

Abstract
Gene editing has shown promise for correcting or bypassing dystrophin mutations in Duchenne muscular dystrophy (DMD). However, preclinical studies have focused on young animals with limited muscle fibrosis and wasting, thereby favoring muscle transduction, myonuclear editing, and prevention of disease progression. Here, we explore muscle-specific dystrophin gene editing following intramuscular delivery of AAV6:CK8e-CRISPR/SaCas9 in 3- and 8-year-old dystrophic CXMD dogs and provide a qualitative comparison to AAV6:CK8e-micro-dystrophin gene replacement at 6 weeks post-treatment. Gene editing restored the dystrophin reading frame in ∼1.3% of genomes and in up to 4.0% of dystrophin transcripts following excision of a 105-kb mutation containing region spanning exons 6–8. However, resulting dystrophin expression levels and effects on muscle pathology were greater with the use of micro-dystrophin gene transfer. This study demonstrates that our muscle-specific multi-exon deletion strategy can correct a frequently mutated region of the dystrophin gene in an aged large animal DMD model, but underscores that further enhancements are required to reach efficiencies comparable to AAV micro-dystrophin. Our observations also indicate that treatment efficacy and state of muscle pathology at the time of intervention are linked, suggesting the need for additional methodological optimizations related to age and disease progression to achieve relevant clinical translation of CRISPR-based therapies to all DMD patients.
Keywords: CRISPR, Cas9, AAV, CXMD, dogs, Duchenne, gene editing, DMD, micro-dystrophin, muscle
Graphical abstract
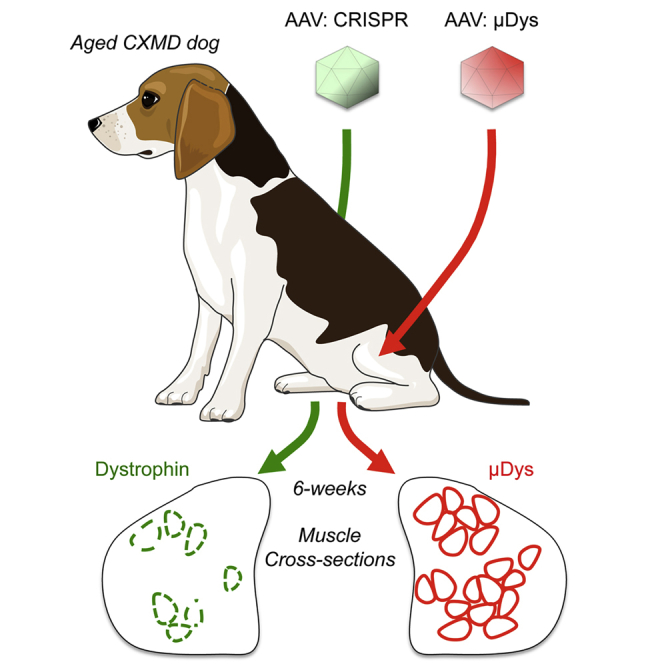
Comparisons of CRISPR/Cas9 gene editing and micro-dystrophin gene replacement in old dystrophic dogs highlight the promise and limitations of AAV-mediated gene therapy in older subjects with advanced muscle pathology.
Introduction
Duchenne muscular dystrophy (DMD) is a devastating X-linked muscle degenerative condition affecting the majority of muscle groups throughout the body.1 DMD is caused by the lack of functional dystrophin at the sarcolemma, which is critical for maintaining muscle integrity during contraction. In its absence, skeletal muscle fibers experience repeated bouts of necrosis and regeneration, eventually resulting in significant loss of muscle mass and function, which together with cardiomyopathy reduces life expectancy. Three ongoing clinical trials in the United States, conducted by Solid Biosciences (NCT03368742), Sarepta Therapeutics (NCT03375164), and Pfizer (NCT03362502), are aimed at restoring muscle integrity and function following micro-dystrophin (μDys) gene replacement using adeno-associated viral vectors (AAVs). While gene replacement therapies hold great promise for ameliorating DMD pathology, the utilization of a μDys gene, as necessitated by the limited carrying capacity of AAVs, means that some aspects of dystrophin functionality are sacrificed.2 For patients with amenable mutations, AAV-mediated CRISPR/Cas-based gene editing therapies could restore larger dystrophin proteins with greater function.3,4 However, for these benefits to be realized, CRISPR/Cas editing efficacies must rival those of μDys gene replacement. Several promising proof-of-principle studies have demonstrated both feasibility and some efficacy of in vivo gene editing for inducing dystrophin expression and improving muscle pathophysiology in DMD animal models.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 However, except for high-efficiency corrections in very young animals with individually rare and relatively easily correctible mutations affecting single exons,6,7 induced dystrophin levels have frequently been less than 5–10% of wild-type levels and well below μDys levels routinely obtained following gene replacement.16, 17, 18 Also, more than two-thirds of DMD patients carry gene deletions or duplications that are challenging to fully “correct” using current gene editing methods,19 though promising progress have been made to enable correction of duplications.20,21 While encouraging, suboptimal levels of dystrophin expression from more broadly applicable editing strategies may offer only transient therapeutic effects, as partially corrected myofibers may be gradually lost over time as a result of skeletal muscle turnover.8 Loss of CRISPR-corrected dystrophin expression may potentially be mitigated by efficient editing of the dystrophin gene in muscle stem cells (satellite cells [SCs]).22 While AAV-mediated gene editing has been demonstrated in SCs,14,23,24 it is still unclear how readily AAV vectors are able to transduce quiescent and/or activated SCs.25 This potential caveat is also highlighted by contrasting reports of relatively high levels of AAV SC targeting using Cre-reporter systems against the very low levels of dystrophin gene correction in SCs of dystrophic hosts (<0.05%) following ubiquitous (CMV-mediated) in vivo editing,24 which indicates a need for significant technological/methodological improvements for SC editing to have a beneficial therapeutic impact.
Notably, most studies to date have focused on treating young animals that exhibit only mild disease pathology, including minimal levels of fibrosis, adipogenic tissue accumulation, and muscle wasting. Effective treatment of older animals, and future patients with more progressed pathophysiology, may require higher vector doses to enable sufficient vector diffusion through fibrotic and adipogenic muscle infiltrates to transduce the remaining muscle mass. If so, this may represent a significant hurdle, as AAV clinical trials using very high doses have in some cases shown adverse events ranging from thrombocytopenia to acute kidney damage and cardiopulmonary insufficiency. Encouragingly, recent advances in AAV vector design have yielded synthetically modified vector capsids (AAVMYO) that exhibit significantly enhanced muscle transduction (up to 10-fold),26,27 which could effectively reduce vector doses while improving therapeutic efficacies.
