oilgo) at the 5′ end that are compatible with the BsaI restriction site were synthesized. Equal amount (1 μl of 100 pmole) of the two single strand gRNAs were mixed, heated to 95°C for 5 min and cooled down to 25°C with a cooling rate of .2°C/min on an Eppendorf Mastercycler (EP384) to anneal them together to form a double stranded gRNA, which was then ligated to the CRISPR/Cas9 binary vector (Wang et al., 2018) linearized by BsaI digestion using the standard DNA ligation protocol. After confirmation by sequencing, equal amount (2 μl of 50 pmole) of each CRISPR/Cas9‐gRNA plasmid was mixed to make a pool containing a total of 22 plasmids. The pooled plasmids were transformed into A. tumefaciens (LBA4404) by electroporation, which was then used in cotton transformation without confirmation of the presence of every CRISPR/Cas9‐gRNA plasmid.
4.3. Cotton transformation and identification of transgenics
Cotyledon leaf discs of Coker‐315 were used as explants in cotton transformation by following the protocol previously reported with selection on kanamycin sulfate (Murray et al., 1999). Positive T0 transgenics were identified based on amplification of a fragment from the Cas9 gene using Cas9_F and Cas9_R (Table 1). Positive T0 transgenic plantlets were grown to maturity for the collection of T1 seeds in a greenhouse with natural lighting.
4.4. Analysis of gRNA integration and editing events
To check the transformed gRNA(s) in each T0 transgenic plant, an ~300‐bp fragment was amplified using GhU6F1 and gRNAR1 that flank the BsaI restriction site of the construct and sequenced using the GhU6F2 oligo located at downstream of GhU6F1 (Table 1). The transformed gRNA could be easily determined by aligning the sequences to the vector sequence when the transgenic plant contains a single gRNA; however, when two or more sgRNAs were transformed in a transgenic plant, they were indistinguishable based on the sequencing result of PCR product. The PCR products were therefore cloned using the TOPO TA cloning kit (Invitrogen) and ~10 clones were sequenced to identify the transformed individual gRNAs.
For analyzing editing events, a pair of primer matching both the At and Dt subgenomes was designed for each gh‐miR482 isoform (four pairs were designed for gh‐miR482d as a single pair of primer is not enough to distinguish the PCR products from a total of 19 gh‐miR482d loci) (Table 1). Barcodes were added to the 5′ ends of both forward and reverse primers so PCR products amplified from different plants using the same primers could be pooled and sequenced in a cost‐effective way. Approximately 20 amplicons (10 ng/amplicon) were pooled to form a sequencing sample. This approach was used for all T0 transgenics as well as their T1 and T2 progeny. For each sample, ~500 k of 250‐bp paired‐reads were generated using Illumina MiSeq platform. Amplicon sequencing was done by Azenta Life Sciences (Suzhou, China).
The raw sequencing data were checked for their quality using FastQC and de‐barcoded using Barcode Splitter to assign the reads to the plant from which they are derived. The de‐barcoded reads were submitted to Cas‐Analyzer (http://www.rgenome.net/cas-analyzer/#!) to identify editing events, including deletion, insertion, and substitution. Only the reads fully matched (except the region targeted by gRNA) to At or Dt subgenome, which was distinguished by subgenome‐specific nucleotide polymorphisms, were kept for further manual curation to tally the mutations.
4.5. Disease assay
A nondefoliating V. dahliae strain isolated from diseased cotton plants collected from commercial cotton field was cultured in half strength potato dextrose broth (12 g/L) for 7 days (25°C on a shaker, 180 rpm). The spore concentration of the inoculation solution was adjusted to 1 × 107 conidia/ml. T3 progeny from T2 plants that were analyzed for editing events were inoculated with the V. dahliae solution using the root dipping approach. This was done by submerging the roots of cotton seedlings (with two true leaves) into the V. dahliae solution for 5 min and then transplanted the seedlings into soil in 8‐cm pots. Seedlings of Coker‐315, the accession used in cotton transformation, were used as controls. Seedlings dipped in sterile water were used as mock inoculation controls. The assays were done twice independently with three replicates (~30 seedlings per replicate) in each experiment. Disease severity of seedlings were evaluated using disease index (DI) calculated based on disease score (0, 1, 2, 3, and 4) of individuals recorded at 14 days‐post‐inoculation (dpi). The scoring criteria are as follows: 0—no chlorosis symptom on all leaves; 1—symptom seen on one or two cotyledons but not on all true leaves; 2—symptom seen on both cotyledons and one true leaf; 3—symptom seen on both cotyledons and two true leaves; 4—symptom seen on all leaves or seedling died. DI = Σ (disease score x number of seedlings with the corresponding disease score)/[(total number of seedlings) × (maximal disease score)]. The assays were carried in a greenhouse with a temperature of 23°C ± 2°C with natural lighting. Significance was tested by the Student's t‐test (two tailed).
