

Abstract
Background
About half of heart failure (HF) patients, while having preserved left ventricular function, suffer from diastolic dysfunction (so‐called HFpEF). No specific therapeutics are available for HFpEF in contrast to HF where reduced ejection fractions (HFrEF) can be treated pharmacologically. Myocardial titin filament stiffening, endothelial dysfunction, and skeletal muscle (SKM) myopathy are suspected to contribute to HFpEF genesis. We previously described small molecules interfering with MuRF1 target recognition thereby attenuating SKM myopathy and dysfunction in HFrEF animal models. The aim of the present study was to test the efficacy of one small molecule (MyoMed‐205) in HFpEF and to describe molecular changes elicited by MyoMed‐205.
Methods
Twenty‐week‐old female obese ZSF1 rats received the MuRF1 inhibitor MyoMed‐205 for 12 weeks; a comparison was made to age‐matched untreated ZSF1‐lean (healthy) and obese rats as controls. LV (left ventricle) function was assessed by echocardiography and by invasive haemodynamic measurements until week 32. At week 32, SKM and endothelial functions were measured and tissues collected for molecular analyses. Proteome‐wide analysis followed by WBs and RT‐PCR was applied to identify specific genes and affected molecular pathways. MuRF1 knockout mice (MuRF1‐KO) SKM tissues were included to validate MuRF1‐specificity.
Results
By week 32, untreated obese rats had normal LV ejection fraction but augmented E/e′ ratios and increased end diastolic pressure and myocardial fibrosis, all typical features of HFpEF. Furthermore, SKM myopathy (both atrophy and force loss) and endothelial dysfunction were detected. In contrast, MyoMed‐205 treated rats had markedly improved diastolic function, less myocardial fibrosis, reduced SKM myopathy, and increased SKM function. SKM extracts from MyoMed‐205 treated rats had reduced MuRF1 content and lowered total muscle protein ubiquitination. In addition, proteomic profiling identified eight proteins to respond specifically to MyoMed‐205 treatment. Five out of these eight proteins are involved in mitochondrial metabolism, dynamics, or autophagy. Consistent with the mitochondria being a MyoMed‐205 target, the synthesis of mitochondrial respiratory chain complexes I + II was increased in treated rats. MuRF1‐KO SKM controls also had elevated mitochondrial complex I and II activities, also suggesting mitochondrial activity regulation by MuRF1.
Conclusions
MyoMed‐205 improved myocardial diastolic function and prevented SKM atrophy/function in the ZSF1 animal model of HFpEF. Mechanistically, SKM benefited from an attenuated ubiquitin proteasome system and augmented synthesis/activity of proteins of the mitochondrial respiratory chain while the myocardium seemed to benefit from reduced titin modifications and fibrosis.
Keywords: HFpEF, ZSF1, MuRF1, Diastolic dysfunction, Skeletal muscle dysfunction, Muscle atrophy
Introduction
Heart failure with preserved ejection fraction (HFpEF, approximately 50% of all HF cases) is a complex syndrome, with high morbidity and mortality rates. 1 This form of HF is frequently seen in patients suffering from co‐morbidities associated with the metabolic syndrome, namely obesity, hypertension, diabetes mellitus and hyperlipidaemia (for a review, see Adamczak et al. 2 ), suggesting that HFpEF is a systemic disease that includes heart features. Besides structural alterations in the myocardium including myocardial stiffening and its functional impairment, HFpEF is also associated with endothelial dysfunction and myopathy of the peripheral skeletal muscles (SKM). 3 , 4 Consistent with SKM myopathy, exercise intolerance and early fatigue are common features in HFpEF similar as in heart failure patients with reduced ejection fraction (HFrEF). Exercise intolerance is associated with reduced quality of life and increased mortality, 5 and early fatigue is accompanied by SKM atrophy as documented by dual‐energy X‐ray absorptiometry (DEXA) showing a significant reduction of lean muscle mass by ~10%. 6 SKM atrophy and myopathy include molecular alterations like fat infiltration, 7 reduced mitochondrial oxidative capacity, 8 shift in fibre type composition, 9 decreased anabolic factors like IGF‐1, 10 and reduced capillary to fibre ratio 11 in the affected SKM.