The outbred canine X-linked muscular dystrophy (CXMD) model of DMD28 carries a splice acceptor site mutation in intron 6 of the dystrophin gene.29 This results in incorrect pre-messenger RNA (mRNA) splicing, skipping of exon 7, and a disrupted translational reading frame leading to a premature stop codon encoded in exon 8. Our group and others have previously utilized canine models in preclinical AAV-μDys studies30, 31, 32, 33 and in one AAV-mediated gene editing study.7 The deltaE50-MD canine model of DMD used in that gene editing study carries an exon 50 deletion that generates an out-of-frame transcript and loss of dystrophin expression.34 This mutation was addressed by delivering AAV9 vectors carrying SpCas9 and a single guide RNA (sgRNA) designed to disrupt the splice acceptor site and an exonic splicing enhancer for exon 51, resulting in the “skipping” of exon 51 and restoration of an open reading frame (ORF) encoding a slightly smaller dystrophin.7 While results from the deltaE50-MD study are extremely encouraging, the sgRNA approach is amenable only to a subset of DMD mutations in which removing a single exon is sufficient to restore an ORF that encodes a functional dystrophin.35 Also, as these dogs were treated at a very young age when muscle pathophysiology is still developing, both AAV transduction and myonuclear editing were facilitated by the absence of significant muscle loss, inflammation, and fibrosis. Here, we investigated the potential for AAV to induce dystrophin expression in old CXMD dogs using our previously developed muscle-specific expression system for CRISPR-mediated multi-exon deletion5 and compared the results with micro-dystrophin delivery.
Results
To generate a functional dystrophin ORF in the CXMD model of DMD, we adapted our previously developed muscle-restricted,3,5,8 multi-exon deletion strategy to excise exons 6–8 (Figure 1A). This ∼105 kb region encodes significant portions of the N-terminal actin-binding domain as well as part of the first hinge region of the dystrophin protein.36 While this actin-binding domain is considered an important domain for proper function and stability of dystrophin,37 removal of this relatively large segment was necessary because the ORF is shared across exons 6–8, and removal of any one or two exons would fail to encode an in-frame transcript.38 The ability to excise exons 6–8 of the canine dystrophin gene was evaluated following in vitro transfection of primary myogenic progenitor cells, isolated from skeletal muscles of CXMD dogs, with a single plasmid carrying the muscle creatine kinase-based CK8e expression cassette driving expression of SaCas9, along with two ubiquitous U6-driven sgRNA expression cassettes targeting introns 5 and 8 (Figure 1A). PCR analysis of DNA isolated from differentiated myotubes at 5 days post-transfection revealed a unique amplification product corresponding in size to a product lacking the genomic sequence encoded between the target sites (Figures 1B and 1C). Subsequent Sanger sequencing of the product confirmed successful deletion of the targeted genomic region containing exons 6–8, thus demonstrating feasibility of the strategy.
Figure 1.

Generating a dystrophin open reading frame in primary CXMD-derived myoblasts
(A) Outline of the strategy to generate a functional dystrophin open reading frame in CXMD dogs via CRISPR-mediated deletion of exons 6–8, including the splice acceptor mutation in intron 6 (red dot). (B) Primary CXMD-derived myotubes at 5 days post-transfection. (C) Semi-quantitative PCR of DNA isolated from CXMD myotubes at 5 days post-transfection with CRISPR-plasmid using 3.75 and 7.5 μL of lipofectamine. Amplification across the ∼105 kb region targeted for deletion generates a unique Δexons 6–8 deletion product (340 bp; red arrow) in CRISPR-treated samples. The additional products seen likely represent non-specific or incomplete amplification of non-edited genomes due to the large size of the native genomic region being amplified.
To evaluate the potential for amelioration or reversal of advanced dystrophic muscle pathology, we compared the therapeutic potential of our single vector gene editing system (modified to target canine exons 6–8)5 with that of our third-generation micro-dystrophins (μDys5).16 This was accomplished by AAV-mediated intramuscular (IM) administration into the cranial tibialis (CT) muscles of fully immunosuppressed 3- and 8-year-old male CXMD dogs, as previously described.30,31 Expression of SaCas9 and µDys5 was restricted to striated muscle using the mouse muscle creatine kinase (MCK)-based CK8e expression cassette, and the CRISPR (AAV6:CK8e-SaCas9-2XsgRNA) or μDys (AAV6:CK8e-μDys5) vectors (Figure 2A) were delivered into contralateral CT muscles via 25 equally spaced injections each containing 4 × 1011 vector genomes (vg). Treated dogs remained on immunosuppression until the end of the study (6 weeks post-transduction), when AAV-injected and control muscles were harvested and analyzed for dystrophin expression. Immunofluorescence (IF) analysis of samples taken from proximal, medial, and distal regions of the treated CT muscles revealed highly mosaic myofiber expression of both μDys- and CRISPR-generated dystrophin following either treatment (Figure 2C). However, the number of dystrophin-positive fibers was greater in the μDys-injected than in the CRISPR-injected 3-year-old CXMD dogs, whereas such differences were less obvious in 8-year-old CXMD dogs (Figure 2C). Interestingly, while μDys protein was typically found to completely envelope the sarcolemma (∼64% of positive fibers), most dystrophin-positive fibers resulting from CRISPR treatment were not completely enveloped (∼17%); instead, the majority of positive gene-edited myofibers exhibited fenestrated distribution of dystrophin (see Discussion). In line with the observed differences in sarcolemmal dystrophin immunofluorescent staining (Figure 2C), fewer dystrophin-positive myofibers exhibited centrally located nuclei (CN) following μDys treatment (∼26% CN) compared with CRISPR treatment (∼37% CN), indicating that uniformly distributed μDys across the sarcolemma offered better stabilization of the muscle. Histological evaluation was then performed on serial muscle cross sections from regions exhibiting the most uniform expression of dystrophin to explore the effect of the resulting expression levels on amelioration of dystrophic histopathology. Notably, interstitial collagen deposition was not obviously reduced during this short treatment window by either therapeutic strategy (Figure 3), although both μDys- and CRISPR-mediated dystrophin expression were able to protect transduced myofibers from necrosis.
Figure 2.