CONFLICT OF INTEREST
The authors declare no conflict of interest associated with the work described in this manuscript.
AUTHOR CONTRIBUTIONS
Q.‐H. Z. conceived the study; Y. Y., Q.‐H. Z. and I. W. performed the experiments and analyzed the data; S. J. and X. Z. designed the CRISPR/Cas9 binary vector; Q. L. discussed the results; Q.‐H. Z. wrote the manuscript.
Supporting information
FIGURE S1. gRNA38‐guided editing in MIR482a_A04 (1 mismatch) and MIR482c_D04 (0 mismatch) in M482–26‐1 (T0)
FIGURE S2. gRNA35‐guided editing in MIR482b_A13 (0 mismatch) and MIR482b_D13 (0 mismatch) in M482–13‐1 (T0)
FIGURE S3. Comparison of the secondary structures of MIR482a and MIR482c before and after editing by gRNA38.
FIGURE S4. Comparison of the secondary structures of MIR482k before and after editing by gRNA50.
TABLE S1. Details of the T0 transgenic plants.
ACKNOWLEDGMENTS
This work was supported by Cotton Breeding Australia, a joint venture between Cotton Seed Distributors Ltd. Australia and CSIRO Agriculture and Food (CBA04).
Zhu, Q.‐H. , Jin, S. , Yuan, Y. , Liu, Q. , Zhang, X. , & Wilson, I. (2022). CRISPR/Cas9‐mediated saturated mutagenesis of the cotton MIR482 family for dissecting the functionality of individual members in disease response. Plant Direct, 6(6), e410. 10.1002/pld3.410
Contributor Information
Qian‐Hao Zhu, Email: qianhao.zhu@csiro.au.
Shuangxia Jin, Email: jsx@mail.hzau.edu.cn.
DATA AVAILABILITY STATEMENT
All data included in this study are available upon request by contact with the corresponding authors.
REFERENCES
- Bai, M. , Yuan, J. , Kuang, H. , Gong, P. , Li, S. , Zhang, Z. , Liu, B. , Sun, J. , Yang, M. , Yang, L. , Wang, D. , Song, S. , & Guan, Y. (2020). Generation of a multiplex mutagenesis population via pooled CRISPR‐Cas9 in soya bean. Plant Biotechnology Journal, 18(3), 721–731. 10.1111/pbi.13239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto‐Pastor, A. , Santos, B. , Valli, A. A. , Summers, W. , Schornack, S. , & Baulcombe, D. C. (2019). Enhanced resistance to bacterial and oomycete pathogens by short tandem target mimic RNAs in tomato. Proceedings of the National Academy of Sciences of the United States of America, 116(7), 2755–2760. 10.1073/pnas.1814380116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, F. , Zeng, F. , Shen, Q. , Abbas, A. , Cheng, J. , Jiang, W. , Chen, G. , Shah, A. N. , Holford, P. , Tanveer, M. , Zhang, D. , & Chen, Z. H. (2022). Molecular evolution and functional modification of plant miRNAs with CRISPR. Trends in Plant Science. 10.1016/j.tplants.2022.01.009 [DOI] [PubMed] [Google Scholar]
- Deng, Y. , Ning, Y. , Yang, D. L. , Zhai, K. , Wang, G. L. , & He, Z. (2020). Molecular basis of disease resistance and perspectives on breeding strategies for resistance improvement in crops. Molecular Plant, 13, 1402–1419. 10.1016/j.molp.2020.09.018 [DOI] [PubMed] [Google Scholar]
- Gaillochet, C. , Develtere, W. , & Jacobs, T. B. (2021). CRISPR screens in plants: Approaches, guidelines, and future prospects. The Plant Cell, 33(4), 794–813. 10.1093/plcell/koab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. , Li, S. J. , Zhang, S. W. , Feng, T. , Zhang, Z. Y. , Luo, S. J. , Mao, H. Y. , Borkovich, K. A. , & Ouyang, S. Q. (2021). SlymiR482e‐3p mediates tomato wilt disease by modulating ethylene response pathway. Plant Biotechnology Journal, 19, 17–19. 10.1111/pbi.13439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, Y. , Meng, J. , He, X. , Zhang, Y. , Liu, Y. , Zhang, C. , Qi, H. , & Luan, Y. (2021). Editing miR482b and miR482c simultaneously by CRISPR/Cas9 enhanced tomato resistance to Phytophthora infestans . Phytopathology, 111(6), 1008–1016. 10.1094/PHYTO-08-20-0360-R [DOI] [PubMed] [Google Scholar]
