


Abstract
Axonally synthesized proteins support nerve regeneration through retrograde signaling and local growth mechanisms. RNA binding proteins (RBP) are needed for this and other aspects of post-transcriptional regulation of neuronal mRNAs, but only a limited number of axonal RBPs are known. We used targeted proteomics to profile RBPs in peripheral nerve axons. We detected 76 proteins with reported RNA binding activity in axoplasm, and levels of several change with axon injury and regeneration. RBPs with altered levels include KHSRP that decreases neurite outgrowth in developing CNS neurons. Axonal KHSRP levels rapidly increase after injury remaining elevated up to 28 days post axotomy. Khsrp mRNA localizes into axons and the rapid increase in axonal KHSRP is through local translation of Khsrp mRNA in axons. KHSRP can bind to mRNAs with 3’UTR AU-rich elements and targets those transcripts to the cytoplasmic exosome for degradation. KHSRP knockout mice show increased axonal levels of KHSRP target mRNAs, Gap43, Snap25, and Fubp1, following sciatic nerve injury and these mice show accelerated nerve regeneration in vivo. Together, our data indicate that axonal translation of the RNA binding protein Khsrp mRNA following nerve injury serves to promote decay of other axonal mRNAs and slow axon regeneration.
Graphical Abstract
Graphical Abstract.
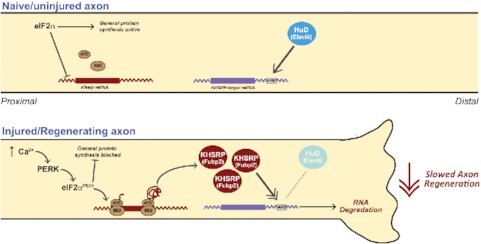
Intra-axonal translation of Khsrp mRNA slows axon regeneration.
INTRODUCTION
Subcellular localization of mRNAs provides polarized cells with means to rapidly respond to environmental stimuli within different domains of those cells. Neurons are highly polarized cells with cytoplasmic processes, axons and dendrites, that extend great distances from the cell body or soma. Proteins synthesized in developing axons drive responses to some axon guidance cues and populations of proteins synthesized in axons changes during synaptogenesis pointing to dynamic post-transcriptional regulation of mRNAs in axons (1–3). In rodents, peripheral and some central nervous system (PNS and CNS, respectively) neurons can extend centimeters from their soma and intra-axonal protein synthesis can bring autonomy from the soma but it also must be tightly regulated (4). Several lines of evidence indicate that translation of mRNAs in mature axons of the peripheral nervous system (PNS) contributes to axon regeneration after injury (5). Since one mRNA can be translated many times over to generate multiple copies of a protein, the survival of an mRNA within an axon can substantially affect the spatial and temporal regulation of that axon's proteome (4). Much has been learned about how mRNAs are transported into and translated within axons over recent years (6–10), and it is clear that the axonal transcriptome is quite extensive in terms of numbers of different mRNAs and dynamic in terms of changes in mRNA populations with different physiological states (11). RNA binding proteins (RBP) and mRNAs assemble into ribonucleoprotein particles (RNP) for transport into axons, and those or other RBPs can also subsequently regulate the translation of interacting mRNAs in the axons or provide a storage depot to sequester the mRNAs until needed (4). However, we know of relatively few axonal RBPs that contribute to these mechanisms. There is also some intrinsic capacity for locally depleting specific mRNAs from axons in addition to regulating their transport, storage, and translation, since both nonsense-mediated decay (NMD) and microRNA (miRNA)-stimulated RNA degradation have been shown to occur in distal axons (12–16). But the extent to which localized mRNAs are subjected to regulation of their decay is not known.
RBP interactions have also been linked to mRNA stability, including axonally localized mRNAs. The neuronal protein HuD (also called ELAVL4) has been known to stabilize mRNAs by binding to AU-rich elements (AREs) in 3’UTRs of target mRNAs (17). HuD protein localizes into distal neurites and has been implicated in transport and translation of some mRNAs (18–20). For Gap43 mRNA, HuD interaction is needed for axonal localization of the mRNA, but it also increases survival of the transcript (20). We previously showed that the KH splicing regulatory protein (KHSRP; also known as KSRP, MARTA1, ZBP2 and FUBP2) decreases neurite growth in cultures of embryonic cortical neurons and competes with HuD for binding to target mRNAs (21). In contrast to HuD interactions, KHSRP binding can promote decay of ARE-containing mRNAs (22,23). Here, we show that KHSRP is one of several RBPs whose levels increase in PNS axons after injury and during regeneration. This increase in axonal KHSRP occurs rapidly after PNS nerve injury through translation of its mRNA in axons. A conventional knockout of the murine KHSRP gene increases axonal levels of the KHSRP target mRNAs, Gap43, Snap25 and Fubp1. KHSRP contains 4 hnRNP K homology (KH) domains of about 70 amino acid residues each and these can bind to single strand DNA or RNA with varying degrees of specificity (22). Interestingly, the increase in Gap43 mRNA requires an intact fourth KH domain in KHSRP that has been linked to promoting mRNA decay by interacting with components of the cytoplasmic exosome (22). In contrast, KHSRP’s modulation of Fubp1 mRNA levels does not require this domain of KHSRP. Selectively deleting KHSRP alleles from only neurons points to a neuron intrinsic mechanism driving the accelerated axon regeneration. Together, our data indicate that neuronal KHSRP slows axon growth and emphasize that localized synthesis of KHSRP in axons provides a means to modulate axonal mRNA levels, which slows nerve regeneration.
MATERIALS AND METHODS
Animal use and survival surgery
The Institutional Animal Care and Use Committee of the University of South Carolina approved all animal procedures. Adult male Sprague Dawley rats (175–250 g) or both male and female Khsrp knockout (Khsrp–/–) (24), wild type (Khsrp+/+) or Khsrpfl/fl mice on C57/Bl6 background were used for all experiments. Wild type animals were typically littermates, and heterozygous animals (Khsrp+/–) were used in several experiments as indicated. Mice for conditional knockout of Khsrp were generated by Biocytogen (Wakefield, MA) using CRISPR/EGE™-based gene editing to insert loxP sites between exons 1 and 2 and exons 6 and 7 that would result in a frameshift upon Cre-driven recombination but were not predicted to affect splicing of the Khsrp RNA transcript prior to any recombination. Insertion of loxP was confirmed by sequencing, and expression of full length Khsrp was confirmed by RT-PCR in founder mice on C57Bl/6 background. Founders were bred to homozygosity after crossing with wild type C57Bl/6 mice.
Isoflurane inhalation was used for anesthesia in all survival surgery experiments (see below). Animals were euthanized by CO2 asphyxiation as indicated in results. For peripheral nerve injury, anesthetized male rats or mice were subjected to sciatic nerve crush at mid-thigh level as previously described (25). Briefly, the nerve at ∼2.5 cm from its origin but proximal to its trifurcation was crushed with # 2 fine jeweler's forceps, twice for 15 s. each; axotomy was monitored by the initial contraction of the hind limb upon applying pressure to the nerve, and then lack of hind paw extension during and upon recovery from anesthesia. For ‘double crush’ injury experiments, a unilateral peripheral sciatic nerve crush was performed at mid-thigh level on day 0, as a ‘conditioning lesion’, and a second crush injury was performed at 0.5 cm proximal to the first crush site following the same procedure. For consistency between animals, a single experimenter performed the crush injuries within each series of animals.
For the Khsrpfl/fl mice, in vivo deletion of the KHSRP was accomplished by injecting 3 μl consisting of 1.32 × 109 AAV2-CMV-Cre-GFP viral particles (Univ. North Carolina Vector Core, Chapel Hill, NC) diluted to 600 mM NaCl into the proximal sciatic nerve just distal to the sciatic notch and at least 1 cm from the crush sites (26).
Sciatic nerve ligations were performed in male rats as described previously as the larger size of these animals provided greater precision in ligation and subsequent crush injuries (27). Briefly, rat sciatic nerve was ligated approximately 1 cm proximal to planned mid-thigh nerve crush site. Immediately after applying 4·0 suture around the nerve and tying the suture to constrict the nerve, the sciatic nerve was crushed distal to the ligation site as above and then animals were euthanized 3–16 h later. Efficacy of the ligation was assessed by immunofluorescence for anterogradely transported amyloid precursor protein (APP) and retrogradely transported signal transducer and activator of transcription 3α (Stat3α).
Cell Culture
Dissociated cultures of adult dorsal root ganglia (DRG) were prepared as described (28). For experiments with naïve DRG neurons, all lumbar, thoracic, and lower cervical DRGs were collected. To study effects of in vivo injury conditioning, L4-6 DRGs were used from ipsi-lateral (injury-conditioned) or contra-lateral (naïve) to the crush injury. DRGs were harvested in Hybernate-A medium (BrainBits, Springfield, IL) and then dissociated with collagenase as described. After centrifugation and three washes in DMEM/F12 (Life Technologies, Grand Island, NY), dissociated ganglia were cultured in complete medium containing DMEM/F12, 1 × N1 supplement (Sigma, St. Louis, MO), 10% fetal bovine serum (Hyclone, Logan, UT), and 10 μM cytosine arabinoside (Sigma) on poly-L-lysine (Sigma) plus laminin (Millipore, Burlington, MA) coated substrates. For imaging, dissociated DRGs were cultured on coated glass coverslips. For analyses of axonal RNA levels or in vitro regeneration assay (see below), dissociated ganglia were cultured on polyethylene-tetrathalate (PET) membrane inserts (1 μm pores; Falcon-Corning, Tewksbury, MA) (29). Axons and CB were isolated from DRGs cultured on PET membranes as described (30).
For transfection, dissociated ganglia were pelleted at 100 × g for 5 min and resuspended in 100 μl ‘Nucleofector solution’ (Rat Neuron Nucleofector kit; Lonza, Alpharetta, GA). 4–6 μg of plasmid was electroporated using the AMAXA Nucleofector device (Neurons Rat DRG, G-013 program; Lonza) before plating and maintained for 48 h.
For in vitro Cre-driven recombination, dissociated DRGs from Khsrpfl/fl mice were incubated with 4.4 × 1012 particles/ml of AAV2-CMV-Cre-GFP or AAV2-CMV-GFP (UNC Vector Core) for 24 h after initial plating. Cultures were analyzed 3 d later. Loss of KHSRP was confirmed at both the mRNA and protein levels.
Plasmid constructs
pAc-GFP-KHSRP and pAc-GFP-KHSRPΔKH4 (KHSRP with deleted KH4 domain) constructs have been published (21). All fluorescent reporter constructs for analyses of RNA translation were based on eGFP with myristoylation element (GFPMYR; originally provided by Dr Erin Schuman, Max-Plank Institute, Frankfurt) (31). cDNAs for the 5’UTR and 3’UTRs of Khsrp mRNA were custom synthesized by Integrated DNA Technologies (Coralville, IA) and GenScript Biotech (Piscataway, NJ), respectively. The 5’UTR was engineered with 5’ Nhe1 and 3’ BamH1 restriction sites and cloned into pGFPMYR5’camk2a/3’actg (30), replacing the 5’UTR of CamK2a [GFPMYR5’khsrp/3’actg]. The 3’UTR sequence was engineered with 5’ Not1 and 3’ Xho1 restriction sites and used to replace the Actg 3’UTR in pGFPMYR5’khsrp/3’actg plasmid [pGFPMYR5’/3’khsrp].
Mass spectrometry for axonal RBPs
Axoplasm from 2 cm segments of sciatic nerve immediately proximal to crush site was extruded into nuclear transport buffer (20 mM HEPES [pH 7.3], 110 mM potassium acetate, 5 mM magnesium acetate) supplemented with protease/phosphatase inhibitor cocktail (Roche) and RNasin Plus (Promega, Madison, WI). Contralateral (uninjured) sciatic nerve of comparable level and length was used for control. Three animals were used for each time point and both naïve and injured sciatic nerve axoplasm. Preparations were cleared by centrifugation at 20 000 × g, 4°C for 30 min, supernatants were diluted in 0.5 ml of TRIzol LS reagent (Invitrogen) and protein was extracted per the manufacturer's protocol. Protein pellets were digested with trypsin as previously described (32).
Parallel reaction monitoring (PRM) was performed on Q Exactive Plus Mass Spectrometer (Thermo-Fisher) online with nanoAcquity UPLC System (Waters, Milford, MA). Digested peptides samples (0.5 μg) were injected onto 200 cm monolithic silica-C18 column (GL Sciences, Tokyo, Japan) and separated using a 6 h reversed phase chromatography gradient as previously described (32). The mass spectrometer was operated in PRM mode with the following parameters: positive polarity, R = 17 500 at 200 m/z, AGC target 1e6, maximum IT 190 ms, MSX count 1, isolation window 3.0 m/z, NCE 35%. PRM data were analyzed in Skyline v. 3.5 (33). Skyline PRM document has been uploaded to PanoramaWeb Public (34) and can be accessed at https://panoramaweb.org/axon-rbps.url.