Induction of dystrophin expression following AAV-mediated CRISPR/Cas9 gene editing and micro-dystrophin gene replacement
(A) Overview of AAV6 vectors expressing SaCas9 under control of the CK8e regulatory cassette and two single gRNA cassettes targeting introns 5 and 8 of the canine dystrophin gene as well as micro-dystrophin-5 (μDys5) under control of CK8e. (B) Schematics depicting structural domains present in normal full-length dystrophin (427 kDa), Δ6–8 dystrophin (∼409 kDa), Δ6–9 dystrophin (∼404 kDa), and μDys5 (147 kDa). (C) Immunofluorescence analysis of well-transduced muscle sample regions from the 3- and 8-year-old dogs injected with AAV6:CK8e-CRISPR/Cas9 (CRISPR) and AAV6:CK8e-μDys5 (μDys). Untreated control samples were derived from non-treated vastus lateralis muscles. Sections were stained with DAPI (top row) and for dystrophin (middle row). Merged images of DAPI and dystrophin are shown in the bottom row. CRISPR-treated sections were stained with antibodies raised against the C-terminus of dystrophin, while untreated control- and μDys-treated samples were stained with antibodies raised against the hinge 1/spectrin-like repeat 1 (H1/R1) region of dystrophin, as μDys5 contains this region but lacks the majority of the C-terminus. Scale bar, 100 μm. Images of control wild-type cranial tibialis muscle sections stained with both C-terminal and H1/R1 dystrophin antibodies are available in Figure S1.
Figure 3.

Partial amelioration of dystrophic muscle histopathology following μDys, but not CRISPR treatment
CRISPR- versus μDys-treated muscle sample cross sections from the 3-year-old dog (exhibiting dystrophin expression in Figure 2), stained with hematoxylin and eosin to reveal myofibers (pink) and cell nuclei (blue) (H&E; left columns) or stained with picrosirius red to reveal myofibers (orange) and collagen/fibrotic tissue (red) (right columns). Images located in the top or bottom rows were acquired from the same muscle samples at 100×- and 400× magnification, respectively. Scale bars, 100 μm.
To evaluate dystrophin gene editing efficiency, nucleic acids were extracted from several CRISPR-treated muscle samples adjacent to those identified (via IF) as exhibiting the most uniform AAV transduction and dystrophin expression. Three of four muscle samples examined showed successful deletion of the targeted region at both the genomic and the transcript levels, as demonstrated by the generation of a unique amplification product following semi-quantitative PCR (Figure 4A) and RT-PCR (Figure 4B). Despite attempting to match adjacent muscle samples shown to contain dystrophin-corrected myofibers via IF, one of the samples from the 8-year-old dog still showed undetectable dystrophin gene editing, presumably as a result of poor AAV transduction, and was excluded from subsequent statistical analysis. Sanger sequencing of the unique PCR and RT-PCR amplicons demonstrated that while the genomic PCR product corresponded to successful deletion of the intended genomic region containing exons 6–8, the main RT-PCR product generated from isolated mRNA corresponded to a deletion of exons 6–9 (Figures 4C and 4D). Dystrophin transcripts lacking exons 6–9 also encode a functional ORF and likely result from a cryptic splice site associated with exon 9, which has previously been detected in both untreated and antisense oligonucleotide-treated GRMD/CXMD muscle extracts.39, 40, 41 As indicated by our results, deleting the genomic region spanning introns 5–8 appears to preferentially result in the production of dystrophin transcripts that lack exons 6–9, which effectively removes the hinge-1 region of the dystrophin protein.
Figure 4.

In vivo gene editing induces a functional dystrophin open reading frame
(A) Semi-quantitative PCR analysis demonstrating presence of unique Δexon 6–8 amplicon in 3 of 4 muscle samples treated with CRISPR/Cas9 (red arrow); bottom amplicon represents GAPDH controls. (B) Semi-quantitative RT-PCR analysis demonstrating presence of unique deletion generated amplicon in 3 of 4 muscle samples treated with CRISPR/Cas9 (red arrow); bottom amplicon represents GAPDH controls. (C and D) Illustrations representing Sanger sequencing results of CRISPR-edited DNA lacking the genomic region flanked by the CRISPR target sites (X) in introns 5–8 (C) and the majority of CRISPR-edited transcripts lacking exons 6–9 as a result of alternative splicing (dashed blue arrow) of exon 9 (D). (E) Digital PCR quantification showing deletion frequency of the genomic region spanning introns 5–8 for CRISPR- versus μDys5-treated muscles as well as for untreated wild-type (WT) and CXMD [(−) control] samples. Note that one of the CRISPR-treated samples demonstrated inefficient gene editing owing to suboptimal AAV transduction (red point) and was hence excluded from statistical analysis (one-way ANOVA with Tukey's multiple comparisons test, ∗∗p < 0.01). (F) Digital RT-PCR quantification showing deletion frequency of exons 6–8 and 6–9 at the transcript level for CRISPR-treated samples compared with untreated controls consisting of one WT and one CXMD muscle sample. The CRISPR-treated muscle sample demonstrating undetectable editing at the genome level also lacked Δexon 6–8 or Δexon 6–9 edited transcripts (red dot) and was excluded from statistical analysis (one-way ANOVA with Tukey's multiple comparisons test, ∗∗p < 0.01, ∗∗∗p < 0.001).
Quantification of genomes and transcripts containing CRISPR-induced deletion of the targeted region was accomplished via digital PCR and RT-PCR. Statistically significant deletion events were detected only in CRISPR-treated samples, in which 1.31 ± 0.17% of total genomes corresponded to deletion of the genomic region spanning introns 5–8 (Figure 4E). At the transcript level, deletion of exons 6–8 was detected in 1.13 ± 0.24% of total dystrophin transcripts, while deletion of exons 6–9 occurred at a significantly higher frequency of 2.93 ± 0.31% (Figure 4F). ICE (Inference of CRISPR Edits) analysis of Sanger sequencing data from PCR amplicons across the individual CRISPR target sites showed that the editing frequency at intron 8 (6%) was significantly higher than that of intron 5 (1.25%) (Table 1). This seemingly asynchronous CRISPR activity likely contributed to the relatively low overall genomic deletion frequency of exons 6–8, though the ICE analysis data reflect only genomes where CRISPR-treatment failed to remove the genomic sequence spanning the two target sites. Individual sample values from ICE analyses, digital PCR (dPCR) and digital RT-PCR (dRT-PCR) are presented in Table 1.
Table 1.
