
Figure 1.

cDTA-seq and genome-wide transcript half-life estimation. (A) cDTA-seq protocol outline. 4tU is added to dozens of quantified samples to label new RNA molecules. Cells are then immediately fixed with a pre-fixed constant amount of spike-in cells (K. lactis yeast in this case). RNA is extracted, 4tU is alkylated and RNA-seq libraries are prepared, resulting in T→C conversions where 4tU was incorporated. The entire process is performed in a 96 sample format. (B) Transcript-level analysis outline. Read conversion statistics per gene are fitted with a binomial mixture model to estimate the percent of recently-transcribed molecules (pr). Assuming a first-order kinetic model (C - number of cells, M—number of mRNA molecules, 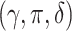 are growth, production, and degradation rates respectively), and assuming steady-state, pr can be translated to transcript half-life given the known labeling period (t). See methods and supplementary material for more details. (C) 4tU conversion is effective, reproducible and measures transcription. Percent of reads (y-axis) along a 4tU time course (x-axis) with a different number of observed T→C conversions (legend, N = 6). Samples exposed to vehicle (DMSO) or the transcription inhibitor thiolutin for 15 min (and labeled with 4tU for 6 min, N = 2). Note that the y-axis begins at 75%, i.e. most reads have no conversions. (D) Binomial Mixture Model (BMM) fits the data. Read conversion statistics are fitted with a 2-component BMM. Each dot represents the number of reads with a certain number of observed Ts and T→C conversions (color as in E). x-axis is the expected number of reads for each (T,T→C) pair assuming the model and the observed #T distribution in the data, the y-axis is the observed number of reads in each (T, T→C) combination. Additional components do not improve the likelihood of the data (Supplementary Figure S1D). (E) Half-life distribution for all yeast transcripts. Assuming steady-state, transcript-specific and global parameters are iteratively fitted, resulting in the half-life of each transcript. The median of the distribution is 8.2', transcripts with a half-life of 45′ or longer are counted in the rightmost bin. (F) Examples of estimated pr along the time course. The individually estimated pr per time point and replicate (N = 6) for two transcripts (Pxr1 and Rpl34A) are shown as red dots along the time course. The data is fitted with a single parameter per gene (degradation rate) resulting in an estimate of 10.9’ half-life for Pxr1 and a 131’ half-life for Rpl34A. Using these estimates, the expected pr along the time course is plotted as a red line with 95% CI as a red shaded area. (G) Half-life estimates correlate with various studies. Half-lives from this and four other published studies are compared to each other and the linear explanatory value (R2) is denoted in the upper diagonal matrix. The scatter depicts a specific example of the comparison between this study and the rates from Baudrimont et al where estimates were not obtained by metabolic labeling.
are growth, production, and degradation rates respectively), and assuming steady-state, pr can be translated to transcript half-life given the known labeling period (t). See methods and supplementary material for more details. (C) 4tU conversion is effective, reproducible and measures transcription. Percent of reads (y-axis) along a 4tU time course (x-axis) with a different number of observed T→C conversions (legend, N = 6). Samples exposed to vehicle (DMSO) or the transcription inhibitor thiolutin for 15 min (and labeled with 4tU for 6 min, N = 2). Note that the y-axis begins at 75%, i.e. most reads have no conversions. (D) Binomial Mixture Model (BMM) fits the data. Read conversion statistics are fitted with a 2-component BMM. Each dot represents the number of reads with a certain number of observed Ts and T→C conversions (color as in E). x-axis is the expected number of reads for each (T,T→C) pair assuming the model and the observed #T distribution in the data, the y-axis is the observed number of reads in each (T, T→C) combination. Additional components do not improve the likelihood of the data (Supplementary Figure S1D). (E) Half-life distribution for all yeast transcripts. Assuming steady-state, transcript-specific and global parameters are iteratively fitted, resulting in the half-life of each transcript. The median of the distribution is 8.2', transcripts with a half-life of 45′ or longer are counted in the rightmost bin. (F) Examples of estimated pr along the time course. The individually estimated pr per time point and replicate (N = 6) for two transcripts (Pxr1 and Rpl34A) are shown as red dots along the time course. The data is fitted with a single parameter per gene (degradation rate) resulting in an estimate of 10.9’ half-life for Pxr1 and a 131’ half-life for Rpl34A. Using these estimates, the expected pr along the time course is plotted as a red line with 95% CI as a red shaded area. (G) Half-life estimates correlate with various studies. Half-lives from this and four other published studies are compared to each other and the linear explanatory value (R2) is denoted in the upper diagonal matrix. The scatter depicts a specific example of the comparison between this study and the rates from Baudrimont et al where estimates were not obtained by metabolic labeling.