RNA isolation and PCR analyses
RNA was isolated from dissociated DRG neurons or cell body/axon compartments collected from insert cultures using RNeasy Microisolation kit (Qiagen, Hilden, Germany). Sciatic nerve was cut in small pieces and digested with collagenase at 37°C for 30 min with intermittent trituration. RNA was isolated from the collagenase-treated nerve using Trizol LS reagent (Invitrogen, Carlsbad CA) according to manufacturer's instructions. RNA concentration was measured by fluorimetry with Ribogreen (Life Technologies) and 10–50 ng of RNA was reverse transcribed with Sensifast cDNA synthesis kit (Bioline, London, UK). DRG axonal purity was assessed by RT-PCR, performed with primers designed to detect cell body-restricted mRNAs (cJun and Map2) and glial cell-specific mRNAs (Gfap). Droplet digital (dd) PCR was performed according to manufacturer's procedure with Evagreen detection (Biorad, Hercules, CA). Mitochondrial 12S ribosomal RNA (Mtrnr1) and Hmgb1 mRNA levels were used for normalizing RNA yields across different isolates. Following primers were used RT-PCR and RTddPCR (all from IDT, listed as 5’ to 3’): Mtrnr1, sense – GGCTACACCTTGACCTAACG and antisense – CCTTACCCCTTCTCGCTAATTC; Actb, sense – CTGTCCCTGTATGCCTCTG and antisense – ATGTCACGCACGATTTCC; cJun, sense – GCAAAGATGGAAACGACCTTCTAC and antisense – AAGCGTGTTCTGGCTATGC Gfap, sense – AGTTACCAGGAGGCACTTG and antisense – GGTGATGCGGTTTTCTTCG; Hmgb1, sense – CATGGGCAAAGGAGATCC and antisense –CTCTGAGCACTTCTTGGAG; Gap43, sense – CAGGAAAGATCCCAAGTCCA and antisense – GAGGAAAGTGGACTCCCACA; Map2, sense – CTGGACATCAGCCTCACTCA and antisense – AATAGGTGCCCTGTGACCTG; Snap25, sense – CAAATTTAACCACTTCCCAGCA and antisense –CAGAATCGCCAGATCGACAG; Fubp1, sense – GCACCAGCTACAACCCAA and antisense – GCCTTTGTATAATCAACCTGTCC ; and Khsrp, sense – CCAGTTGAGAACCAATCGAGTC and antisense – CACCGTGAATAACAACACTCCT.
Immunofluorescent staining
Immunofluorescence was performed as previously described (35) with all steps at room temperatures unless specified otherwise. Coverslips were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 15 min at room temperature and washed 3 times in PBS. PBS washed neurons were permeabilized with 0.3% Triton X-100 in PBS for 15 min and then blocked in 5% BSA for 1 h. Neurons were incubated with primary antibodies overnight in humidified chambers at 4°C. Primary antibodies consisted of chicken anti-NFH, -NFM plus -NFL cocktail (1: 500; Aves Lab, Tigard, OR, NFH # AB_2313552, NFM # AB_2313554, and NFL # AB_2313553), RT97 mouse anti-NF (1:500; Devel. Studies Hybridoma Bank, Iowa City, IA), and rabbit anti-KHSRP (1:200; Novus Biologicals, Centennial, CO, #NBP1-18910). After washes in PBST, coverslips were incubated with combination of FITC-conjugated donkey anti-mouse, Cy5 conjugated donkey anti-chicken (both at 1:500; Jackson ImmunoRes., West Grove, PA) as secondary antibodies for 1 h. After 1 h, coverslips were washed 3 times in PBS, rinsed with distilled H2O, and mounted with Prolong Gold Antifade with DAPI (Thermo-Fisher, Waltham, MA).
For regeneration studies on mouse sciatic nerve and quantifying axonal content of KHSRP in vivo, sciatic nerve segments were fixed for 4 h in 4% PFA and then cryoprotected overnight in 30% sucrose in PBS at 4°C. 10 μm cryostat sections for rat sciatic nerve and 20 μm cryostat sections for mouse sciatic nerve were processed for immunostaining as previously described (35). Primary antibodies consisted of RT97 mouse anti-NF (1:500), rabbit anti-KHSRP (Novus Biologicals, #NBP1-18910), and rabbit anti-Stathmin-2/SCG10 (1:500; Novus Biologicals, #NBP1-49461). Stathmin-2/SCG10 immunofluorescence was used to detect regenerating mouse sciatic nerve axons (36). Cy3-conjugated donkey anti-rabbit and FITC-conjugated donkey anti-mouse in combination were used as secondary antibodies for rat sciatic nerve (both at 1:500, Jackson ImmunoRes.). Cy3-conjugated donkey anti-rabbit antibodies were used on mice sciatic nerve (1:500, Jackson ImmunoRes).
Immunofluorescence for neuromuscular junctions (NMJs) was performed as previously published with minor modifications (37). Briefly, all steps were carried out at room temperature. Gastrocnemius muscle was cleared of any connective tissue, washed in PBS, fixed in 4% PFA, washed in PBS 3 times for 5 min each. Muscle was then dissected into smaller pieces and incubated with 1 μg/ml of Alexa Fluor 488 conjugated α-Bungarotoxin for 4 h with rocking (Thermo-Fisher, #B13422). Tissues were washed with PBS 3 times for 5 min each, treated with methanol at –20°C for 5 min, and rinsed in PBS 3 times for 5 min each with rocking. Tissues were blocked for 1 h with 2% BSA, 0.4% Triton X-100 in PBS. Tissues were then incubated overnight with the following cocktail of primary antibodies to presynaptic components diluted in blocking solution with rocking: rabbit anti-NF 200 (1:200; Millipore-Sigma, #N4142), mouse anti-synaptophysin (1:300; Millipore-Sigma, #MAB5258), and rabbit anti-synapsin-I (1:200; Millipore-Sigma, #AB1543P). The following day, tissues were rinsed 3 times 5 min each in PBS while rocking. Samples were then incubated in Cy3-conjugated donkey anti-rabbit and Cy3-conjugated donkey anti-mouse antibodies (1:500; Jackson ImmunoRes) for 4 h. After rinsing in PBS, muscle fibers were spread into monolayers under a stereomicroscope and affixed to slides using Prolong Gold Antifade; coverslips were sealed with clear nail polish.
All samples were mounted with Prolong Gold Antifade and imaging was performed at room temperature. Samples were analyzed by either epifluorescent or confocal microscopy. Leica DMI6000 epifluorescent microscope with ORCA Flash ER CCD camera (Hamamatsu) was used for epifluorescent imaging. Confocal imaging for immunofluorescence was performed on a Leica SP8X microscope (DMI6000 M platform; Buffalo Grove, IL) fitted with a galvanometer Z stage and HyD detectors; HC PL Apo 63x/1.4 NA objective (oil immersion) was used with acquisition parameters matched for individual experiments using LAS-X software. Z-stack images were post-processed by Leica Lightning Deconvolution integrated into LASX software. Deconvolved image stacks were projected into single plane images. For visualizing NMJs, sequential scanning was used to separate the green and red channels and Z stack at 200 nm intervals.
Fluorescence in-situ hybridization
Single molecule Fluorescence in situ hybridization (smFISH) plus IF was used to detect Khsrp mRNA in DRG and sciatic nerve. We used custom designed Cy3-labelled Stellaris probes (LGC Biosearch Tech, Middlesex, UK) for mouse Khsrp mRNA (Genbank ID # NM_010613.3) with Cy3-labelled scramble probe for control. RT97 mouse anti-NF (1:200) and chicken anti-GFAP (1:200; Millipore-Sigma, #AB5541) were used as primary antibodies; FITC- and Cy5-conjugated donkey anti-mouse and anti-chicken (1:200 each) were used as secondary antibodies. Samples processed without addition of primary antibody served as control for antibody specificity. Samples were mounted with Prolong Gold Anti-fade with DAPI.
smFISH/IF on DRG cultures was performed as described previously (38). Briefly, coverslips were rinsed in PBS and then fixed in buffered 2% PFA for 15 min, with all steps carried out at room temperature unless specified otherwise. Coverslips were rinsed 2 times in PBS, then permeabilized in 0.3% Triton X-100 in PBS for 5 min. Samples were equilibrated for 5 min in hybridization buffer (50% dextran sulphate, 10 μg/ml E. coli tRNA, 10 mM ribonucleoside vanadyl complex, 80 μg BSA, and 10% formamide in 2× SSC), and then incubated with 12.5 μM probe plus mouse anti-NF (1:200) for 12 h at 37°C. Coverslips were then washed in PBS + 0.3% Triton X-100 3 times, followed by incubation with FITC-conjugated donkey anti-mouse for 1 h. After rinse in PBS, post fixation in 2% PFA for 15 min, and a second PBS wash, coverslips were inverted and mounted on glass slides.
Detection of mRNA in mouse tissues was done as previously described (38), with all steps carried out at room temperature unless specified otherwise. Briefly, sciatic nerve segments were fixed for 4 h in 2% PFA, cryoprotected overnight in 30% buffered sucrose at 4°C, and then cryosectioned at 20 μm thickness (sections were stored at -20°C until used). Sections were brought to room temperature, washed three times in PBS for 5 min each, and then treated with 20 mM glycine and fresh 0.25 M NaBH4 in PBS (3 times, 10 min each for both) to quench autofluorescence. Sections were quickly rinsed in 0.1 M Triethylamine (TEA) and then incubated in 0.1 M TEA + 0.25% acetic anhydride for 10 min. Sections were dehydrated in 70, 95 and 100% ethanol (3 min each) and then delipidated in chloroform for 5 min followed by 100 and 95% ethanol (3 min each). After washing in 2× SSC, sections were incubated overnight at 37°C in a humidified chamber with 12.5 μM probe and RT97 anti-NF (1:100) in hybridization buffer. The following day, sections were washed in 2× SSC + 10% formamide at 37°C for 30 min, followed by two incubations in 2× SSC for 5 min each. Sections were then briefly rinsed in PBS + 1% Triton-X100, and then incubated with donkey secondary antibodies diluted in 10 X blocking buffer (1:100; Roche, Penzburg, Germany) + 0.3% Triton-X100 for 1 h. Sections were finally washed in PBS for 5 min, post-fixed in 2% PFA for 15 min, washed 3 times in PBS (5 min each), rinsed in DEPC-treated water, and mounted under glass coverslips.
smFISH and IF signals were imaged using Leica SP8X as above. 63×/NA 1.4 oil immersion objective and pulsed white light laser was used for imaging RNA in both culture and tissue samples. Scramble probe was used to set the image acquisition parameter that would not acquire any nonspecific signal from scramble probe. Taking XYZ image stacks at least two locations in each section scanned nerve sections.
Detection of nascently synthesized proteins
Rat sciatic nerve from three animals per condition was either left naïve or in vitro crushed and incubated in DMEM medium containing, 10% FBS + Cyclosporin A (20 μM; Sigma) + 1% penicillin/streptomycin. Nerves were treated with 200 μg/ml anisomycin or vehicle (0.1% DMSO) for 3 h at 37°C, followed by adding 100 μg/ml O-propargyl-puromycin (OPP; Invitrogen) for 1 h at 37°C. Axoplasm was extruded in 1 ml of transport buffer (20 mM HEPES [pH 7.4], 100 mM sodium acetate, 5 mM magnesium acetate), after extrusion SDS was added to 1%. Protein concentration was quantified by BCA assay and 350 μg of total protein was used for biotin conjugation by click chemistry (100 μM biotin-PEG3-azide) according to manufacturer's instruction. The reaction mix was incubated for 2 h at room temperature on a rotator. Five volumes ice-cold acetone was added to precipitate the protein. Protein pellets were resuspended in PBS containing 1% SDS. Streptavidin pull-down was carried out overnight at 4°C in 1 ml volume containing 60 μl of streptavidin magnetic beads (Thermo-Fisher), 1% NP40, 0.1% SDS and 1× Complete EDTA-free protease inhibitor cocktail (Roche) in PBS. 10% of protein used for pull-down was taken to generate input samples. Beads were washed three times for 10 min with 1% NP40, 0.1% SDS in PBS at room temperature. Proteins were eluted from streptavidin beads by boiling for 10 min in 2× Laemmli sample buffer and then adjusted to 1× with PBS for denaturing polyacrylamide gel electrophoresis (SDS/PAGE).
Immunoblotting
Adult mouse DRG cultures (3 days in vitro, ∼80 000 neurons/well) were lysed in Laemmli sample buffer and denatured by boiling at 95°C × 5 min. Rat axoplasm from naïve and crushed sciatic nerves was extruded in nuclear transport buffer (20 mM HEPES [pH 7.3], 110 mM potassium acetate, 5 mM magnesium acetate, supplemented with protease inhibitors) as previously described (39). Lysates were cleared of debris by centrifugation at 15 000 × g for 15 min at 4°C and then normalized for protein content using Bradford assay (Bio-Rad). Normalized protein lysates were fractionated by 10% SDS/PAGE and transferred onto a PVDF membrane (GE Healthcare Life Sciences, Marlborough, MA). After blocking in 5% non-fat dried milk powder (Bio-Rad) diluted in Tris-buffered saline with 1% Tween 20 (TBST), membranes were probed overnight at 4°C with rabbit anti-KHSRP (1:1000; Novus, # NBP1-18910), rabbit anti-GFP (1:1000; Enquire BioReagents, Littleton, CO, # QAB10298), rabbit anti-GAPDH (1:2000; Cell Signaling Tech, Beverly, MA, # 2118), rabbit anti-α-tubulin (1:1000; Cell Signaling Tech, # 2125); mouse anti-eIF2α (1:1000; Cell Signaling Tech, # 2103) and rabbit anti-eIF2αPS51 (1:1000; Cell Signaling Tech, # 9721) antibodies or streptavidin-HRP (1:10 000, Abcam, # Ab_7403) diluted in blocking buffer. Blots were washed in TBST and then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:2000; Cell Signaling Tech.) diluted in blocking buffer for 1 h at room temperature. Blots were washed in TBST and signals were detected by ECL Prime™ (GE Healthcare Life Sciences).