The pathomechanisms underlying HFpEF are complex and appear to include myocardial stiffening. Myocardial stiffness pathology includes both intracellular cardiomyocyte titin filament and the extracellular matrix collagen stiffening. 12 With regard to SKM myopathy, the activation of the ubiquitin proteasome system (UPS) is a central step in promoting muscle wasting, 13 , 14 most likely including SKM atrophy in HFpEF as in all chronic muscle wasting states. Atrophic stress is promoted by a family of E3 ligases including the muscle ring finger protein 1 (MuRF1) and muscle atrophy F‐box protein (MAFBx). These E3 ligases trigger the ubiquitinylation of contractile proteins thereby marking them for degradation via the UPS and promoting loss of myofibrils. Hence, these E3 ligases are also referred to as atrogins (for a review, see Scalabrin et al. 15 ). The importance of MuRF1 in the development of muscle atrophy is supported by the protection from muscle atrophy after MuRF1 gene knockout in mice. 16 Accordingly, a potential strategy to prevent the development of SKM wasting is to prevent the activation of MuRF1 (reviewed in Scalabrin et al. 15 ), for example, by small molecule inhibition 17 or adenoviral‐mediated knock‐down. 18
In contrast to the wealth of proven therapies for HFrEF, most efforts to improve morbidity and mortality in HFpEF have failed so far (for a review, see Wintrich et al. 19 ). Only the recently published EMPEROR trial using empagliflozin, a SGLT2 inhibitor, reported positive effects on combined risk of cardiovascular death or hospitalization for HFpEF. 20
Lately, our group described small molecules capable of attenuating the development of SKM atrophy and dysfunction in animal models of HFrEF, 21 cardiac cachexia, 22 and tumour associated muscle atrophy 23 by targeting MuRF1: These compounds down‐regulate MuRF1 E3 ligase functions by interfering with its recognition of other muscle proteins, including titin. 21 In the present study, we tested if this compound class can also prevent SKM atrophy in HFpEF. For this, we selected the ZSF1 rat model that is commonly used to investigate interventional strategies in HFpEF. 3 , 24 , 25 As compound, we selected MyoMed‐205 that has improved serum stability when compared with initial lead compound ID#704946 21 , 22 (see Supporting Information, Figure S1).
Materials and methods
Study design
ZSF1‐lean (n = 25) and ZSF1‐obese (n = 40) animals were purchased from Charles River (Figure 1A). At an age of 20 weeks, 10 animals from each group were withdrawn from the study cohort to assess the development of HFpEF by echocardiography and invasive haemodynamic measurements and to document SKM and endothelial dysfunction. The remaining ZSF1‐obese animals were randomized either into a placebo group receiving normal rat chow (obese, n = 15) or a treatment group (n = 15) receiving rat chow containing MyoMed‐205 (obese + MyoMed‐205; 0.1% w/w). MyoMed‐205 is a close structural analogue of the previously described compound ID#704946 21 , 22 but has improved serum stability and bioavailability to muscle (see Figure S1). The ZSF1‐lean animals served as healthy control group (lean, n = 15). After 12 weeks of treatment, functional measurements on the myocardium (echocardiography and invasive haemodynamic measurements) were performed. At the end of the invasive haemodynamic measurement, the animals were sacrificed by bilateral thoracotomy. While the rats were under a surgical plane of anaesthesia, the extensor digitorum longus (EDL), the soleus muscle (SO), and the carotid artery were prepared for functional analyses. In addition, organ weights of SKM [SO, EDL, and tibialis anterior (TA)] and heart were determined and organs were snap frozen in liquid nitrogen. All experiments and procedures were approved by the local animal research council, TU Dresden and the Landesbehörde Sachsen (TVV 42/2018).
Figure 1.