Quantification of CRISPR-editing efficiency
| Samples | ICE (intron 5) |
ICE (intron 8) |
dPCR (genomic) |
dRT-PCR (transcript) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % indels | R2 | % indels | R2 | Δ6–8 (%) | CI (%) | Δ6–8 (%) | CI (%) | Δ6–9 (%) | CI (%) | |
| Untreated control | Ref | N/A | Ref | N/A | 0.006 | 0.003–0.044 | 0.002 | 0.0002–0.0091 | 0.002 | 0.0002–0.0093 |
| WT control | 0 | 1 | – | – | 0.012 | 0.011–0.036 | 0.002 | 0.0002–0.0086 | 0.005 | 0.0014–0.0140 |
| CRISPR (D1-S1) | 2 | 1 | 9 | 0.99 | 1.419 | 1.183–1.698 | 1.443 | 1.2950–1.5990 | 3.222 | 2.9630–3.4890 |
| CRISPR (D1-S2) | 1 | 1 | 6 | 1 | 0.981 | 0.796–1.204 | 0.654 | 0.5570–0.7620 | 2.320 | 2.0770–2.5710 |
| CRISPRa (D2-S1) | 1 | 1 | 1 | 1 | 0.089 | 0.051–0.153 | 0.067 | 0.0458–0.0974 | 0.139 | 0.1060–0.1810 |
| CRISPR (D2-S2) | 1 | 1 | 8 | 1 | 1.537 | 1.326–1.776 | 1.306 | 1.1650–1.4550 | 3.261 | 2.9930–3.5380 |
| μDys5 (D1-S1) | 0 | 1 | 0 | 1 | 0.006 | 0.001–0.033 | 0.002 | 0.0004–0.0134 | – | – |
| μDys5 (D1-S2) | – | – | – | – | 0.055 | 0.028–0.109 | 0 | – | – | – |
| μDys5 (D2-S1) | – | – | – | – | 0.016 | 0.004–0.057 | 0.006 | 0.0021–0.0164 | – | – |
| μDys5 (D2-S2) | – | – | – | – | 0.007 | 0.001–0.042 | 0 | – | – | – |
Quantification of editing frequency in CRISPR- versus μDys-treated muscle samples (S1-2) acquired from the 3-year-old dog (D1) and the 8-year-old dog (D2), as presented in Figure 4. ICE analysis reveals estimated cutting frequency at the individual genomic target sites within introns 5 and 8 and where the coefficient of determination (R2) represents the fit of the indel distribution to the Sanger sequences of the edited versus untreated negative control sample for ICE analyses. Note: ICE analysis of amplicons generated across the individual target sites is possible only for genomes where gene editing failed to remove the targeted genomic region. Digital PCR [dPCR (genomic)] and dRT-PCR (transcript) columns represent percent Δ6–8 deletion genomes per total genomes and percent Δ6–8 or Δ6–9 transcripts per total dystrophin transcripts, respectively. Confidence intervals (CI) for individual sample levels of corrected genomes and transcripts following dPCR and dRT-PCR are generated as part of the automated analysis performed using the Thermo Fisher Scientific Cloud-based dPCR analysis platform. CI, confidence interval; Ref, reference; N/A, not applicable.
CRISPR-treated muscle sample exhibiting near-undetectable gene editing in Figure 4.
Successful generation of a functional dystrophin open reading frame led to near-full-length dystrophin expression above background levels in three out of the four analyzed CRISPR-treated samples, as determined via western blotting of muscle lysates (Figure 5). Low levels of background expression of near-full-length dystrophin was observed in untreated and μDys-treated samples, likely reflecting the selective clonal preservation of dystrophin revertant myofibers with age.41, 42, 43 Expression of μDys5 was reliably detected using antibodies raised against the region of dystrophin that is encoded by exons 10 and 11, as μDys5 lacks the majority of the C-terminal domain that was used to detect CRISPR-generated dystrophin.
Figure 5.

Expression of dystrophin in treated muscle samples
(A) Western blotting of muscle sample lysates reveals increased near-full-length dystrophin expression over background/revertant levels in samples receiving CRISPR treatment (Dys (CT), 427 kDa band). (B) Expression of μDys5 (∼147 kDa) was visualized using antibodies raised against H1/R1 of dystrophin [Dys (Ex10/11)]. The slightly weaker product seen between the 147 kDa μDys5 band and the 427 kDa full-length dystrophin band (possibly reflecting dystrophin-revertant fibers) likely represents μDys5 duplexes that were insufficiently reduced during sample preparation. (C) Expression of SaCas9 was observed only in samples receiving CRISPR/Cas9, visualized using antibodies against a triple HA epitope fused at the 3′ end of SaCas9. (D) Detection of GAPDH expression to visualize total protein load for the various samples. Note that CRISPR-treated sample 1 from the 8-year-old dog exhibits reduced expression of near-full-length dystrophin and almost undetectable expression of SaCas9, likely reflecting a poorly transduced sample from the IM injection protocol applied. The positive control (WT) lanes were loaded with normal (wild-type) canine muscle extracts. The negative control sample (−) was acquired from the vastus lateralis muscle of the 3-year-old dog. Select additional molecular size markers are provided in gray italics (130 and 250 kDa).
Discussion
The difference in sarcolemmal distribution of dystrophin following CRISPR versus μDys treatment (Figure 2B) may reflect differences in the mechanisms by which the two methods produce dystrophin. With gene editing, individual myonuclei must accumulate both Cas9 and two sgRNAs, which must then act in synchrony to cleave the dystrophin gene twice and enable joining of the two ends. In contrast, μDys can be generated following transcription of individual AAV vector genomes without the need for multiple endonuclease and ligation events. Also, a correctly edited dystrophin gene must be transcribed across a >2 MB genomic region from its endogenous promoter, whereas μDys is generated by transcription of a <4 kb cDNA using the powerful CK8e-regulatory cassette. Previous studies have also shown that individual proteins produced in myofibers display discreet “nuclear domains” of expression, with small proteins diffusing considerably greater distances from their nuclei of origin than larger proteins, including full-length dystrophin.44 While our micro-dystrophins have been carefully developed to provide both stability and functionality, it is also possible that deleting exons 6–8 (or 6–9) through gene editing might produce unstable, less functional, or mis-localized dystrophin isoforms. Dystrophins lacking exons 6–9 carry all 24 spectrin-like repeats, but they lack the C-terminal portion of the N-terminal actin-binding domain and much of hinge-1. Previous studies have shown that this same deletion, when induced by morpholino antisense oligos, in this same dog model led to a clear improvement in performance.40 In contrast, all micro-dystrophins include the full N-terminal actin-binding domain (ABD) and hinge-1, portions of which are encoded by exons 6–9.16 Additionally, previous studies of Becker muscular dystrophy (BMD) patients with deletions in the ABD showed that many of these proteins were unstable.45,46 Though available in vivo data are sparse, iPSC-derived cardiomyocytes lacking dystrophin exons 6–9 exhibited no aberrant dystrophin expression patterns and displayed improved calcium handling compared with dystrophic controls, albeit not to the level of wild-type controls.47 Also, many deletions in the N-terminal actin-binding domain have been associated with BMD,48 and in particular, a mildly affected BMD patient with a deletion of exons 3–9 was not diagnosed until age 67.49 As one of the two deletion “hotspots” of the DMD gene overlap the N-terminal actin-binding domain, widespread use of gene editing for mutations in this part of the gene will need to consider effects on the stability of the edited dystrophin proteins.50
The overall observation of mosaic transduction and expression of delivered genes following IM administration may result from limited AAV vector diffusion from the injection site and is a major consideration in using systemic versus local delivery methods in clinical trials.51 Vector diffusivity throughout aged dystrophic muscle (whether following IM or systemic delivery) may be particularly concerning for treating older patients, as increased fat and connective tissue deposition could present significant obstacles for efficient AAV transduction of remaining muscle fibers. As we previously demonstrated in mice, achieving sufficient levels and uniform distribution of dystrophin expression is essential for stabilizing dystrophic skeletal muscle and has important implications for the longevity of DMD gene therapies.8 The results seen here align with those studies, suggesting that turnover of inadequately stabilized skeletal muscle contributes to the loss of CRISPR-corrected dystrophin expression over time. While Cas9-mediated immune responses were recently shown to cause rapid and significant loss of gene-edited dystrophin correction,52 our dogs were maintained on an extensive immunosuppression regimen31,53 for the duration of the study for the specific purpose of avoiding elicitation of host immune responses toward a likely highly immunogenic transgene product, thus making it unlikely that Cas9 immune responses played any significant role in the resulting outcomes.