Fluorescence recovery after photobleaching
Fluorescence recovery after photobleaching (FRAP) experiments were conducted at 37°C, 5% CO2 as previously described (40). Briefly, dissociated adult mouse DRG cultures transfected with the GFPMYR5’/3’khsrp were equilibrated in culture medium as above except phenol red was excluded. A region of interest (ROI) in the most distal axon of dissociated DRG neurons was photobleached with 488 nm argon laser set at 100% power for 80 frames at 0.65 s each. Pre-bleach and post-bleach signals were captured using 70% power for 488 nm laser line every 30 s (2 for pre-bleach and 30 for post-bleach). Translation dependence for recovery was tested by pre-treating DRG cultures with 100 μg/ml anisomycin (Sigma) or 150 μg/ml cycloheximide (Sigma) 20 min prior to photobleaching. For testing Ca2+-dependent translation by FRAP, transfected DRG cultures were pretreated with 1 μM thapsigargin (Sigma), 3 μM BAPTA-AM (Sigma), 90 μM GSK260614 (Bio-Techne Corp/Tocris, Minneapolis, MN) or 50 μM Sephin1 (Apexbio, Houston, TX). Leica SP8X confocal microscope was used for imaging at 37°C, 5% CO2 with 63X/NA 1.4 oil immersion objective. Pinhole was set to 3 Airy units for pre-bleach, bleach, and post-bleach sequences to ensure full thickness excitation of the axon. ROIs were 40 × 40 μm and at least 250 μm from the soma.
Image analyses
ImageJ was used to quantify protein and RNA levels in sciatic nerve tissues from optical planes of XYZ scans. Axon only signal was extracted via Colocalization plug-in that extracted only protein or RNA signals that overlapped with axonal marker (NF) in each plane. Extracted ‘axon only signal’ was projected as a separate channel (38). Signal intensities were then calculated from each XY plane of these axon only channels. NF immunoreactivity area was used to normalize signal intensities across the individual XY planes. The relative signal intensity was then averaged for all tiles in each biological replicate.
To assess regeneration in vivo, tile scans were post-processed by Straighten plug-in for ImageJ (http://imagej.nih.gov/ij/). SCG10 fluorescence intensity was measured along the length of the nerve using ImageJ. Regeneration index was calculated by measuring the average SCG10 intensity in bins across at least 3 mm distal to the crush site. The crush site was defined by the position along the nerve length with maximal SCG10 intensity (secondarily confirmed by DAPI signals and DIC images). For analyses of axon growth in vitro, dissociated DRGs were immunostained with NF antibodies as described above. Images from 36 or 48 h cultures were used for neurite length and branching parameters (neurites/cell body and branch density) using WIS-Neuromath (41). For NMJs, confocal Z stacks from muscle were projected as single XY images using ImageJ. Nerve terminal and endplate (AchR) areas were calculated by ImageJ and fractional occupancy of NMJ was calculated by dividing nerve terminal area to endplate AChR area as described (37).
For FRAP image analyses, raw images sequences were analyzed for recovery in the bleached ROI using Leica confocal software package. Recovery was determined relative to pre-bleach and post-bleach signals, which were set at 100 and 0% to allow for comparisons between experiments and between neurons.
Statistical analyses
GraphPad Prism software package (La Jolla, CA) was used for statistical analyses. One-way ANOVA with Tukey post-hoc was used to compare between data points and Student's t-test was used to compare two independent groups for most experiments. For FRAP studies, two-way ANOVA with Tukey post-hoc was used, where control values were compared to cycloheximide- and anisomycin-treated cultures for each time point. All experiments were performed in at least triplicate. P ≤ 0.05 was considered as statistically significant.
RESULTS
Peripheral nerve injury changes the axonal RNA binding protein population
Protein synthesis in PNS axons has been shown to facilitate nerve regeneration after injury (5). Transport and translation of the mRNA templates needed for this intra-axonal protein synthesis are driven by proteins interacting with those mRNAs (4). We recently showed that many RBPs that were thought to have exclusively nuclear roles localize into PNS axons by an RNA affinity mass spectrometry (RAMS) approach (42). The levels of some of these RBPs increased after axotomy, which pointed to functions in growing axons. As this RAMS assay focused on interactions with axonal mRNA localization motifs, we wanted to gain a more systematic and unbiased view of the axonal RBP population. With the limited amount of protein obtained from PNS axoplasm coupled with the likely low abundance of RBPs relative to cytoskeletal components in the axons, we turned to a targeted mass spectrometry approach using PRM to profile axonal RBPs in naïve, injured and regenerating sciatic nerve axoplasm. By mining the UniProt database (https://www.uniprot.org) for proteins with ‘RNA binding’ in functional or domain descriptions and validated expression in the nervous system (cross-referencing each for RNA expression in neurons using the GeneCards database (https://www.genecards.org), we arrived at 357 proteins to test. Of these, 196 were represented in a reference MS library that had been generated from rat nervous system tissues (including sciatic nerve, dorsal root ganglion (DRG), and spinal cord) and were included in a PRM-MS method that targeted 511 unique precursor peptides (26). Using this PRM method, we quantified the abundance of 84 RBPs represented by 184 precursor peptides in adult rat sciatic nerve axoplasm. Several of the RBPs showed increased or decreased axoplasm levels over 3–28 days post injury compared to the contralateral uninjured sciatic nerve (Figures 1A, B; Supplemental Figure S1). Initial validation by immunoblotting from naïve and 7 days post-crush injury sciatic nerve axoplasm showed that FXR1, hnRNP A3, hnRNP AB, hnRNP H1 and KHSRP increased in the 7 days samples (Figure 1C). We have previously reported axonal levels of hnRNP H1 similarly increase following nerve crush injury (42).
Figure 1.

Peripheral nerve injury changes the axonal RNA binding protein populations.(A, B) Sciatic nerve axoplasm harvested proximal to the injury site from 3–28 days post-crush lesion was trypsin-digested and processed for liquid chromatography and mass spectrometry using parallel reaction monitoring (PRM) to detect proteins with known RNA binding activity. Levels of proteins from spectral counts relative to uninjured (naïve) axoplasm shown are Log2 fold-change as indicated in (A) (N = 3 for each time point). (B) shows volcano plot for PRM results for 7 days crush versus naïve samples graphed as log2 fold-change versus negative log10P value for differences. Also see Supplemental Figure S1 for graphical representation of full time course. (C) Representative immunoblot for naïve and 7 days injured sciatic nerve axoplasm confirms the increase in FXR1, hnRNP A3, hnRNP AB, hnRNP H1 and KHSRP. ERK 1/2 and GAPDH show relatively equivalent loading of the lysates.(D, E)Representative immunoblots for kinetics of KHSRP elevation in sciatic nerve axoplasm over 0–28 days post-crush lesion are shown in (D). Quantification of KHSRP immunoreactivity across multiple animals is shown in (E) as mean fold-change relative to naïve ± standard error of the mean (SEM; N = 3 mice for each time point; *** P ≤ 0.001 for versus 0 day, †P ≤ 0.05, †††P ≤ 0.005, †††P ≤ 0.001 and ††††P ≤ 0.0005 versus 3 days, &&&P ≤ 0.001 and &&&&P ≤ 0.0005 versus 7 days, and $$ P ≤ 0.01 and $$$$P ≤ 0.0005 versus 14 days by one-way ANOVA with Tukey's post-hoc analysis). (F) Representative confocal images for KHSRP protein in naïve and 7 days post-crush sciatic nerve. Upper panels of each pair show XY images of merged neurofilament (NF) + KHSRP and DAPI + KHSRP; lower panels show KHSRP signals overlapping with NF or DAPI in individual Z planes projected as an XYZ image [scale bar = 5 μm].
It is appealing to speculate that the increased levels of these RBPs in injured and regenerating peripheral nerve axoplasm would serve to promote regeneration. For example, hnRNP H1 was identified by the RAMS approach as binding to Hmgb1 mRNA’s axonal localization motif (42), and local translation of Hmgb1 mRNA promotes axons growth (43). The increase in KHSRP after injury (Figure 1D-E) was surprising as this RBP has been shown to promote mRNA decay (22,23), including targeting Gap43 mRNA, whose encoded protein has long been linked to axon growth promotion, for degradation (21). KHSRP also has other cytoplasmic functions including RNA transport and microRNA (miRNA) processing that could support axon regeneration (44,45). Thus, we examined KHSRP more closely to determine which function the increased axoplasmic KHSRP might serve in the injured sciatic nerve. Intriguingly, the axoplasm KHSRP levels were significantly higher at 21–28 days post injury than earlier time points (Figure 1D, E). With the mid-thigh sciatic nerve crush used, rats here begin to regain lower hind limb function over 21–28 days post injury indicative of some target reinnervation. Thus, KHSRP levels appeared to be increased in axons after injury and continued to elevate across the duration of axon regeneration at least until target reinnervation begins.
While the axoplasm preparation used here is highly enriched for axonal proteins, the preparation does contain some glial constituents (46). Since KHSRP is ubiquitously expressed (47,48), we used confocal microscopy to determine if the elevations in KHSRP levels seen by PRM and immunoblotting derived from axonal KHSRP. Consistent with the ubiquitous KHSRP expression, KHSRP protein immunofluorescence was seen in both axons and the adjacent non-neuronal cells; however, extraction of the KHSRP immunofluorescent signals overlapping with neurofilament signals across individual optical planes, showed markedly increased intra-axonal KHSRP signals at 7 days post injury compared to uninjured nerve (Figure 1F). Interestingly, DAPI co-staining showed that the signals in non-neuronal cells within the nerve were predominantly nuclear, likely representing Schwann cell nuclei in the nerve (Figure 1F). Thus, axotomy increases axonal KHSRP levels in both axons and glia in the PNS, but glial KHSRP is predominantly intra-nuclear compared to the cytoplasmic signals of the axons.
Loss of KHSRP enhances axon growth in the peripheral nervous system through an axon-intrinsic mechanism
Since axonal mRNA translation supports both axon development and regeneration (4) and KHSRP has multiple functions (22,24,45,48,49), we asked if axonal KHSRP might play a role in axon growth in adult neurons. To address this, we took advantage of a constitutive KHSRP knockout mouse line, where both KHSRP alleles are deleted (47). Cultures of dissociated lumbar (L) 4–6 DRG neurons from KHSRP knockout (Khsrp–/–) mice showed significantly increased axon lengths compared to those from wild type (Khsrp+/+) mice (Figure 2A, B), but there were no differences in axon branching between the genotypes (Supplemental Figure S2A, B). This raised the possibility that KHSRP attenuates axon growth in adult DRG neurons as we had previously reported for embryonic CNS neurons (21).
Figure 2.

KHSRP depletion enhances axonal growth from naïve but not injury-conditioned DRG neurons. (A) Representative images of 24 h DRG neuron cultures from naïve and 7 days injury-conditioned Khsrp+/+ and Khsrp–/– mice immunostained for NF are shown [scale bar = 100 μm]. (B) Quantification of total axon length per neuron from cultures as in (A) shows significantly greater axon growth in Khsrp–/– compared to Khsrp+/+ DRGs. However, there is no significant difference in axon length between the genotypes when cultures were prepared 7 days after nerve crush injury (i.e. ‘injury-conditioned’ neurons). Data are expressed as mean ± SEM; refer to Supplemental Figure S2 for axon branching analyses (N ≥ 75 neurons analyzed per condition in three independent culture preparations; * P ≤ 0.05 and ** P ≤ 0.01 by one-way ANOVA with Tukey's post-hoc analysis). (C) Representative immunoblot shows signal for KHSRP in cell body and axonal preparations from DRG cultures from indicated mice. There is no KHSRP band in the Khsrp–/– cell body or axonal preparations, despite GAPDH showing more protein in those samples than in the Khsrp+/+ samples.
Cultures of dissociated L4–6 DRGs from mice that have been primed or conditioned by an in vivo sciatic nerve crush injury exhibit increased growth that is transcription-independent, but translation-dependent (25,50). The in vivo conditioning injury activates transcription of growth-associated genes, whose mRNA products are then translationally regulated after the second injury that is brought by the DRG dissociation at the time of culture (25). With KHSRP’s roles in post-transcriptional regulation (51), we tested whether the in vitro axon growth from in vivo ‘injury-conditioned’ L4–6 DRGs is further increased in the absence of KHSRP. In contrast to the naïve DRG cultures, there was no significant difference in axon lengths comparing the injury-conditioned DRGs from Khsrp–/– versus Khsrp+/+ mice (Figure 2A, B). To be certain that KHSRP was indeed absent from these neurons, we assessed KHSRP levels in soma and axon preparations from L4–6 DRG neurons cultured on a porous membrane for separation of axons (52). KHSRP immunoreactive bands were clearly present in the Khsrp+/+ soma and axon isolates, but were not detected in the Khsrp–/– samples even with extended exposures (Figure 2C). These data suggest that removing KHSRP can increase axon growth, but in vivo injury conditioning appears to overcome KHSRP’s growth attenuating effects when those neurons are dissociated and cultured.
The comparable axon growth from injury-conditioned DRG neurons from the Khsrp–/– and Khsrp+/+ mice could reflect a ceiling effect for KHSRP deletion in the DRG neurons, with the growth promoting effects of KHSRP loss mitigated by the in vivo conditioning lesion. Our initial data from Figure 1 pointed to an increase of KHSRP within the axons in vivo after peripheral nerve injury, and those axons are notably stripped away from the soma by the dissociation for culturing the neurons. Thus, an alternative hypothesis is that axon-intrinsic functions of KHSRP underlie its growth-attenuating functions. That is, when the axons are sheared from the soma during in vitro culture preparation the intra-axonal increase in KSHRP seen in Figure 1 could have been lost or minimized. To test this alternate hypothesis, we compared in vivo axon regeneration in the Khsrp–/– and Khsrp+/+ mice. Although, there was no significant difference in axonal profiles extending beyond the crush site comparing Khsrp–/– and Khsrp+/+ mice at 7 days after crush injury, the Khsrp–/– mice showed higher regeneration indices than the Khsrp+/+ mice at 10 days post-injury and the Khsrp–/– mice showed significantly higher regeneration indices 14 days post-injury (Figure 3A). This suggests that the growth-attenuating effects seen from the injury-induced increase in axonal KHSRP after axotomy can accumulate over time after injury, which is consistent with the increasing axoplasm levels of this protein shown in Figure 1.