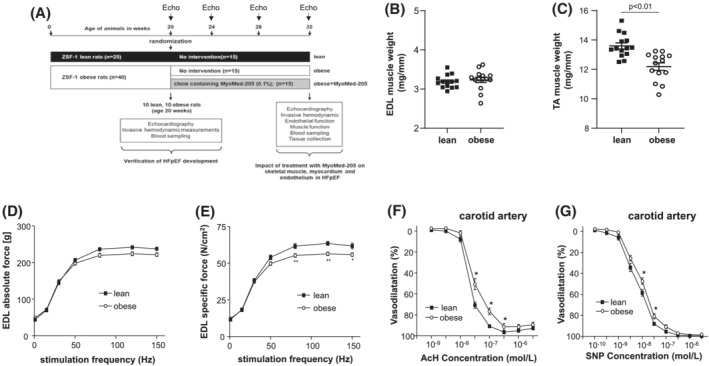
Study design and animal characteristics at 20 weeks. A schematic drawing of the study design is depicted (A). ZSF1‐lean and ZSF1‐obese were included into the study. At an age of 20 weeks, 10 animals of each group were removed from the study to confirm the development of HFpEF. The remaining ZSF1‐lean animals served as controls (lean), whereas the ZSF1‐obese animals were randomized into a placebo group with no intervention (obese) or a group receiving rat chow containing MyoMed‐205 (obese‐MyoMed‐205). Twelve weeks after randomization, animals were subjected to echocardiography and invasive haemodynamic measurements, and collected tissues were used for functional, histological, and molecular analyses. Before randomization, skeletal muscle mass (B,C), skeletal muscle function (D,E), and endothelial function (F,G) was assessed in ZSF1‐lean and ZSF1‐obese animals. *P < 0.05, **P < 0.01 versus lean.
Echocardiography
Rats were anaesthetised by isoflurane (1.5–2%), and transthoracic echocardiography was performed using a Vevo 3100 system and a 21‐MHz transducer (Visual Sonic, Fujifilm) to assess cardiac function as recently described. 24 , 25 For a detailed description, see supporting information.
Invasive haemodynamic measurements
Invasive haemodynamic pressure measurements were performed as the terminal procedure as recently described. 25 For a detailed description, see supporting information.
Skeletal muscle trophicity and function
Determination of total muscle weights, normalization to tibia length, and determination of myofibrillar cross‐sectional areas (CSAs) in dissected TA were performed as described previously. 21 For force measurements, the right EDL and the left soleus were dissected and mounted vertically in a Krebs–Henseleit buffer‐filled organ bath between a hook and a force transducer. Force and fatigue measurements were performed as described earlier. 25 For a detailed description, see supporting information.
Measurement of carotid artery function
Vessel function of carotid artery rings (2–3 mm) was analysed in vitro (DMT multimyograph System 620 M). For a detailed description, see supporting information.
Western blot analysis
Frozen muscle samples were homogenized in Relax buffer (90 mMol/L HEPES, 126 mMol/L KCl, 36 mMol/L NaCl, 1 mMol/L MgCl, 50 mMol/L EGTA, 8 mMol/L ATP, 10 mMol/L creatinphosphate; pH 7.4) containing a protease inhibitor mix (Inhibitor mix M, Serva, Heidelberg, Germany). Protein quantification by western blot analysis was performed as recently described 21 , 26 using the specific antibodies listed in Table S1.
Enzyme activity measurements
Tibialis anterior was homogenized in relaxing buffer and aliquots were used for enzyme activity measurements. Enzyme activities for citrate synthase (CS) and mitochondrial complex I were measured spectrophotometrically as reported earlier. 21 Enzyme activity data are presented as the fold change versus lean.
RNA isolation and quantitative real‐time polymerase chain reaction (PCR)
Total RNA was isolated from LV tissue and real‐time PCR was performed using the CFX384™ Real Time PCR System (BioRad, USA) and Maxima SYBR Green qPCR Kit (Thermo Scientific, Germany) as recently described. 25 Specific primer sequences are listed in Table S2. The expression of specific genes was normalized to its expression in lean. The CFX Maestro program (BioRad, USA) was used to calculate relative gene expression.
Analysis of mtDNA/nDNA ratio
The content of mitochondrial (mt‐DNA) and nuclear DNA (nDNA) was assessed by PCR as described by Quiros et al., 27 and the relative expression was calculated using the CFX Maestro program (BioRad). For amplification of mtDNA, sequences of 16S rRNA and ND1 were used. Foxo3, Sirt1, and Myod1 specific sequences were amplified for nuclear DNA and used as housekeeping genes for standardization. Specific primer sequences are listed in Table S2.
Quantification of perivascular fibrosis
Paraffin sections were stained with Picrosirius red and perivascular fibrosis around arteries, expressed as perivascular fibrosis ratio (PFR) was quantified as described by Dai and colleagues. 28 PFR was defined as the area of perivascular fibrosis divided by the area of the vascular wall, averaged over all quantifiable images of arteries taken from a section.