Our results show the feasibility of increasing therapeutic dystrophin levels in aged dystrophic skeletal muscle using both CRISPR gene correction and micro-dystrophin replacement strategies. However, pronounced dystrophic pathology may present significant hurdles for both efficient vector transduction and subsequent dystrophin expression via either approach. While our data demonstrate the promise of in vivo gene editing for correcting dystrophin expression in advanced DMD stages, it is currently less efficient than micro-dystrophin gene replacement. Importantly, though, mutations that can be edited to enable production of large, highly functional dystrophins following gene editing are likely to be functionally superior to micro-dystrophins. Nonetheless, as with the example here involving truncation of the N-terminal actin-binding domain, some larger dystrophins may lack a subset of functional domains or may have improperly phased spectrin-like repeats that could impact their overall stability or functionality such that rationally designed, synthetic micro-dystrophins could display better expression, stability, or protection from contraction-induced injury.2,9,16 Further improvements to both approaches (or combinatorial treatments) may significantly improve therapeutic outcomes, especially in older patients. In this regard, we previously showed that co-administration of vectors that mediate both micro-dystrophin and gene editing in systemic treatments of young mdx4cv mice provided higher levels of dystrophin correction.8
Materials and methods
Cloning and vector production
Plasmids containing regulatory cassettes for expression of Cas9 and sgRNAs flanked by AAV serotype 2 inverted terminal repeats (ITRs) were generated using standard cloning techniques. A single AAV6 vector system was designed to express Cas9 from Staphylococcus aureus (SaCas9),54 under control of a synthetic muscle-specific creatine kinase (CK8e) gene regulatory cassette as well as two U6-driven sgRNA expression cassettes targeting introns 5 and 8 of the canine genome. We have previously designed and validated a similar single vector system for use in the mdx4cv mouse model of DMD using sgRNAs targeting introns 51 and 53 of the mouse genome.5 Both canine sgRNA spacer sequences demonstrated no predictable genomic off-target sites containing three or fewer nucleotide mismatches based on in silico modeling using Cas-OFFinder (CRISPR RGEN Tools, http://www.rgenome.net/cas-offinder/). Additional sgRNAs were not evaluated, as the selected pair showed clear editing in vitro. Some improvements in editing activity might be achieved by screening additional target sites, as we have done in previous and related studies. However, while we sometimes find that some sgRNAs exhibit very low to undetectable activities, the ones that do show clear activity rarely vary drastically from each other when applied in vivo. Plasmids for producing AAV6 vectors that express micro-dystrophin-5 (μDys5)16 under control of the CK8e muscle-specific expression cassette were also generated. Both plasmids were co-transfected with pDGM6 packaging plasmid into subcloned HEK293 cells (American Type Culture Collection) using calcium phosphate-mediated transfection to generate AAV6 vectors that were harvested, purified via heparin-affinity chromatography, and concentrated using sucrose gradient centrifugation.55,56 Resulting titers were determined by Southern analyses and qPCR using probes specific to CK8e.
Primary canine myogenic cell isolation and in vitro transfection
Primary myogenic cells were obtained via explant cultures of muscle samples harvested from a CXMD dog using previously described protocols.57 Resulting cells were seeded in culture vessels coated with 10% Matrigel (growth factor reduced; Invitrogen) containing myogenic proliferation media (DMEM containing 4.5 g/L L-glutamine, 10% fetal bovine serum, penicillin, and streptomycin) and allowed to grow to ∼75% confluency before transfection with pAAV-CK8eSaCas9-K9ΔX6-8 plasmid according to manufacturer's specifications (Lipofectamine 3000; Invitrogen). At 24 h post-transfection myogenic growth media were switched to differentiation media (DMEM containing 4,500 mg/L L-glutamine, 2% horse serum, penicillin, and streptomycin) and allowed to differentiate into myotubes for an additional 4 days before harvest of DNA and RNA (DNeasy and RNeasy kits; Qiagen).
Animals
Dogs were handled according to principles outlined in the NIH Guide for the Care and Use of Laboratory Animals and as approved by the University of Washington Institutional Animal Care and Use Committee (IACUC). The canine X-linked muscular dystrophy (CXMD) dog model of DMD carries the GRMD splice acceptor site mutation in intron 6, causing skipping of exon 7 and production of an out-of-frame dystrophin transcript.28 Dogs were housed in a temperature- and humidity-monitored environment, maintained between 18°C and 26°C. Dogs were fed twice daily, and fresh drinking water was provided ad libitum. Animals were fasted as required by specific procedures (e.g., involving sedation or anesthesia). Enrichment toys and treats were routinely supplied, and trained staff exercised the animals.