Figure 3.

KHSRP deletion increases in vivo axon regeneration after a conditioning sciatic nerve injury. (A) Regeneration indices calculated as fraction of SCG10 axonal profiles at injury site (0 μm) are shown as mean ± SEM as indicated. There was no significant differences between Khsrp–/– and Khsrp+/+ mice at 7 and 10 days post-injury, but significant differences are seen between the genotypes at 14 days post-injury (n = 6 mice per genotype and timepoint, * P ≤ 0.05, ** P ≤ 0.01 and *** P ≤ 0.001 by two-way ANOVA with Bonferroni post-hoc analysis). (B) Representative SCG10 immunostained images of sciatic nerves after single (3 days; upper panels) or injury-conditioned (7 + 3 days; lower panels) sciatic nerve crush injuries for Khsrp+/+ and Khsrp–/– are shown. Proximal is on the left and distal on the right; the dashed line indicates the injury site, with the second injury for the double crush injured animals placed at ∼0.5 cm proximal to the initial injury site [scale bar = 500 μm]. (C) Regeneration indices calculated as fraction of SCG10 axonal profiles relative to the injury site (0 μm) are shown as mean ± SEM as indicated. There was no significant difference in the regeneration after the single injury, but the injury-conditioned Khsrp–/– mice show significantly higher regeneration indices. Refer to Supplemental Figure S3B for regeneration index comparisons of naïve versus injury-conditioned nerves within genotypes (N = 5 mice per genotype and condition; * P ≤ 0.05, ** P ≤ 0.01 and *** P ≤ 0.001 by two-way ANOVA with Bonferroni post-hoc analysis). (D) Confocal XYZ images of gastrocnemius muscles of the injury-conditioned Khsrp+/+ and Khsrp–/– mice at 14 days after second nerve crush are shown. NMJs are detected by post-synaptic (α-bungarotoxin; green) and pre-synaptic markers (cocktail of anti-NF, -synapsin I and -synaptophysin; red) signals showing higher matching of pre- and post-synaptic markers in Khsrp–/– than Khsrp+/+ mice. Inset panels on lower right of both rows show higher magnification of the NMJs outlined by dashed boxes [scale bars = 20 μm for main panels and 5 μm for insets]. (E) Quantification of NMJ occupancy (% presynaptic area/postsynaptic area) shows significantly greater occupancy in the injury-conditioned Khsrp–/– than in Khsrp+/+ mice but no difference between genotypes was seen with the single crush lesion (injury-conditioned = 7 + 14 days; single nerve crush = 14 days). Data are expressed as mean ± SEM (N ≥ 15 NMJs quantified in three animals per condition per genotype; ** P ≤ 0.01 by Student's t test).
Considering this delayed effect of axonal KHSRP on nerve regeneration, we asked if the growth attenuation from KHSRP might be seen sooner if axotomy occurred in the setting of already increased axonal KHSRP. To address this possibility, we used an in vivo injury-conditioning paradigm where an initial sciatic nerve crush injury is performed and then 7 days later the nerve is crushed a second time proximal to the initial injury site (53). For comparison, the contralateral sciatic nerve underwent single crush at the same time and same approximate level as the second lesion. Three days later, nerve regeneration contralateral to the conditioning lesion (i.e. single crush injured nerves) showed no significant difference between Khsrp–/– and Khsrp+/+ mice (Figure 3B, C). In sharp contrast, the injury-conditioned Khsrp–/– nerves (i.e. double crush injured nerves) showed a dramatic increase in axon regeneration compared to the injury-conditioned Khsrp+/+ mice (Figure 3B, C; Supplemental Figure S3A). To determine if this increased axon growth affected target reinnervation, we analyzed neuromuscular junctions (NMJ) in the gastrocnemius muscle 14 days after crush injury (Figure 3D). NMJ occupancy, based on percentage of presynaptic compared postsynaptic area, showed no differences between Khsrp+/+ and Khsrp–/– mice at 14 days in the single crush injured nerves (Figure 3E). However, the double crush injured nerves showed significantly greater NMJ occupancy in Khsrp–/– versus Khsrp+/+ at 14 days after the second crush lesion (Figure 3D, E).
The difference between in vivo nerve regeneration and in vitro axon growth after injury-conditioning is intriguing since the injury-conditioning is thought to drive a change in gene expression programs, with injury-conditioned neurons already having a growth-associated gene expression program active at the time of second injury (50,54). Since axons are stripped from soma in the cultures used in Figure 2, axon-intrinsic roles of KHSRP for dampening the full effects of these changes in the injury-conditioned neurons could explain the difference between in vivo and in vitro axon growth. To test this possibility, we developed an in vitro axotomy model where DRGs were initially allowed to extend axons and then the axon shafts were severed. By severing the axon shafts rather than stripping axons from the soma, this allowed us to assess regeneration initiating from the axon shaft rather than initiation of new axon growth from the soma. For this, DRG neurons from Khsrp–/– and Khsrp+/+ mice were cultured on porous membrane filters for 36 h, and then the lower surface of the membrane was scraped to remove axons by severing the axon shafts as they exited the pores of the membrane (Supplemental Figure S3B). After an additional 72 h in culture, neurofilament immunostained axons were traced along the membrane's lower surface and total axon lengths were quantified (Supplemental Figure S3C). Consistent with increased regeneration from injury-conditioned nerves in vivo, the Khsrp–/– DRGs showed more axon regeneration than the Khsrp+/+ DRGs after their axon shafts were severed in vitro (Supplemental Figure S3D). Taken together, these data suggest that the increase in axonal KHSRP slows extension of the regenerating axon.
The axotomy-induced increase in axonal KHSRP occurs via axon-intrinsic mRNA translation
We next asked what mechanisms might underlie the increase in axonal KHSRP upon injury. mRNA translation in axons provides a means to rapidly change protein content in the axons (4). For example, injury-induced translation of axonal Calr mRNA was recently shown to support early regrowth of severed axons (55) and translation of axonal Kpnb1 mRNA generates a retrograde signal that triggers transcriptional changes in the neuronal soma (54). Thus, we wondered whether the change in axonal KHSRP levels might be driven by localized translation of its mRNA. We had previously detected Khsrp mRNA in RNA-seq analyses of sciatic nerve axoplasm (42); however, these axoplasm preparations obviously can contain non-neuronal contents (46), particularly for mRNAs isolated from injured nerve as Lee et al. used (42). To overcome this limitation, we used smFISH/IF to ask whether sensory axons contain Khsrp mRNA. Dissociated DRG cultures from Khsrp+/+ mice showed prominent Khsrp mRNA signals in soma and axons, with granular appearing axonal Khsrp mRNA signals (Figure 4A–C). There was no Khsrp mRNA signal in the axons of the Khsrp–/– DRG cultures (Figure 4C). smFISH signals for the soma of the DRGs from Khsrp–/– mice did show a faint but consistent signal with the Khsrp mRNA probes that was significantly greater than with scrambled probes (Figure 4A, B). The Khsrp–/- mice were generated by deleting exons 1–13 (of 18 exons) of the murine KHSRP gene (24), and the Stellaris smFISH Khsrp mRNA probes used here hybridize to sequences across the full exons comprising the mature Khsrp mRNA. Considering the absence of KHSRP protein in these cultures by immunoblotting (Figure 2C), we suspect that the faint Khsrp mRNA signal in soma of the Khsrp–/– DRGs reflects RNA transcription of exons 14–18 remaining in the Khsrp–/– mice or portions thereof, as confirmed by RNA-sequencing of Khsrp–/– adult mouse brain and embryonic cortical neuron cultures (data not shown). The absence of Khsrp mRNA signals in the axons of Khsrp–/– DRGs here and lack of any detectable KHSRP by immunoblotting in the Khsrp–/– DRG lysates shown in Figure 2C suggest that any transcript from remaining exons 14–18 of KHSRP in the Khsrp–/– mice does not localize into axons or generate detectable protein.
Figure 4.

Khsrp mRNA is transported into PNS axons. (A) Representative confocal images for smFISH/IF for Khsrp mRNA (grey) and NF (magenta) in dissociated DRG cultures are shown as indicated. There is a clear signal for Khsrp mRNA in the cell body (left and middle columns) and distal axons (right column) of DRGs from Khsrp+/+ mice. Axons of Khsrp–/– DRGs show Khsrp mRNA signals comparable to the scrambled probe; however, there was faint Khsrp signal in the soma of the Khsrp–/– cultures that was consistently above the scrambled probe signal [scale bar = 10 μm].(B, C)Quantification smFISH signals for Khsrp mRNA in soma (B) and axons (C) is shown as mean ± SEM for scramble (Khsrp+/+) and Khsrp mRNA (Khsrp+/+ and Khsrp–/–) probes; in each case, scramble probe was hybridized to Khsrp+/+ DRG cultures as in panel A (N ≥ 16 neurons over three separate cultures; *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001 by one way ANOVA with Tukey's post-hoc analysis). (D) Representative confocal images for smFISH/IF for Khsrp mRNA (grey) and NF (magenta) in uninjured sciatic nerve are shown as indicated. Left column shows XYZ projections from eight optical planes taken at 0.2 μm Z step intervals; right column shows ‘axon only’ Khsrp mRNA signals generated by extracting FISH signals overlapping with NF in individual Z sections and projecting those as an ‘Axonal Khsrp mRNA’ XYZ image [scale bar = 5 μm]. (E) Quantification of axonal Khsrp mRNA signals from (D) are shown as mean ± SEM (N = 6 animals per genotype; ** P ≤ 0.01 by Student's t-test). (F) Representative matched exposure smFISH/IF images for Khsrp mRNA, NF, GFAP and DAPI in naïve versus 7 days post-crush injured sciatic nerves as indicated. Upper row shows merged signals as single XY planes; lower two rows show XYZ projections for Khsrp mRNA colocalizing with NF (middle row) and GFAP (lower row). Representative matched exposure images for scramble probe are shown in Supplemental Figure S3E [scale bar = 5 μm]. (G) Quantification of axonal smFISH signals for Khsrp mRNA in naive and 7 days regenerating sciatic nerve axons (N = 6 animals; no significant differences detected by Student's t-test).
smFISH/IF performed on sciatic nerve sections also showed prominent Khsrp mRNA signal in the axons of the Khsrp+/+ mice that was completely absent in Khsrp–/– mice (Figure 4D, E). Axonal Khsrp mRNA signals showed no significant differences in Khsrp+/+ mice comparing naïve and 7 days injured sciatic nerve (Figure 4F, G; Supplemental Figure S3E), suggesting that the elevation of KHSRP protein seen in Figure 1 is not from increased transport or survival of Khsrp mRNA following sciatic nerve crush injury. Notably, Khsrp mRNA was also detected in Schwann cells (Figure 4F), which is consistent with the strong nuclear KHSRP signals seen in the Schwann cells in Figure 1F.
The RNA analyses above suggest that KHSRP can be synthesized locally in axons. To determine if the PNS nerve injury-induced increase in KHSRP is intrinsic to the nerve or results from KHSRP transported from proximal nerve and soma, we ligated the sciatic nerve to restrict anterograde transport and performed a crush injury distal to the ligation (Figure 5A). Adult rats were used for these analyses since the larger sciatic nerve could be ligated more consistently than in the mouse. Immunofluorescence for amyloid precursor protein (APP) and signal transducer and activator of transcription 3α (Stat3α) confirmed that the ligation attenuated both anterograde and retrograde transport, as APP accumulated proximal to and Stat3α accumulated distal to the ligation site (Supplemental Figure S4). As expected, KHSRP signals were detected both in axons and adjacent Schwann cells (Figure 5B, C). Colocalization with DAPI indicated that the non-axonal KHSRP is predominantly nuclear as seen in Figure 1F (data not shown). Axonal KHSRP Immunofluorescence showed increased signals at both the proximal and distal ligation sites as well as the crush site relative to axons in naïve nerve; however, the increased axonal KHSRP signals at the crush site were significantly greater than the proximal and distal ligation sites and continued to elevate over time after injury (Figure 5D). These results are consistent with local synthesis of KHSRP in the injured nerve, as opposed to anterograde transport of KHSRP from the soma or more proximal axons where the signal would have been greater at the proximal ligation site than crush and distal ligation sites. Of note, ligation causes a degree of nerve injury, and the increase in KHSRP levels both proximal to and distal to the ligation are consistent with such an injury response.
Figure 5.