- Jacobs, T. B. , Zhang, N. , Patel, D. , & Martin, G. B. (2017). Generation of a collection of mutant tomato lines using pooled CRISPR libraries. Plant Physiology, 174(4), 2023–2037. 10.1104/pp.17.00489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, N. , Meng, J. , Cui, J. , Sun, G. , & Luan, Y. (2018). Function identification of miR482b, a negative regulator during tomato resistance to Phytophthora infestans . Horticulture Research, 5, 9. 10.1038/s41438-018-0017-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. F. , Norville, J. E. , Aach, J. , McCormack, M. , Zhang, D. , Bush, J. , Church, G. M. , & Sheen, J. (2013). Multiplex and homologous recombination‐mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nature Biotechnology, 31(8), 688–691. 10.1038/nbt.2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T. G. , Wang, B. L. , Yin, C. M. , Zhang, D. D. , Wang, D. , Song, J. , Zhou, L. , Kong, Z. Q. , Klosterman, S. J. , Li, J. J. , Adamu, S. , Liu, T. L. , Subbarao, K. V. , Chen, J. Y. , & Dai, X. F. (2019). The Gossypium hirsutum TIR‐NBS‐LRR gene GhDSC1 mediates resistance against Verticillium wilt. Molecular Plant Pathology, 20(6), 857–876. 10.1111/mpp.12797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, H. , Wang, L. , Ma, N. , Zhou, C. M. , Han, L. , Zhang, T. Q. , & Wang, J. W. (2021). Redundant and specific roles of individual MIR172 genes in plant development. PLoS Biology, 19, e3001044. 10.1371/journal.pbio.3001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. J. , Jian, L. , Xu, J. , Zhang, Q. , Zhang, M. , Jin, M. , Peng, Y. , Yan, J. , Han, B. , Liu, J. , Gao, F. , Liu, X. , Huang, L. , Wei, W. , Ding, Y. , Yang, X. , Li, Z. , Zhang, M. , Sun, J. , … Yan, J. (2020). High‐throughput CRISPR/Cas9 mutagenesis streamlines trait gene identification in maize. The Plant Cell, 32(5), 1397–1413. 10.1105/tpc.19.00934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y. , Ye, X. , Guo, R. , Huang, J. , Wang, W. , Tang, J. , Tan, L. , Zhu, J. K. , Chu, C. , & Qian, Y. (2017). Genome‐wide targeted mutagenesis in rice using the CRISPR/Cas9 system. Molecular Plant, 10(9), 1242–1245. 10.1016/j.molp.2017.06.007 [DOI] [PubMed] [Google Scholar]
- Meng, X. , Yu, H. , Zhang, Y. , Zhuang, F. , Song, X. , Gao, S. , Gao, C. , & Li, J. (2017). Construction of a genome‐wide mutant library in rice using CRISPR/Cas9. Molecular Plant, 10(9), 1238–1241. 10.1016/j.molp.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Murray, F. , Llewellyn, D. , McFadden, H. , Last, D. , Dennis, E. S. , & Peacock, W. J. (1999). Expression of the Talaromyces flavus glucose oxidase gene in cotton and tobacco reduces fungal infection, but is also phytotoxic. Molecular Breeding, 5, 219–232. 10.1023/A:1009625801909 [DOI] [Google Scholar]
- Nekrasov, V. , Staskawicz, B. , Weigel, D. , Jones, J. D. , & Kamoun, S. (2013). Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA‐guided endonuclease. Nature Biotechnology, 31(8), 691–693. 10.1038/nbt.2655 [DOI] [PubMed] [Google Scholar]
- Ngou, B. P. M. , Ding, P. , & Jones, J. D. (2022). Thirty years of resistance: Zig‐Zag through the plant immune system. The Plant Cell, 34(5), 1447–1478. 10.1093/plcell/koac041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Maoileidigh, D. S. , van Driel, A. D. , Singh, A. , Sang, Q. , Le Bec, N. , Vincent, C. , de Olalla, E. B. G. , Vayssieres, A. , Romera Branchat, M. , Severing, E. , Martinez Gallegos, R. , & Coupland, G. (2021). Systematic analyses of the MIR172 family members of Arabidopsis define their distinct roles in regulation of APETALA2 during floral transition. PLoS Biology, 19, e3001043. 10.1371/journal.pbio.3001043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, S. , Park, G. , Atamian, H. S. , Han, C. S. , Stajich, J. E. , Kaloshian, I. , & Borkovich, K. A. (2014). MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum . PLoS Pathogens, 10, e1004464. 