AAV serotype 6 vectors encoding CK8e-CRISPR/Cas9 or CK8e-μDys5 were injected into the left and right cranial tibialis (CT) muscles, respectively, of 3- and 8-year-old CXMD dogs under anesthesia (n = 2 dogs). Briefly, longitudinal skin incisions were made to expose the CT muscles before administering a total of 1 × 1013 vg per CT via 25 equally spaced 100 mL injections (∼ 10 mm apart). AAV vectors were diluted in phosphate-buffered saline (PBS) and injected using a ½-in 30G needle. Saline-soaked gauze was used to cover the CT for 5 min after the injection and before the incision was sutured closed. Carprofen and/or buprenorphine were administered for post-surgical pain management. Two days before AAV injection, dogs were started on an immunosuppressive regimen of cyclosporine (up to 7.5 mg/kg b.i.d.), and on the day of injection, mycophenolate mofetil (MMF) (up to 5 mg/kg b.i.d.) was added to the regimen. Additionally, diphenhydramine was administered 30 min before the injections. AAV-treated dogs remained on the immunosuppression regimen (cyclosporine and MMF) for the duration of the study. This regimen has been shown to block cellular immune responses against dystrophin and AAV vectors in the canine model of DMD and is based on clinical immunosuppression regimens used in human bone marrow transplantation.30,31,53,58, 59, 60
Tissue harvest and processing
Treated CT muscles were harvested at 6 weeks post-transduction and compared with wild-type CT samples or non-injected control muscle samples. Treated CT muscles were divided into proximal, medial, and distal sampling regions, from which three sequential cross-sectional slices were obtained. Each slice was further subdivided in quarters (3 × 12 total samples from each muscle), and serial portions of each quarter were used for immunohistochemical, RNA/DNA, and protein analyses. For immunohistochemical analyses, muscle samples were embedded in an optimal cutting temperature compound (O.C.T. Compound; VWR International) and fresh frozen in liquid-nitrogen-cooled isopentane for IF analysis. Corresponding serial quadrant samples were snap frozen in liquid nitrogen and ground to a powder under liquid nitrogen in a mortar kept on dry ice for subsequent RNA, DNA, and protein extraction.
Immunohistochemical analyses
Muscle cross sections (10 μm) were co-stained with antibodies raised against the C-terminal domain of dystrophin (331/332 rabbit polyclonal Ab, 1:500 dilution; a kind gift from Dr. Stanley Froehner at the University of Washington Department of Physiology and Biophysics). Expression of μDys5 was detected using antibodies raised against the hinge-1 (H1)/spectrin-like repeat 1 (R1) region (exons 10 and 11) of dystrophin (clone 1011b, mouse monoclonal IgG2a, developmental studies hybridoma bank), as μDys5 lacks the majority of the C-terminal domain and exhibits only limited immunoreactivity with certain C-terminal antibodies that share partial epitope homology. Slides were mounted using ProLong Gold with DAPI (Thermo Fisher Scientific) and imaged using an Olympus E1000 fluorescent microscope running SlideBook 6 acquisition software (3i, Denver, CO). Images were processed and assembled into figures using Photoshop CS5 (Adobe, San Jose, CA).
Nucleic acid and protein analyses
Nucleic acids were isolated from homogenized muscle tissue using TRIzol Reagent (Invitrogen), and RNA was extracted according to the manufacturer's recommendations. DNA was extracted from the TRIzol suspension using back-extraction with 4 M guanidine thiocyanate, 50 mM sodium citrate, and 1 M Tris base. Analysis of dystrophin transcripts by RT-PCR was performed on cDNA produced from muscle lysates using the Superscript IV VILO with ezDNase master mix kit (Thermo Fisher Scientific). Semi-quantitative PCR and RT-PCR amplicons across the targeted region were generated using Phusion high-fidelity polymerase (New England Biolabs) and separated by gel electrophoresis on 2% agarose gels. Unique deletion PCR and RT-PCR amplicons were excised following gel electrophoresis, purified, and submitted for Sanger sequencing (Eurofins Genomics). PCR amplicons were also generated across the individual cut sites in introns 5 and 8 and submitted for Sanger sequencing. The resulting reads were subjected to ICE CRISPR Analysis Tool (https://www.synthego.com/products/bioinformatics/crispr-analysis), where the Sanger sequencing data from treated samples were compared with an untreated control sample to establish editing frequency at each target site. Deletion of the entire targeted region at the genome and transcript levels was quantified using a QuantStudio 3D digital PCR system (Thermo Fisher Scientific) using the manufacturer's proprietary reagents in combination with primer/probe sets detecting either native unmodified dystrophin alleles/transcripts or alleles/transcripts lacking the sequence spanning the two target sites. Data analysis was performed on the Thermo Fisher Scientific Cloud Software Connect platform using dPCR AnalysisSuite.
Muscle proteins were extracted in radioimmunoprecipitation assay (RIPA) buffer supplemented with 5 mM EDTA and 4% protease inhibitor cocktail (MilliporeSigma, catalog no. P8340), for 1 h on ice with gentle agitation every 15 min. Total protein concentration was determined using Pierce BCA assay kit (Thermo Fisher Scientific). Muscle lysates from wild-type (WT) (3 and 0.3 μg), untreated (30 μg), and treated (30 μg) tissue samples were denatured at 100°C for 10 min, quenched on ice, and separated via gel electrophoresis after loading onto Bolt 4–12% Bis-Tris polyacrylamide gels (Invitrogen). Protein transfer to 0.45 μm polyvinylidene fluoride (PVDF) membranes was performed overnight at constant 46 V at 4°C in Towbin's buffer containing 20% methanol. Blots were blocked for 1 h at room temperature (RT) in 5% non-fat dry milk (NFDM) before overnight incubation with antibodies raised against the C-terminal domain of dystrophin (rabbit polyclonal, 1:15,000; Froehner Lab), 1011b anti-H1/R1 of dystrophin (DSHB; University of Iowa, Iowa city, IA), anti-HA (rat monoclonal-HRP conjugated, 1:2,000; Roche) for detection of hemagglutin antigen (HA)-tagged saCas9 and GAPDH (rabbit polyclonal, 1:100,000; MilliporeSigma). Horseradish-peroxidase-conjugated secondary antibody staining (1:50,000) was performed for 1 h at RT before signal development using Clarity Western ECL Substrate (Bio-Rad) and visualization using a Chemidoc MP imaging system (Bio-Rad).
Statistical analyses
Except where otherwise noted, data values are represented as mean ± SEM and were analyzed in Excel (Microsoft) and Prism 6 (GraphPad). Measurements were analyzed for statistical significance using one-way analysis of variance (ANOVA) multiple comparison tests with Turkey's post hoc tests unless otherwise stated. Statistical significance was set to p < 0.05.
Acknowledgments
We thank the Viral Vector Core of the Seattle Wellstone Muscular Dystrophy Specialized Research Center (P50AR065139) for generating AAV vectors and James Allen and Christine Halbert for their advice and assistance. We thank Jessica Snyder of UW Veterinary Services for assistance with our canine colony and David Mack, Maura Parker, and Stephen Tapscott for their assistance in the isolation and culturing of primary CXMD myoblasts. This research was supported by NIH grants R01AR44533 and R01AR40864 (to J.S.C. and S.D.H.), by grant RRG 715234 from the Muscular Dystrophy Association (to J.S.C.), and by NIH P30DK017047.