Khsrp mRNA is rapidly translated in axons after injury. (A) Schematic of nerve ligation model used for panels B and C. Proximal nerve is on the left and distal on the right as indicated. The nerve was ligated and then immediately crushed ∼1 cm distal to the ligation.(B, C) Confocal images for KHSRP protein in naïve (B) and post-crush injury (3 and 16 h; (C). Upper rows of image pairs show XYZ projections of merged signals for KHSRP (grey) and NF (magenta), while lower rows show ‘axonal KHSRP’ signals as from individual Z planes that were projected as an XYZ image. As in Figure 1E, the strong signals that are outside of the axons are in Schwann cell nuclei based on DAPI co-labeling (data not shown). Representative images for ligation efficiency and KHSRP signals proximal and distal to ligation are shown in Supplemental Figure S4 [scale bar = 5 μm]. (D) Quantification of the axonal KHSRP immunoreactivity from ligation proximal and distal and crush sites are shown as mean ± SEM (N = 3 animals per time point; * P ≤ 0.05 and ** P ≤ 0.01 for indicated time points versus naïve nerve, ††P ≤ 0.01 for indicated time points versus ligation proximal, and && P ≤ 0.01 for indicated time points versus ligation distal by Student's t-test; ligation proximal versus distal have no significance). (E) Representative immunoblots for ex vivo puromycinylated naïve versus crushed sciatic nerve segments are shown as indicated. Protein synthesis inhibition with anisomycin completely blocks the puromycinylation of KHSRP in axoplasm samples extruded from the nerve segments, and GAPDH shows relatively equivalent protein loading across the lanes. Note that total KHSRP levels also increases with crush injury and this was attenuated by anisomycin. (F) Quantification of puromycinylated KHSRP signals from (D) is shown as mean ± SEM. Crush injury significantly increases axonal KHSRP synthesis and this blocked by anisomycin (N = 3; *** P ≤ 0.001 by one way ANOVA with Tukey's post-hoc analysis). (G) FRAP analyses for distal axons of neurons transfected with GFPMYR5’/3’khsrp (schematic above graph) is shown as normalized average % recovery ± SEM. Pre-treatment with anisomycin or cycloheximide significantly attenuates the GFP recovery, indicating protein synthesis dependent recovery for GFPMYR fluorescence in the axons (N ≥ 10 neurons over three culture preparations; * P ≤ 0.05, ** P ≤ 0.01 and *** P ≤ 0.005 by two-way ANOVA with Bonferroni post-hoc analyses for indicated time points versus control). Representative images sequences for FRAP are shown in Supplemental Figure S5. (I) Schematic for proposed signaling pathway for axotomy induced increase in axoplasmic Ca2+ leading to eIF2α phosphorylation is shown. GSK260614 was used as a specific PERK inhibitor and Sephin1 as an inhibitor of eIF2α dephosphorylation. (H) FRAP analyses for distal axons of neurons transfected with GFPMYR5’/3’khsrp reporter is as normalized average % recovery ± SEM. Treatment with thapsigargin increased and BAPTA-AM decreased recovery over control conditions. The thapsigargin-induced increase was blocked by GSK260614, while the BAPTA-AM-induced decrease was partially blocked by Sephin1 (N ≥ 17 neurons over three culture preparations; # P ≤ 0.05, ## P ≤ 0.01, ### P ≤ 0.005 and #### P ≤ 0.001 for thapsigargin or BAPTA-AM versus corresponding control time points and +P ≤ 0.05, ++P ≤ 0.01, +++P ≤ 0.005 and ++++P ≤ 0.001 for thapsigargin or BAPTA-AM versus corresponding thapsigargin + GSK260614 or BAPTA-AM + Sephin1 time points by two-way ANOVA with Bonferroni post-hoc analyses for indicated time points versus control; for data points appearing to error bars, the SEM is too small to show).
To more directly test if KHSRP is translated in sciatic nerve axons, we exploited an ex vivo nerve injury model where we used puromycin incorporation to detect newly synthesized peptides in axoplasm extruded from the nerve (56). For this, we excised segments of rat sciatic nerve and placed these into culture medium with cyclosporin A included to delay Wallerian degeneration (57) and OPP for puromycinylation of nascently synthesized polypeptides (58,59). There was a significant increase in puromycinylated KHSRP in the axoplasm isolates at 4 h after ex vivo nerve crush that was attenuated by the protein synthesis inhibitor anisomycin (Figure 5E, F). Notably, an increase in overall KHSRP levels was also seen in the crushed nerve axoplasm and this was similarly attenuated by pretreatment with anisomycin (Figure 5E), confirming that axonal injury increases KHSRP in sciatic nerve axoplasm through mRNA translation.
To further test for translation of Khsrp mRNA in axons, we fused 5’ and 3’ untranslated regions (UTR) of rodent Khsrp mRNA to the coding sequence of a diffusion-limited GFP reporter cDNA (GFPMYR5’/3’khsrp; Figure 5G) as a surrogate for axonal localization and translation of Khsrp mRNA. Co-translational myristoylization of this GFP reporter allows for membrane attachment to limit the protein's diffusion (31,40), and the UTRs of axonal mRNAs typically contain motifs for axonal mRNA targeting and translational regulation (4). By fluorescence recovery after photobleaching (FRAP), DRG neurons expressing GFPMYR5’/3’khsrp showed rapid fluorescent recovery that was significantly attenuated by pretreatment with anisomycin or a second protein synthesis inhibitor cycloheximide (Figure 5G; Supplemental Figure S5A). Importantly, the bleached regions of interests (ROI) were separated from the soma by >250 μm, indicating that this fluorescence recovery occurs faster than can be accounted for by anterograde transport of reporter synthesized in the soma.
The above data support that the axotomy-induced increase in axonal KHSRP is derived from localized translation of Khsrp mRNA in axons, but these did not address the mechanism underlying this translational increase. Axon injury is known to increase axoplasmic Ca2+ and activate a localized intrinsic stress or unfolded protein response (60). Although increased cytoplasmic Ca2+ is well known to block generalized protein synthesis through an inhibitory phosphorylation of the translation factor eIF2α (Figure 5H), some injury-response mRNAs show a paradoxic increase in their translation with Ca2+ despite eIF2α phosphorylation (61,62). We used thapsigargin to simulate the injury-induced increase in axoplasmic Ca2+ (59). Recovery of axonal GFPMYR5’/3’khsrp fluorescence was significantly increased by thapsigargin treatment; conversely, chelating Ca2+ with BAPTA-AM attenuated recovery of the axonal GFPMYR5’/3’khsrp fluorescence (Figure 5I). Inhibition of protein kinase R-like ER kinase (PERK) prevented the increase in axonal GFPMYR5’/3’khsrp fluorescence recovery seen with thapsigargin treatment (Figure 5H, I). Conversely, pretreatment with Sephin1 that prevents dephosphorylation of eIF2α partially reversed the BAPTA-AM dependent decrease in axonal GFPMYR5’/3’khsrp fluorescence recovery (Figure 5H-I). Immunoblotting confirmed that the increase in eIF2αPS51 in response to thapsigargin was prevented by the GSK260614 PERK inhibitor and decrease in basal eIF2αPS51 seen with BAPTA-AM was reversed by treatment with Sephin1 to inhibit eIF2α phosphatase (Supplemental Figure S5B). Thus the axotomy-induced increase in axonal KHSRP is derived through activation of Khsrp mRNA via Ca2+ → PERK → eIF2αPS51.
KHSRP’s ARE-binding KH4 domain attenuates axon growth
KHSRP was previously shown to bind to the ARE in Gap43 mRNA’s 3’UTR and promote decay of the transcript (21). The GAP43 gene is transcriptionally activated following PNS nerve injury (63), and axonal Gap43 mRNA levels increase in regenerating PNS axons (20). Given that Gap43 mRNA and protein expression are typically associated with axon growth (64), we tested the effect of Khsrp knockout on the sciatic nerve levels of this KHSRP target mRNA. RTddPCR from sciatic nerve samples of Khsrp+/+ mice showed an increase in Gap43 mRNA at 7 days after nerve crush injury and this was significantly greater in the Khsrp–/– versus Khsrp+/+ mice (Figure 6A). Given that transcription of the GAP43 gene is increased during axon regeneration, there were only modest, non-significant increases in nerve Gap43 mRNA of the uninjured Khsrp–/– than Khsrp+/+ mice (Figure 6A). Since KHSRP can potentially bind to many different axonal mRNAs, we asked if axonal Snap25 and Fubp1 mRNAs might be affected by Khsrp knockout. Both mRNAs are elevated in cortex and hippocampus of Khsrp–/– mice and the neurites of cortical neurons cultured from embryonic day 18 Khsrp–/– versus Khsrp+/+ mice (65). The Hengst lab reported translation of Snap25 mRNA in central nervous system (CNS) axons promotes synaptogenesis (66), but to our knowledge, potential roles for FUBP1 in neurons have not been tested. Snap25 and Fubp1 mRNAs were detected in sciatic nerve, and their levels were significantly increased in the injured sciatic nerves of Khsrp–/– compared to Khsrp+/+ mice (Figure 6A). In contrast, Hmgb1 mRNA (also called Amphoterin), an mRNA that we have previously shown localizes to axons and whose local translation also supports axon growth (43), did not show any significant difference between Khsrp–/– and Khsrp+/+ nerves (Figure 6A). Notably, Hmgb1 mRNA did not show binding to KHSRP in RIP-Seq analyses (65). Thus, increased levels of some mRNAs encoding proteins linked to axon growth accompany the increased axon regeneration seen in injury-conditioned Khsrp–/– mice, but this does not extend to all mRNAs encoding growth-associated proteins indicating a level of specificity to KHSRP’s role in modulating target neuronal mRNA levels.
Figure 6.

Increased levels of KHSRP target mRNAs in Khsrp–/– neurons. (A) Sciatic nerve levels of Gap43, Snap25 and Fubp1 mRNAs are increased 7 days after sciatic nerve crush in Khsrp–/– compared to Khsrp+/+ mice. In contrast, sciatic nerve Hmgb1 mRNA levels show no change comparing the Khsrp–/– versus Khsrp+/+ mice. Values shown as mean ± SEM (N = 5 mice per genotype; * P ≤ 0.05, ** P ≤ 0.01 and NS for indicated pairs across genotype within condition and & P ≤ 0.05 and && P ≤ 0.01 for crush versus naïve within genotype by Student's t-test). (B) Schematic of expression constructs used for rescue experiments shown in panels C–F. (C) Analyses of soma and axon Gap43 and Fubp1 mRNA levels in Khsrp–/– DRG neurons transfected with GFP, GFP-KHSRP and GFP-KHSRPΔKH4 is shown as mean mRNA copies/ng of total RNA ± SEM after normalization to mitochondrial 12S RNA. Supplemental Figure S6A shows expression levels for GFP, GFP-KHSRP and GFP-KHSRPΔKH4 and Supplemental Figure S6B shows RNA levels for transfected Khsrp+/+ DRG cultures (N = 3 per condition; * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001 and NS = not significant for indicated pairs by Student's t test).(D, E)Representative images of DRG neurons transfected with GFP, GFP-KHSRP or GFP-KHSRPΔKH4 are shown for Khsrp+/+ and Khsrp–/– (schematics for constructs above image columns) are shown in (D) as indicated. Quantification of total axon length/neuron for transfected DRG neurons is shown in (E) as mean ± SEM. Expression of GFP-KHSRP decreases axon outgrowth in both Khsrp+/+ and Khsrp–/– DRGs, but GFP-KHSRPΔKH4 had no significant effect on Khsrp+/+ and only a modest decrease in axon length in Khsrp–/– neurons (N ≥ 30 neurons over three different culture preparations; * P ≤ 0.05, ** P ≤ 0.01 and *** P ≤ 0.001 by one-way ANOVA with Tukey's post-hoc analysis for indicated comparisons) [scale bar = 100 μm].
Previous work has shown that the KH3 and KH4 domains of KHSRP are needed for ARE binding and to promote decay of ARE-containing mRNAs via the cytoplasmic exosome complex (22), and deletion of KH4 attenuated the effects of KHSRP on neurite growth in embryonic CNS neuron cultures (21). To determine if loss of KHSRP’s function in promoting mRNA decay is responsible for the elevations in KHSRP target mRNAs in the Khsrp–/– mice, we transfected Khsrp–/– DRG cultures with full length GFP, GFP-KHSRP, GFP-KHSRPΔKH4 (Figure 6B) and evaluated levels of Gap43 and Fubp1 mRNAs in soma and axon preparations. Gap43 and Fubp1 mRNAs were chosen for these analyses since they showed relatively consistent changes in the sciatic nerve with KHSRP deletion. Immunoblotting confirmed the presence of bands at the predicted molecular weights for GFP, GFP-KHSRP and GFP-KHSRPΔKH4 proteins (data not shown) and comparable expression levels in the transfected DRG neurons based on GFP fluorescence (Supplemental Figure S6A). Gap43 mRNA levels in the Khsrp–/– DRG axons and soma significantly declined with GFP-KHSRP expression, but no significant change was seen in the axons with GFP-KHSRPΔKH4 expression while there was a significant increase in soma Gap43 mRNA levels with GFP-KHSRPΔKH4 expression (Figure 6C). Similarly, both axonal and soma Fubp1 mRNA levels were decreased in Khsrp–/– DRG neurons expressing in GFP-KHSRP. In contrast to Gap43 mRNA, GFP-KHSRPΔKH4 expression significantly decreased both axonal and soma Fubp1 mRNA levels in the Khsrp–/– DRG cultures (Figure 6C). Gap43 and Fubp1 mRNA levels in cell bodies and axons of transfected Khsrp+/+ DRGs followed similar trends with Gap43 mRNA decreased by transfection GFP-KHSRP but not GFP-KHSRPΔKH4 and Fubp1 mRNA decreased by both GFP-KHSRP and GFP-KHSRPΔKH4 (Supplemental Figure S6B). These data indicate that KHSRP promotes Gap43 mRNA decay in both axons and soma, but other functions of KHSRP account for its depletion of Fubp1 mRNA in the DRG neurons.