10.1371/journal.ppat.1004464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan, M. , Alariqi, M. , Ma, Y. , Li, Y. , Liu, Z. , Zhang, R. , Jin, S. , Min, L. , & Zhang, X. (2021). Efficient CRISPR/Cas9 mediated pooled‐sgRNAs assembly accelerates targeting multiple genes related to male sterility in cotton. Plant Methods, 17, 16. 10.1186/s13007-021-00712-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, Q. , Wang, Y. , Li, J. , Zhang, Y. , Chen, K. , Liang, Z. , Zhang, K. , Liu, J. , Xi, J. J. , Qiu, J. L. , & Gao, C. (2013). Targeted genome modification of crop plants using a CRISPR‐Cas system. Nature Biotechnology, 31(8), 686–688. 10.1038/nbt.2650 [DOI] [PubMed] [Google Scholar]
- Shen, E. , Chen, T. , Zhu, X. , Fan, L. , Sun, J. , Llewellyn, D. J. , Wilson, I. , & Zhu, Q. H. (2020). Expansion of MIR482/2118 by a class‐II transposable element in cotton. The Plant Journal, 103(6), 2084–2099. 10.1111/tpj.14885 [DOI] [PubMed] [Google Scholar]
- Shivaprasad, P. V. , Chen, H. M. , Patel, K. , Bond, D. M. , Santos, B. A. , & Baulcombe, D. C. (2012). A microRNA superfamily regulates nucleotide binding site‐leucine‐rich repeats and other mRNAs. The Plant Cell, 24(3), 859–874. 10.1105/tpc.111.095380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya, N. M. , Zhu, Q. H. , & Bhat, R. S. (2011). Transposon insertional mutagenesis in rice. Methods in Molecular Biology, 678, 147–177. 10.1007/978-1-60761-682-5_12 [DOI] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Yuan, D. , Zhu, D. , Shen, C. , Li, J. , Liu, F. , Pei, L. , Wang, P. , Zhao, G. , Ye, Z. , Huang, H. , Yan, F. , Ma, Y. , Zhang, L. , Liu, M. , You, J. , Yang, Y. , Liu, Z. , … Zhang, X. (2019). Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense . Nature Genetics, 51, 224–229. 10.1038/s41588-018-0282-x [DOI] [PubMed] [Google Scholar]
- Wang, P. , Zhang, J. , Sun, L. , Ma, Y. , Xu, J. , Liang, S. , Deng, J. , Tan, J. , Zhang, Q. , Tu, L. , Daniell, H. , Jin, S. , & Zhang, X. (2018). High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnology Journal, 16, 137–150. 10.1111/pbi.12755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Hu, Y. , Jiang, W. , Fang, L. , Guan, X. , Chen, J. , Zhang, J. , Saski, C. A. , Scheffler, B. E. , Stelly, D. M. , Hulse‐Kemp, A. M. , Wan, Q. , Liu, B. , Liu, C. , Wang, S. , Pan, M. , Wang, Y. , Wang, D. , Ye, W. , … Chen, Z. J. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM‐1) provides a resource for fiber improvement. Nature Biotechnology, 33, 531–537. 10.1038/nbt.3207 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Malzahn, A. A. , Sretenovic, S. , & Qi, Y. (2019). The emerging and uncultivated potential of CRISPR technology in plant science. Nature Plants, 5(8), 778–794. 10.1038/s41477-019-0461-5 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Waseem, M. , Zeng, Z. , Xu, J. , Chen, C. , Liu, Y. , Zhai, J. , & Xia, R. (2022). MicroRNA482/2118, a miRNA superfamily essential for both disease resistance and plant development. The New Phytologist, 233(5), 2047–2057. 10.1111/nph.17853 [DOI] [PubMed] [Google Scholar]
- Zhu, Q. H. , Fan, L. , Liu, Y. , Xu, H. , Llewellyn, D. , & Wilson, I. (2013). miR482 regulation of NBS‐LRR defense genes during fungal pathogen infection in cotton. PLoS ONE, 8(12), e84390. 10.1371/journal.pone.0084390 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. gRNA38‐guided editing in MIR482a_A04 (1 mismatch) and MIR482c_D04 (0 mismatch) in M482–26‐1 (T0)
FIGURE S2. gRNA35‐guided editing in MIR482b_A13 (0 mismatch) and MIR482b_D13 (0 mismatch) in M482–13‐1 (T0)
FIGURE S3. Comparison of the secondary structures of MIR482a and MIR482c before and after editing by gRNA38.
FIGURE S4. Comparison of the secondary structures of MIR482k before and after editing by gRNA50.
TABLE S1. Details of the T0 transgenic plants.
Data Availability Statement
All data included in this study are available upon request by contact with the corresponding authors.