Author contributions
Conceptualization, N.E.B. and J.S.C.; methodology, N.E.B., J.M.C., J.K., C.L.H., S.D.H., and J.S.C; investigation, N.E.B., J.M.C., J.K., and S.D.H.; formal analysis, N.E.B., J.M.C., and J.S.C.; writing, N.E.B., J.M.C., J.K., S.D.H., and J.S.C.; supervision, N.E.B., S.D.H., and J.S.C.
Declaration of interests
N.E.B., S.D.H., and J.S.C. are inventors on patents covering muscle-specific gene editing; J.S.C. and S.D.H. are inventors on patents covering muscle-specific gene regulatory cassettes, micro-dystrophins, and systemic delivery of AAV. J.S.C. holds equity in, and is a member of the scientific advisory board of, Solid Biosciences.
Published: June 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.02.003.
Supplemental information
References
- 1.Emery A.E.H., Muntoni F. 3rd edn. Oxford University Press; 2003. Duchenne Muscular Dystrophy. [Google Scholar]
- 2.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., Chamberlain J.S. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 3.Bengtsson N.E., Seto J.T., Hall J.K., Chamberlain J.S., Odom G.L. Progress and prospects of gene therapy clinical trials for the muscular dystrophies. Hum. Mol. Genet. 2016;25:R9–R17. doi: 10.1093/hmg/ddv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young C.S., Pyle A.D., Spencer M.J. CRISPR for neuromuscular disorders: gene editing and beyond. Physiology. 2019;34:341–353. doi: 10.1152/physiol.00012.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Corrigendum: muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:16007. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D., et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M., et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bengtsson N.E., Tasfaout H., Hauschka S.D., Chamberlain J.S. Dystrophin gene-editing stability is dependent on dystrophin levels in skeletal but not cardiac muscles. Mol. Ther. 2021;29:1070–1085. doi: 10.1016/j.ymthe.2020.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duchêne B.L., Cherif K., Iyombe-Engembe J.P., Guyon A., Rousseau J., Ouellet D.L., Barbeau X., Lague P., Tremblay J.P. CRISPR-induced deletion with SaCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol. Ther. 2018;26:2604–2616. doi: 10.1016/j.ymthe.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hakim C.H., Wasala N.B., Nelson C.E., Wasala L.P., Yue Y., Louderman J.A., Lessa T.B., Dai A., Zhang K., Jenkins G.J., et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3:e124297. doi: 10.1172/jci.insight.124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K., et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X., et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A., et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyombe-Engembe J.P., Ouellet D.L., Barbeau X., Rousseau J., Chapdelaine P., Lagüe P., Tremblay J.P. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol. Ther. Nucleic Acids. 2016;5:e283. doi: 10.1038/mtna.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramos J.N., Hollinger K., Bengtsson N.E., Allen J.M., Hauschka S.D., Chamberlain J.S. Development of novel micro-dystrophins with enhanced functionality. Mol. Ther. 2019;27:623–635. doi: 10.1016/j.ymthe.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregorevic P., Allen J.M., Minami E., Blankinship M.J., Haraguchi M., Meuse L., Finn E., Adams M.E., Froehner S.C., Murry C.E., Chamberlain J.S. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregorevic P., Blankinship M.J., Allen J.M., Chamberlain J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 2008;16:657–664. doi: 10.1038/mt.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koenig M., Beggs A.H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Müller C.R., Lindlöf M., Kaariainen H., et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 20.Maino E., Wojtal D., Evagelou S.L., Farheen A., Wong T.W.Y., Lindsay K., Scott O., Rizvi S.Z., Hyatt E., Rok M., et al. Targeted genome editing in vivo corrects a Dmd duplication restoring wild-type dystrophin expression. EMBO Mol. Med. 2021;13:e13228. doi: 10.15252/emmm.202013228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lattanzi A., Duguez S., Moiani A., Izmiryan A., Barbon E., Martin S., Mamchaoui K., Mouly V., Bernardi F., Mavilio F., Bovolenta M. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol. Ther. Nucleic Acids. 2017;7:11–19. doi: 10.1016/j.omtn.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein J.M., Tabebordbar M., Zhu K., Wang L.D., Messemer K.A., Peacker B., Ashrafi Kakhki S., Gonzalez-Celeiro M., Shwartz Y., Cheng J.K.W., et al. In situ modification of tissue stem and progenitor cell genomes. Cell Rep. 2019;27:1254–1264.e7. doi: 10.1016/j.celrep.2019.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon J.B., Ettyreddy R.B., Vankara A., Bohning J.D., Delvin G., Hauschka S.D., Asokan A., Gersbach C.A. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2020;19:320–329. doi: 10.1016/j.omtm.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnett A.L., Konieczny P., Ramos J.N., Hall J., Odom G., Yablonka-Reuveni Z., Chamberlain J.R., Chamberlain J.S. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev. 2014;1:14038. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinmann J., Weis S., Sippel J., Tulalamba W., Remes A., El Andari J., Herrmann A.K., Pham Q.H., Borowski C., Hille S., et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020;11:5432. doi: 10.1038/s41467-020-19230-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabebordbar M., Lagerborg K.A., Stanton A., King E.M., Ye S., Tellez L., Krunnfusz A., Tavakoli S., Widrick J.J., Messemer K.A., et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell. 2021;184:4919–e22 e4922. doi: 10.1016/j.cell.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimatsu Y., Katagiri K., Furuta T., Nakura M., Tanioka Y., Yuasa K., Tomohiro M., Kornegay J.N., Nonaka I., Takeda S. Canine X-linked muscular dystrophy in Japan (CXMDJ) Exp. Anim. 2003;52:93–97. doi: 10.1538/expanim.52.93. [DOI] [PubMed] [Google Scholar]