Since KHSRP’s KH4 domain destabilized the growth associated Gap43 mRNA, we asked if the KH4 domain is needed for KHSRP’s axon growth attenuating effects. While GFP-KHSRP expression reversed the axon growth promoting effects of KHSRP deletion in the Khsrp–/– DRGs, GFP-KHSRPΔKH4 expression only modestly decreased axon growth in the Khsrp–/– DRGs (Figure 6D). Consistent with effects of these constructs on KHSRP target mRNA levels in the Khsrp+/+ DRGs, GFP-KHSRP expression decreased axon growth in the Khsrp+/+ DRGs but expression GFP-KHSRPΔKH4 had no effect on axon growth in these wild type DRG neurons (Figure 6E). Taken together these data indicate that the RNA degradation activity of KHSRP contributes to slowing of axon growth by KHSRP and decreased axonal levels of Gap43 mRNA but not Fubp1 mRNA.
Effects of KHSRP knockout on axon regeneration are neuron-intrinsic
The studies above suggest a neuron-intrinsic role for KHSRP in slowing PNS axon regeneration. However, we relied on a constitutive KHSRP knockout mouse (24), where Khrsp expression is lost in every tissue and cell type. Since KHSRP is ubiquitously expressed and previous studies have shown altered interleukin and cytokine expression in Khsrp–/– mice in non-neuronal cells (24,47), we could not completely exclude that the accelerated regeneration in Khsrp–/– mice derived through KHSRP’s roles in non-neuronal cells. To test for neuronal specific functions in vivo, we generated a conditional Khsrp knockout mouse with LoxP sites surrounding exons 2–6 of the mouse KHSRP gene (Khsrpfl/fl). DRGs cultured from the Khsrpfl/fl mice were transduced with AAV2-CMV-Cre virus in vitro for KHSRP deletion. This resulted in approximately 50% reduction in Khsrp mRNA in the DRG cultures by RTddPCR (Figure 7A). In our hands, the AAV2 preparations show preferential transduction of the neurons in these murine DRG cultures that contain sensory neurons, Schwann cells, and some fibroblasts. Consistent with this, immunofluorescence of the DRG cultures showed near complete loss of KHSRP in the neurons but continued expression of KHSRP in the non-neuronal cells (Figure 7B, C). In keeping with findings in the Khsrp–/– DRG cultures shown in Figure 2, the AAV2-CMV-Cre-GFP transduced Khsrpfl/fl DRG cultures showed significantly increased axon lengths compared to AAV2-CMV-GFP transduced DRGs but no change in axon branching (Figure 7D, E). Thus, selectively depleting KHSRP from adult sensory neurons increases axon growth, indicating neuron-intrinsic effects for KHSRP in slowing axon growth rates in vitro.
Figure 7.

Axon growth promotion from loss of neuronal KHSRP. (A–C) DRGs cultured from Khsrpfl/fl mice in (A) show reduced Khsrp mRNA by RTddPCR after transduction with AAV2-CMV-GFP-Cre compared to AAV2-CMV-GFP. AAV2-CMV-GFP-Cre transduction of wild type DRGs had no effect on Khsrp mRNA levels. By immunofluorescence where only neuronal KHSRP levels were assessed in (B), the AAV2-CMV-GFP-Cre transduced Khsrpfl/fl DRGs showed greater than 80% reduction in KHSRP signals. Representative immunofluorescent images in (C) show relative absence of KHSRP signals in neuronal cell body and axons of DRGs, but prominent signals in adjacent Schwann cells of the AAV2-CMV-GFP-Cre transduced Khsrpfl/fl dissociated DRG culture (N = 6 culture preparations for each condition for (A) and N = 29 neurons in three separate transfections for (B); *** P ≤ 0.005 and **** P ≤ 0.001 for indicated pairs by Students t-test) [scale bar = 10 μm].(D, E)Representative immunofluorescent images for NF in AAV2-CMV-GFP versus AAV2-CMV-GFP-Cre transduced Khsrpfl/fl mice is shown in (D). Quantification of axon length and axon branching (E) as mean ± SEM indicate show significantly increased axon growth with Cre-driven deletion of KHSRP in the Khsrpfl/fl mouse DRGs (N ≥ 75 neurons over three separate culture preparations/transductions for each condition; *** P ≤ 0.005 and NS = not significant by Student's t-test) [scale bar = 100 μm]. (F) Schematic with time line for viral transduction of Khsrpfl/fl mice followed by double sciatic nerve crush lesion as used in Figure 3. Not that the AAV2-CMV-GFP-Cre injection site is separated from both nerve crush sites by ∼0.75 and ∼1.25 cm. The animals were transduced on day 0, underwent distal nerve crush on day 10 (crush # 1), and underwent proximal nerve crush on day 17 (crush # 2). Regeneration was evaluated 3 days later. (G, H) Representative confocal images for KHSRP and NF with DAPI staining of sciatic nerve from AAV2-CMV-GFP-Cre transduced wild type and Khsrpfl/fl mice after nerve crush injury # 2 are shown in (G) as indicated. (H) shows regeneration indices for AAV2-CMV-GFP-Cre transduced wild type and Khsrpfl/fl mice 3 days after nerve crush injury # 2. Supplemental Figure S7 shows SCG10 and GFP immunofluorescence to compare regeneration between the AAV2-CMV-GFP-Cre transduced wild type and Khsrpfl/fl mice (N = 6 mice per genotype; * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.005 and **** P ≤ 0.001 by two-way ANOVA with Bonferroni post-hoc analysis) [scale bar = 10 μm].
We used an in vivo AAV2-CMV-Cre-GFP transduction to determine if deletion of neuronal Khsrp alleles from adult mice affects nerve regeneration. For this, the AAV2 preparations were injected into the proximal sciatic nerve in a high salt solution that facilitates retrograde transport of the viral particles (26). We used the same conditioned-crush lesion as shown in Figure 3, with initial crush injury performed 10 days after AAV transduction and placing the first crush site ∼1.25 cm from the transduction site plus the second crush site at ∼0.75 cm distal from the transduction site (Figure 7F). Immunofluorescence showed prominent KHSRP signals in the NF+/GFP- axons and no detectable KHSRP signals in the SCG10+/GFP+ axons; notably, Schwann cells in the nerve showed prominent KHSRP signals in their nuclei indicating the AAV2-CMV-Cre-GFP did not transduce these non-neuronal cells distant from the injection site (Figure 7G). Consistent with findings in the Khsrp–/– mice shown in Figure 3, axons of the Cre-GFP expressing neurons of the Khsrpfl/fl mice showed significantly higher regeneration indices compared to wild type mice transduced with the same AAV2-CMV-Cre-GFP preparations (Figure 7H; Supplemental Figure S7). Taken together, these data indicate that the local increase in axonal KHSRP attenuates PNS axon regeneration selectively through neuron-intrinsic mechanisms.
DISCUSSION
RBPs play critical roles in determining neuronal protein levels through post-transcriptional control mechanisms. Since one mRNA can generate multiple copies of a protein, RNA stability needs to be tightly regulated. HuD and KHSRP both bind to ARE-containing mRNAs, with HuD increasing mRNA stability and KHSRP promoting mRNA decay (51). Here, we show that PNS axons contain many different RBPs whose levels change after injury, including KHSRP. Axonal KHSRP protein levels rapidly increase in peripheral nerves following axotomy through intra-axonal translation of Khsrp mRNA. Since Khsrp–/– mice show increased axonal levels of Gap43, Snap25 and Fubp1 mRNAs compared to Khsrp+/+ mice, the increase in KHSRP levels following injury decreases the levels of KHSRP target mRNAs in injured sciatic nerves. Interestingly, we find that the decline in Gap43 mRNA levels with KHSRP expression requires an intact KH4 domain that is essential for the RNA degradation role of KHSRP (22), while Fubp1 mRNA levels appear to be regulated by other functions of KHSRP. Deletion of the Khsrp gene increases axon growth. Axon regeneration was further accelerated in vivo in the absence of neuronal KHSRP comparing Khsrp deleted to wild type mice under injury-conditioned settings, where KHSRP is elevated in axons. Neuronal specific knockout confirmed that KHSRP’s growth effects are neuron intrinsic. The growth attenuating functions of KHSRP also require an intact KH4 domain, pointing KHSRP’s RNA decay promoting functions for regulating axon growth. Together, our data point to injury-induced increase in axonal KHSRP as an axon-intrinsic mechanism that serves to slow axon regeneration by affecting mRNA stability.
The targeted proteomics approach used here uncovered 84 RBPs in sciatic nerve axoplasm, with many beyond KHSRP showing increased or decreased levels after nerve injury and during regeneration. Since the axoplasm isolates used here rely on extrusion in detergent-free conditions (46), we likely missed many RBPs that are associated with cytoskeleton or the axoplasmic membrane. HuD/ELAVL4 has previously been reported to fractionate with the cytoskeleton in rat hippocampus (67). So, this may explain the decrease in HuD/ELAVL4 levels detected in the sciatic nerve axoplasm after injury shown in Figure 1 compared to our previous immunofluorescent analyses showing that axonal HuD/ELAVL4 levels increase during regeneration (20). Despite limitations in methodology and amount of starting materials for the axoplasm, we have substantially increased the number of known axonal RBPs with the PRM approach used here. Other axoplasm RBPs identified here as increasing or decreasing with sciatic nerve injury and during regeneration could contribute to axon growth. For example, fragile X-related protein 1 (FXR1) showed a remarkable increase after sciatic nerve crush in Figure 1A-C, and this protein has been shown to associate with RNA granules containing fragile x mental retardation protein (FMRP) in CNS axons (68,69). Though not identified here, FMRP is well characterized as a translational modulator in dendrites and it regulates translation of microtubule-associated protein 1b (MAP1b) in axonal growth cones (70). Additionally, a number of hnRNPs linked to RNA splicing were previously shown to localize into axons and bind to axonal mRNA localization motifs, including hnRNP H1 and F that bind to Hmgb1 mRNA localization motif and whose depletion from adult DRGs decreases axon outgrowth (42). Our PRM data also show a decline in Splicing Factor Proline and Glutamine Rich (SFPQ) that has been linked to survival of axons through its role in transport of Bclw mRNA into axons (71,72). It will be of high interest to determine how the different RBPs impact axon growth after injury as well as the mechanisms underlying their change in axonal levels. Notably analyses of axoplasm hnRNP H1 and F immunoprecipitates pointed to existence of mRNA regulons encoding growth-associated proteins binding to axonal hnRNP H1 and F (42). So, it is likely that the axonal RBPs reported here can post-transcriptionally regulate many axonal mRNAs. It is appealing to hypothesize that those increased RBPs in injured and regenerating axons support axon growth, so it is intriguing that KHSRP does the opposite. It has been reported that intra-axonal translation decreases as axons reach their target tissues and form synapses (73). Further, ribosome profiling for axonal mRNAs has shown changes in which mRNAs are translated in retinal ganglion cell axons as they reach the optic tectum and form synapses (1). Thus, one can speculate that the elevated KHSRP levels contribute to changes in axonal mRNA populations associated with different stages of regeneration, with the nascently synthesized axonal KHSRP locally interacting with target mRNAs and possibly displacing other RBPs from those transcripts.
Much interest has been devoted to understanding how mRNAs are transported into axons and dendrites. RBPs that drive subcellular mRNA localization sometimes also regulate translation of those mRNA cargos (4). To our knowledge, KHSRP is the first example for localized translational regulation of an RBP mRNA whose protein product goes on to modify axon growth potential. This emphasizes that localized translation can be used to modify the fate of other subcellular mRNAs. The initial increase in axonal KHSRP protein through translational upregulation of its axonal mRNA shows similar kinetics to translational induction of injury-associated mRNAs following axotomy. mRNAs encoding Importin β1, Vimentin, RanBP1, Stat3, mTor and Calreticulin (CALR) are translated in axons within the first six hours following PNS nerve crush injury (39,40,56,74,75). Some of those locally synthesized injury-associated proteins help to signal retrogradely to the soma for changing gene expression, including transport of transcription factors, to support the injured neuron's survival and axon regeneration (41). A retrograde calcium wave has also been shown to support axon regeneration by altering neuronal gene expression after PNS injury through epigenetic mechanisms (76). Together, these changes in gene expression are thought to contribute to the enhanced regeneration seen in injury-conditioned neurons. Consistent with this, the accelerated axon growth seen upon culture of in vivo injury-conditioned DRG neurons requires mRNA translation but not new gene expression (25,50). Notably, translation of Calr mRNA in injured axons is needed for initiating axon growth so the axonal CALR protein serves a localized function in injured axons (55). Our data show that axonal Khsrp is an injury-response mRNA whose intra-axonal translation is activated through Ca2+ → PERK → eIF2αPS51 pathway similar to Calr mRNA (55,77). In contrast to axonal Calr mRNA, the axonal Khsrp protein product attenuates rather than facilitates axon regeneration. This nerve regeneration slowing effect of KHSRP undoubtedly requires other factors in the nerve including components of the cytoplasmic exosome for RNA degradation and possibly other mediators interacting with KHSRP.
The difference between in vivo axon regeneration for the Khsrp deleted compared to wild type mice was most apparent in the injury-conditioned setting where axonal KHSRP levels are already increased in the wild type mice at the time of the second injury. Though axon growth from the cultured DRGs is overall increased by neuronal Khsrp deletion, in vivo injury-conditioning diminished the differences between the Khsrp deleted and wild type DRGs when cultured. This could represent a ceiling or threshold effect, where the transcriptional changes occurring after injury-conditioning are able to overcome growth-attenuating effects of KHSRP. Consistent with this notion, exogenously manipulating neuronal levels of several growth-associated proteins that are part of the transcriptional response to injury-conditioning has been shown to induce corresponding changes in axon growth potential, with the injury-conditioning effect blocked by genetic knockouts and depletions or increased axon growth from naive neurons upon overexpression of growth-associated proteins (78). For example, overexpression of GAP43 can increase axon growth in naive neurons (79). Notably, increasing axonally targeted Gap43 mRNA levels in naive DRG neurons shifts them to an elongating growth phenotype typical of injury-conditioned DRG neurons (80). Though axonal levels of Gap43, Snap25 and Fubp1 mRNAs all increased in wild type mice after nerve injury, this clearly was not sufficient to accelerate axon growth considering that loss of KHSRP exaggerated these increases and accelerates axon growth above what is seen in injury-conditioned mice.
Rather than a transcriptional effect, our data support the hypothesis that axonal KHSRP levels accumulate after axotomy through post-transcriptional mechanisms. This constitutes a novel axon-intrinsic mechanism to slow axon regeneration over time. Consistent with this idea of accumulating effects of KHSRP, there was no significant difference between single injury Khsrp–/– and wild type mice at 7 days but the single injury Khsrp–/– mice showed significantly greater regeneration at 14 d than the wild type mice (Figure 3A). Additionally, in vitro axon regeneration was much greater in Khsrp–/– and wild type DRG cultures when axon shafts were severed (Supplementary Figure S3B, C) as opposed to initiation of axon growth that is seen after dissociation of the ganglia. Naive DRG neurons are known to transition to the elongating axon growth phenotype typical of the injury-conditioned neurons if provided a 12–24 h in vitro period for new gene expression after culturing (50). The assessment for regeneration of sheared axons in cultured DRGs used here in Supplementary Figure S3B-C was designed with this in vitro priming period in mind. Others have used a similar in vitro priming and then replated the DRGs after the priming from dissociation and culture (81). In contrast to our approach, replated neurons reinitiate axon growth from the soma. So, the replated neurons are starting over without the effect of axons with elevated KHSRP, similar to the culture of injury-conditioned neurons used in Figure 2. Taken together, our data support a model where accumulation of KHSRP in the axonal compartment slows axon regeneration through post-transcriptional mechanisms. The increase in axon regeneration from injury-conditioned KHSRP deleted mice raises the possibility of targeting localized post-transcriptional mechanisms to increase axon regeneration rates beyond the accelerated rates normally seem in injury-conditioned neurons.
KHSRP has been shown to play roles in RNA splicing, trafficking and degradation as well as miRNA biogenesis (22,44,45,48). Our analyses of KHSRP target mRNA levels in sciatic nerves of the Khsrp–/– mice indicate that loss of KHSRP increases axonal levels of those mRNAs. miRNAs are clearly linked to stability of mRNAs, and their precursors (pre-miRNAs) have been shown to localize into PNS axons, with PNS nerve injury triggering localized processing of some axonal pre-miRNAs into mature miRNAs (82). So, the increase in axonal KHSRP could affect axonal mRNA levels by promoting miRNA maturation. In our hands, levels of previously reported KHSRP target miRNAs (45) showed no changes when comparing DRG cultures from Khsrp–/– vs. Khsrp+/+ mice (data not shown). However, our data argue for a direct effect of KHSRP on mRNA stability rather than an indirect effect through miRNAs or other mediators underlying the axon growth attenuation by KHSRP. KHSRP’s KH3 and KH4 are implicated in promoting mRNA decay by binding to ARE-containing mRNAs and recruiting components of the cytoplasmic exosome complex used for RNA degradation (22). We find that KHSRP’s function in slowing axon regeneration and decreasing axonal Gap43 mRNA in adult DRG neurons require an intact KH4 domain that has been shown to bind to ARE-containing mRNAs. As we see with Fubp1 mRNA levels being depleted by KHSRPΔKH4 expression, KHSRP can clearly affect neuronal mRNA levels through other mechanisms. A recent combination of RNA profiling studies of Khsrp–/– vs. Khsrp+/+ mice brains with mRNAs co-immunoprecipitating with KHSRP from mouse brain uncovered 527 KHSRP bound mRNAs whose levels are elevated in KHSRP knockout brain, including Gap43, Fubp1 and Snap25 mRNAs (65). Thus, KHSRP likely binds to many other axonal mRNAs beyond those tested here to regulate their stability, and the proteins encoded by those mRNAs could also impact axon growth capacity. Recent work in neonatal DRG neurons showed that KHSRP binds to the long non-coding RNA ALAE in axons and this interaction prevents KHSRP’s binding to Gap43 mRNA. In contrast to our observations, translation of axonal Gap43 mRNA was altered by the KHSRP-ALAE RNA competion (83) rather than increased Gap43 mRNA levels that we see with deletion of KHSRP. We cannot completely exclude secondary effects from KHSRP deletion used here as contributing to axon growth promotion. Consistent with this possibility, the work from Olguin et al. reported ∼1400 mRNAs showing altered levels in brains of Khsrp–/– but no binding to KHSRP in RIP assays (65). Some of these mRNAs may represent indirect effects from the protein products altered by KHSRP mRNA decay promotion. Also, it should be noted that KHSRP deletion does not affect levels of all regeneration-associated mRNAs, since it did not affect Hmgb1 mRNA levels and intra-axonal translation of Hmgb1 enhances axon growth (43). RNA profiling studies will be needed to determine the RNA regulons that are directly regulated by KHSRP’s decay-promoting domains vs. those that are regulated by other domains of KHSRP or indirectly modified by changing levels of proteins encoded by KHSRP mRNA targets. Nonetheless, our data point to the KHSRP’s role in promoting mRNA decay as a key determinant of axon regeneration in vivo.
DATA AVAILABILITY
Proteomics data for axonal RBPs has been uploaded to PanoramaWeb Public and can be accessed at https://panoramaweb.org/axon-rbps.url.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs Brian Popko and Yanan Cheng for guidance on testing the role of eIF2α phosphorylation in regulation of Khsrp mRNA translation. J.L.T. is the incipient South Carolina SmartState Chair in Childhood Neurotherapeutics at the University of South Carolina.
Notes
Present address: Priyanka Patel, St. Jude Children's Research Hospital, Memphis, TN 38105, USA.
Present address: Seung Joon Lee, Biogen, Cambridge, MA 02142, USA.
Present address: Devon Cassidy, School of Medicine, University of Michigan, Ann Arbor, MI 48109, USA.
Present address: Amar N. Kar, GlaxoSmithKline, Collegeville, PA 19426, USA.
Contributor Information
Priyanka Patel, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Courtney N Buchanan, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Matthew D Zdradzinski, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Pabitra K Sahoo, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Amar N Kar, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Seung Joon Lee, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Lauren S Vaughn, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Anatoly Urisman, Department of Pharmaceutical Sciences, University of California, San Francisco, CA 94143, USA.
Juan Oses-Prieto, Department of Pharmaceutical Sciences, University of California, San Francisco, CA 94143, USA.
Michela Dell’Orco, Department of Neurosciences, University of New Mexico School of Health Sciences, Albuquerque, NM 87131, USA.
Devon E Cassidy, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Irene Dalla Costa, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Sharmina Miller, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Elizabeth Thames, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Terika P Smith, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
Alma L Burlingame, Department of Pharmaceutical Sciences, University of California, San Francisco, CA 94143, USA.
Nora Perrone-Bizzozero, Department of Neurosciences, University of New Mexico School of Health Sciences, Albuquerque, NM 87131, USA.
Jeffery L Twiss, Department of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wings for Life Spinal Cord Injury Research Foundation [WFL-US-09/18 to P.P.]; National Institutes of Health [R01-NS089633 to J.L.T., N.P.B., K01-NS105879 to T.P.S.]; Dr Miriam and Sheldon G. Adelson Medical Research Foundation (to J.L.T. and A.L.B.); South Carolina Spinal Cord Injury Research Fund [2019 PD-02 to P.K.S.]; SC EPSCoR Stimulus Grant Program [18-SR04 to J.L.T.]. Funding for open access charge: NIH + internal funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Shigeoka T., Jung H., Jung J., Turner-Bridger B., Ohk J., Lin J.Q., Amieux P.S., Holt C.E.. Dynamic axonal translation in developing and mature visual circuits. Cell. 2016; 166:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong H.H., Lin J.Q., Strohl F., Roque C.G., Cioni J.M., Cagnetta R., Turner-Bridger B., Laine R.F., Harris W.A., Kaminski C.F.et al.. RNA docking and local translation regulate site-specific axon remodeling in vivo. Neuron. 2017; 95:852–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koppers M., Cagnetta R., Shigeoka T., Wunderlich L.C., Vallejo-Ramirez P., Qiaojin Lin J., Zhao S., Jakobs M.A., Dwivedy A., Minett M.S.et al.. Receptor-specific interactome as a hub for rapid cue-induced selective translation in axons. Elife. 2019; 8:e48718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dalla Costa I., Buchanan C., Zdradzinski M.D., Sahoo P.K., Smith T.P., Thames E., Kar A.N., Twiss J.L. Functional platforms for organizing axonal mRNA transport and translation. Nat. Rev. Neurosci. 2020; 22:77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith T.P., Sahoo P.K., Kar A.N., Twiss J.L.. Intra-axonal mechanisms driving axon regeneration. Brain Res. 2020; 1740:146864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hafner A.S., Donlin-Asp P.G., Leitch B., Herzog E., Schuman E.M.. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 2019; 364:eaau3644. [DOI] [PubMed] [Google Scholar]
- 7. Poulopoulos A., Murphy A.J., Ozkan A., Davis P., Hatch J., Kirchner R., Macklis J.D.. Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature. 2019; 565:356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baumann S., Komissarov A., Gili M., Ruprecht V., Wieser S., Maurer S.P.. A reconstituted mammalian APC-kinesin complex selectively transports defined packages of axonal mRNAs. Sci. Adv. 2020; 6:eaaz1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cioni J.M., Lin J.Q., Holtermann A.V., Koppers M., Jakobs M.A.H., Azizi A., Turner-Bridger B., Shigeoka T., Franze K., Harris W.A.et al.. Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell. 2019; 176:56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shigeoka T., Koppers M., Wong H.H., Lin J.Q., Cagnetta R., Dwivedy A., de Freitas Nascimento J., van Tartwijk F.W., Strohl F., Cioni J.M.et al.. On-Site ribosome remodeling by locally synthesized ribosomal proteins in axons. Cell Rep. 2019; 29:3605–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kar A., Lee S., Twiss J.. Expanding axonal transcriptome brings new functions for axonally synthesized proteins in health and disease. Neuroscientist. 2018; 24:111–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aschrafi A., Kar A.N., Natera-Naranjo O., MacGibeny M.A., Gioio A.E., Kaplan B.B.. MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery. Cell Mol. Life Sci. 2012; 69:4017–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colak D., Ji S.J., Porse B.T., Jaffrey S.R.. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell. 2013; 153:1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hengst U., Cox L.J., Macosko E.Z., Jaffrey S.R.. Functional and selective RNA interference in developing axons and growth cones. J. Neurosci. 2006; 26:5727–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murashov A.K., Chintalgattu V., Islamov R.R., Lever T.E., Pak E.S., Sierpinski P.L., Katwa L.C., Van Scott M.R.. RNAi pathway is functional in peripheral nerve axons. FASEB J. 2007; 21:656–670. [DOI] [PubMed] [Google Scholar]
- 16. Vargas J.N., Kar A.N., Kowalak J.A., Gale J.R., Aschrafi A., Chen C.Y., Gioio A.E., Kaplan B.B.. Axonal localization and mitochondrial association of precursor microRNA 338. Cell Mol. Life Sci. 2016; 73:4327–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beckel-Mitchener A.C., Miera A., Keller R., Perrone-Bizzozero N.I.. Poly(A) tail length-dependent stabilization of GAP-43 mRNA by the RNA-binding protein huD. J. Biol. Chem. 2002; 277:27996–28002. [DOI] [PubMed] [Google Scholar]
- 18. Bolognani F., Merhege M.A., Twiss J., Perrone-Bizzozero N.I.. Dendritic localization of the RNA-binding protein HuD in hippocampal neurons: association with polysomes and upregulation during contextual learning. Neurosci. Lett. 2004; 371:152–157. [DOI] [PubMed] [Google Scholar]
- 19. Smith C.L., Afroz R., Bassell G.J., Furneaux H.M., Perrone-Bizzozero N.I., Burry R.W.. GAP-43 mRNA in growth cones is associated with HuD and ribosomes. J. Neurobiol. 2004; 61:222–235. [DOI] [PubMed] [Google Scholar]
- 20. Yoo S., Kim H.H., Kim P., Donnelly C.J., Kalinski A.L., Vuppalanchi D., Park M., Lee S.J., Merianda T.T., Perrone-Bizzozero N.I.et al.. A hud-ZBP1 ribonucleoprotein complex localizes GAP-43 mRNA into axons through its 3' untranslated region AU-rich regulatory element. J. Neurochem. 2013; 126:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bird C.W., Gardiner A.S., Bolognani F., Tanner D.C., Chen C.Y., Lin W.J., Yoo S., Twiss J.L., Perrone-Bizzozero N.. KSRP modulation of GAP-43 mRNA stability restricts axonal outgrowth in embryonic hippocampal neurons. PLoS One. 2013; 8:e79255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gherzi R., Lee K.Y., Briata P., Wegmuller D., Moroni C., Karin M., Chen C.Y.. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell. 2004; 14:571–583. [DOI] [PubMed] [Google Scholar]
- 23. Chou C.F., Mulky A., Maitra S., Lin W.J., Gherzi R., Kappes J., Chen C.Y.. Tethering KSRP, a decay-promoting AU-rich element-binding protein, to mRNAs elicits mRNA decay. Mol. Cell Biol. 2006; 26:3695–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin W.J., Zheng X., Lin C.C., Tsao J., Zhu X., Cody J.J., Coleman J.M., Gherzi R., Luo M., Townes T.M.et al.. Posttranscriptional control of type i interferon genes by KSRP in the innate immune response against viral infection. Mol. Cell Biol. 2011; 31:3196–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Twiss J.L., Smith D.S., Chang B., Shooter E.M.. Translational control of ribosomal protein L4 mRNA is required for rapid neurite regeneration. Neurobiol. Dis. 2000; 7:416–428. [DOI] [PubMed] [Google Scholar]