- 29.Valentine B.A., Cooper B.J., de Lahunta A., O'Quinn R., Blue J.T. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J. Neurol. Sci. 1988;88:69–81. doi: 10.1016/0022-510X(88)90206-7. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z., Kuhr C.S., Allen J.M., Blankinship M., Gregorevic P., Chamberlain J.S., Tapscott S.J., Storb R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 2007;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z., Storb R., Halbert C.L., Banks G.B., Butts T.M., Finn E.E., Allen J.M., Miller A.D., Chamberlain J.S., Tapscott S.J. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol. Ther. 2012;20:1501–1507. doi: 10.1038/mt.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin J.H., Pan X., Hakim C.H., Yang H.T., Yue Y., Zhang K., Terjung R.L., Duan D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol. Ther. 2013;21:750–757. doi: 10.1038/mt.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Guiner C., Servais L., Montus M., Larcher T., Fraysse B., Moullec S., Allais M., François V., Dutilleul M., Malerba A., et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017;8:16105. doi: 10.1038/ncomms16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walmsley G.L., Arechavala-Gomeza V., Fernandez-Fuente M., Burke M.M., Nagel N., Holder A., Stanley R., Chandler K., Marks S.L., Muntoni F., et al. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS One. 2010;5:e8647. doi: 10.1371/journal.pone.0008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chemello F., Bassel-Duby R., Olson E.N. Correction of muscular dystrophies by CRISPR gene editing. J. Clin. Invest. 2020;130:2766–2776. doi: 10.1172/JCI136873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koenig M., Kunkel L.M. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- 37.Abmayr S., Chamberlain J. In: Winder S.J., editor. Landes Biosciences; 2006. The structure and function of dystrophin; pp. 14–34. (The Molecular Mechanisms of Muscular Dystrophies). Ch. 2. [Google Scholar]
- 38.Sharp N.J., Kornegay J.N., Van Camp S.D., Herbstreith M.H., Secore S.L., Kettle S., Hung W.Y., Constantinou C.D., Dykstra M.J., Roses A.D. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13:115–121. doi: 10.1016/0888-7543(92)90210-j. [DOI] [PubMed] [Google Scholar]
- 39.McClorey G., Moulton H.M., Iversen P.L., Fletcher S., Wilton S.D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006;13:1373–1381. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- 40.Yokota T., Lu Q.L., Partridge T., Kobayashi M., Nakamura A., Takeda S., Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schatzberg S.J., Anderson L.V., Wilton S.D., Kornegay J.N., Mann C.J., Solomon G.G., Sharp N.J. Alternative dystrophin gene transcripts in golden retriever muscular dystrophy. Muscle Nerve. 1998;21:991–998. doi: 10.1002/(sici)1097-4598(199808)21:8<991::aid-mus2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 42.Echigoya Y., Lee J., Rodrigues M., Nagata T., Tanihata J., Nozohourmehrabad A., Panesar D., Miskew B., Aoki Y., Yokota T. Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice. PLoS One. 2013;8:e69194. doi: 10.1371/journal.pone.0069194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crawford G.E., Lu Q.L., Partridge T.A., Chamberlain J.S. Suppression of revertant fibers in mdx mice by expression of a functional dystrophin. Hum. Mol. Genet. 2001;10:2745–2750. doi: 10.1093/hmg/10.24.2745. [DOI] [PubMed] [Google Scholar]
- 44.Kinoshita I., Vilquin J.T., Asselin I., Chamberlain J., Tremblay J.P. Transplantation of myoblasts from a transgenic mouse overexpressing dystrophin prduced only a relatively small increase of dystrophin-positive membrane. Muscle Nerve. 1998;21:91–103. doi: 10.1002/(sici)1097-4598(199801)21:1<91::aid-mus12>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 45.Beggs A.H., Hoffman E.P., Snyder J.R., Arahata K., Specht L., Shapiro F., Angelini C., Sugita H., Kunkel L.M. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am. J. Hum. Genet. 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- 46.Corrado K., Rafael J.A., Mills P.L., Cole N.M., Faulkner J.A., Wang K., Chamberlain J.S. Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a "mild Becker" phenotype. J. Cell Biol. 1996;134:873–884. doi: 10.1083/jcb.134.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kyrychenko V., Kyrychenko S., Tiburcy M., Shelton J.M., Long C., Schneider J.W., Zimmermann W.H., Bassel-Duby R., Olson E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight. 2017;2:e95918. doi: 10.1172/jci.insight.95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wein N., Vulin A., Falzarano M.S., Szigyarto C.A., Maiti B., Findlay A., Heller K.N., Uhlén M., Bakthavachalu B., Messina S., et al. Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat. Med. 2014;20:992–1000. doi: 10.1038/nm.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heald A., Anderson L.V., Bushby K.M., Shaw P.J. Becker muscular dystrophy with onset after 60 years. Neurology. 1994;44:2388–2390. doi: 10.1212/wnl.44.12.2388. [DOI] [PubMed] [Google Scholar]
- 50.Chamberlain J.S., Chamberlain J.R., Fenwick R.G., Ward P.A., Caskey C.T., Dimnik L.S., Bech-Hansen N.T., Hoar D.I., Richards S., Covone A.E., et al. Diagnosis of Duchenne and Becker muscular dystrophies by polymerase chain reaction. A multicenter study. JAMA. 1992;267:2609–2615. doi: 10.1001/jama.1992.03480190051030. [DOI] [PubMed] [Google Scholar]
- 51.Gregorevic P., Blankinship M.J., Allen J.M., Crawford R.W., Meuse L., Miller D.G., Russell D.W., Chamberlain J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hakim C.H., Kumar S.R.P., Pérez-López D.O., Wasala N.B., Zhang D., Yue Y., Teixeira J., Pan X., Zhang K., Million E.D., et al. Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat. Commun. 2021;12:6769. doi: 10.1038/s41467-021-26830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Z., Storb R., Lee D., Kushmerick M.J., Chu B., Berger C., Arnett A., Allen J., Chamberlain J.S., Riddell S.R., Tapscott S.J. Immune responses to AAV in canine muscle monitored by cellular assays and noninvasive imaging. Mol. Ther. 2010;18:617–624. doi: 10.1038/mt.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blankinship M.J., Gregorevic P., Allen J.M., Harper S.Q., Harper H., Halbert C.L., Miller A.D., Miller D.A., Chamberlain J.S. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 56.Halbert C.L., Allen J.M., Chamberlain J.S. AAV6 vector production and purification for muscle gene therapy. Methods Mol. Biol. 2018;1687:257–266. doi: 10.1007/978-1-4939-7374-3_18. [DOI] [PubMed] [Google Scholar]
- 57.Parker M.H., Kuhr C., Tapscott S.J., Storb R. Hematopoietic cell transplantation provides an immune-tolerant platform for myoblast transplantation in dystrophic dogs. Mol. Ther. 2008;16:1340–1346. doi: 10.1038/mt.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z., Allen J.M., Riddell S.R., Gregorevic P., Storb R., Tapscott S.J., Chamberlain J.S., Kuhr C.S. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum. Gene Ther. 2007;18:18–26. doi: 10.1089/hum.2006.093. [DOI] [PubMed] [Google Scholar]
- 59.Arnett A.L., Garikipati D., Wang Z., Tapscott S., Chamberlain J.S. Immune responses to rAAV6: the influence of canine parvovirus vaccination and neonatal administration of viral vector. Front. Microbiol. 2011;2:220. doi: 10.3389/fmicb.2011.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z., Tapscott S.J., Chamberlain J.S., Storb R. Immunity and AAV-mediated gene therapy for muscular dystrophies in large animal models and human trials. Front. Microbiol. 2011;2:201. doi: 10.3389/fmicb.2011.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.