- 26. Sahoo P.K., Lee S.J., Jaiswal P.B., Alber S., Kar A.N., Miller-Randolph S., Thames E., Taylor E.E., Smith T., Singh B.et al.. Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Commun. 2018; 9:3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cavalli V., Kujala P., Klumperman J., Goldstein L.S.. Sunday driver links axonal transport to damage signaling. J. Cell Biol. 2005; 168:775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Twiss J., Smith D., Chang B., Shooter E.. Translational control of ribosomal protein L4 is required for rapid neurite extension. Neurobiol. Dis. 2000; 7:416–428. [DOI] [PubMed] [Google Scholar]
- 29. Zheng J.Q., Kelly T.K., Chang B., Ryazantsev S., Rajasekaran A.K., Martin K.C., Twiss J.L.. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J. Neurosci. 2001; 21:9291–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willis D.E., van Niekerk E.A., Sasaki Y., Mesngon M., Merianda T.T., Williams G.G., Kendall M., Smith D.S., Bassell G.J., Twiss J.L.. Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J. Cell Biol. 2007; 178:965–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aakalu G., Smith W.B., Nguyen N., Jiang C., Schuman E.M.. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001; 30:489–502. [DOI] [PubMed] [Google Scholar]
- 32. Urisman A., Levin R.S., Gordan J.D., Webber J.T., Hernandez H., Ishihama Y., Shokat K.M., Burlingame A.L.. An optimized chromatographic strategy for multiplexing in parallel reaction monitoring mass spectrometry: insights from quantitation of activated kinases. Mol. Cell Proteomics. 2017; 16:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J.. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010; 26:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma V., Eckels J., Schilling B., Ludwig C., Jaffe J.D., MacCoss M.J., MacLean B.. Panorama public: a public repository for quantitative data sets processed in skyline. Mol. Cell Proteomics. 2018; 17:1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merianda T.T., Gomes C., Yoo S., Vuppalanchi D., Twiss J.L.. Axonal localization of neuritin/CPG15 mRNA in neuronal populations through distinct 5' and 3' UTR elements. J. Neurosci. 2013; 33:13735–13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin J.E., Miller B.R., Babetto E., Cho Y., Sasaki Y., Qayum S., Russler E.V., Cavalli V., Milbrandt J., DiAntonio A.. SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3696–E3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maimon R., Ionescu A., Bonnie A., Sweetat S., Wald-Altman S., Inbar S., Gradus T., Trotti D., Weil M., Behar O.et al.. miR126-5p Downregulation facilitates axon degeneration and NMJ disruption via a non-cell-autonomous mechanism in ALS. J. Neurosci. 2018; 38:5478–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kalinski A.L., Sachdeva R., Gomes C., Lee S.J., Shah Z., Houle J.D., Twiss J.L.. mRNAs and Protein synthetic machinery localize into regenerating spinal cord axons when they are provided a substrate that supports growth. J. Neurosci. 2015; 35:10357–10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hanz S., Perlson E., Willis D., Zheng J.Q., Massarwa R., Huerta J.J., Koltzenburg M., Kohler M., van-Minnen J., Twiss J.L.et al.. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003; 40:1095–1104. [DOI] [PubMed] [Google Scholar]
- 40. Yudin D., Hanz S., Yoo S., Iavnilovitch E., Willis D., Gradus T., Vuppalanchi D., Segal-Ruder Y., Ben-Yaakov K., Hieda M.et al.. Localized regulation of axonal rangtpase controls retrograde injury signaling in peripheral nerve. Neuron. 2008; 59:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rishal I., Fainzilber M.. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014; 15:32–42. [DOI] [PubMed] [Google Scholar]
- 42. Lee S., Sahoo P., Oses-Prieto J., Kawaguchi R., Kar A., Oliver D., Chand S., Shtutman M., Rozenbaum M., Miller-Randolph S.et al.. hnRNPs binding to the axonal localization motifs of Nrn1 and HMGB1 mRNAs define growth-associated RNA regulons. Mol. Cell Proteomics. 2018; 17:2091–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Merianda T.T., Coleman J., Kim H.H., Kumar Sahoo P., Gomes C., Brito-Vargas P., Rauvala H., Blesch A., Yoo S., Twiss J.L.. Axonal amphoterin mRNA is regulated by translational control and enhances axon outgrowth. J. Neurosci. 2015; 35:5693–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pan F., Huttelmaier S., Singer R.H., Gu W.. ZBP2 facilitates binding of ZBP1 to beta-actin mRNA during transcription. Mol. Cell Biol. 2007; 27:8340–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trabucchi M., Briata P., Garcia-Mayoral M., Haase A.D., Filipowicz W., Ramos A., Gherzi R., Rosenfeld M.G.. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009; 459:1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rishal I., Michaelevski I., Rozenbaum M., Shinder V., Medzihradszky K.F., Burlingame A.L., Fainzilber M.. Axoplasm isolation from peripheral nerve. Dev. Neurobiol. 2010; 70:126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li X., Lin W.J., Chen C.Y., Si Y., Zhang X., Lu L., Suswam E., Zheng L., King P.H.. KSRP: a checkpoint for inflammatory cytokine production in astrocytes. Glia. 2012; 60:1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Min H., Turck C.W., Nikolic J.M., Black D.L.. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 1997; 11:1023–1036. [DOI] [PubMed] [Google Scholar]
- 49. Briata P., Chen C.Y., Ramos A., Gherzi R.. Functional and molecular insights into KSRP function in mRNA decay. Biochim. Biophys. Acta. 2013; 1829:689–694. [DOI] [PubMed] [Google Scholar]
- 50. Smith D.S., Skene P.. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 1997; 17:646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. García-Mauriño S.M., Rivero-Rodríguez F., Velázquez-Cruz A., Hernández-Vellisca M., Díaz-Quintana A., De la Rosa M.A., Díaz-Moreno I.. RNA binding protein regulation and cross-talk in the control of AU-rich mRNA fate. Front. Mol. Biosci. 2017; 4:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Willis D.E., Twiss J.L.. Profiling axonal mRNA transport. Methods Mol. Biol. 2011; 714:335–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McQuarrie I.G., Grafstein B., Gershon M.D.. Axonal regeneration in the rat sciatic nerve: effect of a conditioning lesion and of dbcAMP. Brain Res. 1977; 132:443–453. [DOI] [PubMed] [Google Scholar]
- 54. Perry R.B., Doron-Mandel E., Iavnilovitch E., Rishal I., Dagan S.Y., Tsoory M., Coppola G., McDonald M.K., Gomes C., Geschwind D.H.et al.. Subcellular knockout of importin beta1 perturbs axonal retrograde signaling. Neuron. 2012; 75:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pacheco A., Merianda T.T., Twiss J.L., Gallo G.. Mechanism and role of the intra-axonal calreticulin translation in response to axonal injury. Exp. Neurol. 2020; 323:113072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Terenzio M., Koley S., Samra N., Rishal I., Zhao Q., Sahoo P.K., Urisman A., Marvaldi L., Oses-Prieto J.A., Forester C.et al.. Locally translated mTOR controls axonal local translation in nerve injury. Science. 2018; 359:1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barrientos S.A., Martinez N.W., Yoo S., Jara J.S., Zamorano S., Hetz C., Twiss J.L., Alvarez J., Court F.A.. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 2011; 31:966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Forester C.M., Zhao Q., Phillips N.J., Urisman A., Chalkley R.J., Oses-Prieto J.A., Zhang L., Ruggero D., Burlingame A.L.. Revealing nascent proteomics in signaling pathways and cell differentiation. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:2353–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sahoo P., Kar A., Samra N., Terenzio M., Patell P., Lee S., Miller S., Thames E., Jones B., Kawaguchi R.et al.. A translational switch drives axonal stress granule disassembly through casein kinase 2α. Curr. Biol. 2020; 30:4882–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Onate M., Catenaccio A., Martinez G., Armentano D., Parsons G., Kerr B., Hetz C., Court F.A.. Activation of the unfolded protein response promotes axonal regeneration after peripheral nerve injury. Sci. Rep. 2016; 6:21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brostrom M.A., Brostrom C.O.. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium. 2003; 34:345–363. [DOI] [PubMed] [Google Scholar]
- 62. Harding H.P., Zhang Y., Bertolotti A., Zeng H., Ron D.. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000; 5:897–904. [DOI] [PubMed] [Google Scholar]
- 63. Van der Zee C.E., Nielander H.B., Vos J.P., Lopes da Silva S., Verhaagen J., Oestreicher A.B., Schrama L.H., Schotman P., Gispen W.H.. Expression of growth-associated protein B-50 (GAP43) in dorsal root ganglia and sciatic nerve during regenerative sprouting. J. Neurosci. 1989; 9:3505–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Skene J.H. Axonal growth-associated proteins. Annu. Rev. Neurosci. 1989; 12:127–156. [DOI] [PubMed] [Google Scholar]
- 65. Olguin S.L., Patel P., Dell’Orco M., Gardiner A.S., Cole R., Buchanan C., Sundara A., Mudge J., Allan A.M., Ortinski P.et al.. The RNA binding protein KHSRP attenuates axonal and dendritic growth, synaptic transmission, and memory consolidation via dysregulation of neuronal gene expression. 2020; bioRxiv doi:25 October 2020, preprint: not peer reviewed 10.1101/2020.10.25.354076. [DOI]
- 66. Batista A.F.R., Martinez J.C., Hengst U.. Intra-axonal synthesis of SNAP25 is required for the formation of presynaptic terminals. Cell Rep. 2017; 20:3085–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pascale A., Gusev P.A., Amadio M., Dottorini T., Govoni S., Alkon D.L., Quattrone A.. Increase of the RNA-binding protein HuD and posttranscriptional up-regulation of the GAP-43 gene during spatial memory. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Akins M.R., Leblanc H.F., Stackpole E.E., Chyung E., Fallon J.R.. Systematic mapping of fragile x granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J. Comp. Neurol. 2012; 520:3687–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chyung E., LeBlanc H.F., Fallon J.R., Akins M.R.. Fragile x granules are a family of axonal ribonucleoprotein particles with circuit-dependent protein composition and mRNA cargos. J. Comp. Neurol. 2018; 526:96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li C., Bassell G.J., Sasaki Y.. Fragile x mental retardation protein is involved in protein synthesis-dependent collapse of growth cones induced by semaphorin-3A. Front. Neural Circuits. 2009; 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cosker K.E., Fenstermacher S.J., Pazyra-Murphy M.F., Elliott H.L., Segal R.A.. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 2016; 19:690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pease-Raissi S.E., Pazyra-Murphy M.F., Li Y., Wachter F., Fukuda Y., Fenstermacher S.J., Barclay L.A., Bird G.H., Walensky L.D., Segal R.A.. Paclitaxel reduces axonal bclw to initiate IP3R1-Dependent axon degeneration. Neuron. 2017; 96:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Costa R.O., Martins H., Martins L.F., Cwetsch A.W., Mele M., Pedro J.R., Tome D., Jeon N.L., Cancedda L., Jaffrey S.R.et al.. Synaptogenesis stimulates a proteasome-mediated ribosome reduction in axons. Cell Rep. 2019; 28:864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ben-Yaakov K., Dagan S.Y., Segal-Ruder Y., Shalem O., Vuppalanchi D., Willis D.E., Yudin D., Rishal I., Rother F., Bader M.et al.. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012; 31:1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Perlson E., Hanz S., Ben-Yaakov K., Segal-Ruder Y., Seger R., Fainzilber M.. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005; 45:715–726. [DOI] [PubMed] [Google Scholar]
- 76. Cho Y., Sloutsky R., Naegle K.M., Cavalli V.. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013; 155:894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vuppalanchi D., Merianda T.T., Donnelly C., Pacheco A., Williams G., Yoo S., Ratan R.R., Willis D.E., Twiss J.L.. Lysophosphatidic acid differentially regulates axonal mRNA translation through 5'UTR elements. Mol. Cell Neurosci. 2012; 50:136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Senger J.B., Verge V.M.K., Chan K.M., Webber C.A.. The nerve conditioning lesion: a strategy to enhance nerve regeneration. Ann. Neurol. 2018; 83:691–702. [DOI] [PubMed] [Google Scholar]
- 79. Bomze H.M., Bulsara K.R., Iskandar B.J., Caroni P., Skene J.H.P.. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat. Neurosci. 2001; 4:38–43. [DOI] [PubMed] [Google Scholar]
- 80. Donnelly C.J., Park M., Spillane M., Yoo S., Pacheco A., Gomes C., Vuppalanchi D., McDonald M., Kim H.H., Merianda T.T.et al.. Axonally synthesized beta-actin and GAP-43 proteins support distinct modes of axonal growth. J. Neurosci. 2013; 33:3311–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rishal I., Kam N., Perry R.B., Shinder V., Fisher E.M., Schiavo G., Fainzilber M.. A motor-driven mechanism for cell-length sensing. Cell Rep. 2012; 1:608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kim H.H., Kim P., Phay M., Yoo S.. Identification of precursor microRNAs within distal axons of sensory neuron. J. Neurochem. 2015; 134:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wei M., Huang J., Li G.W., Jiang B., Cheng H., Liu X., Jiang X., Zhang X., Yang L., Bao L.et al.. Axon-enriched lincRNA ALAE is required for axon elongation via regulation of local mRNA translation. Cell Rep. 2021; 35:109053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data for axonal RBPs has been uploaded to PanoramaWeb Public and can be accessed at https://panoramaweb.org/axon-rbps.url.