Assessment of titin phosphorylation
Homogenized myocardial samples (20 μg) were separated using a 2% agarose gel as described elsewhere. 29 After O/N fixation (50% methanol/10% acid acid) gels were stained with Pro‐Q Diamond phosphoprotein stain (ThermoFisher, Waltham, Ma, USA) for 1 h and subsequently with SYPRO Ruby (ThermoFisher, Waltham, Ma, USA) overnight. Phosphorylation signals for titin proteins on Pro‐Q Diamond‐stained gels were normalized to SYPRO Ruby‐stained total protein signals.
MMP zymography
MMP2 activity in myocardial tissue was assessed as recently described. 3 , 24 For detailed information, see supporting information.
Quantification of NT‐proBNP, cGMP, and triglyceride content
The plasma concentration of NT‐proBNP and the concentration of cGMP in LV tissue were determined using enzyme‐linked immunosorbent assays (NT‐proBNP: MBS, BIOZOL, Germany; cGMP: CUSABIO, Wuhan, China) according to the manufacturer's protocols.
Triglyceride concentration in the TA muscle was measured using a triglyceride quantification kit (Sigma) according to the manufacturer's protocol.
Proteomic analysis
Mass spectrometry based proteomic analysis was performed as recently described 21 , 23 using five TA muscle from each group (ZSF1‐lean, ZSF1‐obese, ZSF1‐obese treated with MyoMed‐205, respectively). For detailed analysis, see supporting information.
MuRF1 knockout animals
The mice used in the present study are all on a clean C57/BL6 background. Details on the gene inactivation of MuRF1 is given in Witt et al. 30 At an age of 10 weeks, C57/BL6 and MuRF1‐KO animals were sacrificed, and SKM tissue (TA muscle) was snap frozen for molecular analyses.
Statistical analyses
Data are presented as mean ± SEM. One‐way analysis of variance (ANOVA) followed by Bonferroni post hoc was used to compare groups, while two‐way repeated measures ANOVA followed by Bonferroni post hoc was used to assess contractile function (GraphPad Prism). Significance was accepted as P < 0.05. For proteome analysis, P values were corrected for multiple hypothesis testing using the R implementation of Benjamini–Hochberg's FDR.
Results
At randomization ZSF1‐obese rats present with a HFpEF syndrome including SKM atrophy and endothelial dysfunction
Comparison of ZSF1‐obese and ZSF1‐lean animals at week 20 already detected a significant increase in body weight, E/e′, left ventricular end‐diastolic pressure (LVEDP), and the development of cardiac hypertrophy, accompanied by an increase of systemic NT‐proBNP levels. In contrast, left ventricular ejection fraction was not significantly different between both groups. For detailed results, see Table S3. Based on the echocardiographic and invasive haemodynamic evaluation, the ZSF1‐obese animals developed HFpEF before they were randomized either into the MyoMed‐205 treatment or placebo group, respectively. In accordance with earlier studies, 3 , 25 ZSF1‐obese animals also developed SKM atrophy in the TA muscle (Figure 1B,C), and muscle dysfunction was evident when measuring specific muscle force in the EDL muscle (Figure 1D,E). Furthermore, endothelial dysfunction was evident in the carotid artery (Figure 1F,G).
MuRF1 based intervention to HFpEF by feeding with MyoMed‐205 spiked food
Food supplementation with compound at a dosage of 0.1% w/w was well tolerated by the animals, and no complications were observed (i.e. no deaths, change in food‐up take, activity, and grooming). Furthermore, feeding of MyoMed‐205 to healthy wild type Sprague Dawley rats had no impact on SKM atrophy, SKM and myocardial function (Figure S2), and MMP2 activity in the myocardium (Figure S3). Consistent with this, toxicology assessment of MyoMed‐205 chow in rats according to the OECD‐423 dose escalation protocol (for detailed information on the OECD‐423 protocol, see https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd_gl423.pdf) indicated that dosages of 0.3–2 g/kg were tolerated without detectable side effects. Food uptake was not different between ZSF1‐obese animals receiving normal chow or MyoMed‐205 chow (control chow: 26.0 ± 0.8 vs. MyoMed‐205 chow: 26.7 ± 1.0 g food per animal per day; P = NS). In contrast, body weight gain was significantly greater during the 12 weeks in the animals fed with MyoMed‐205 chow (control chow: 83.5 ± 3.3 vs. MyoMed‐205 chow: 110 ± 3.8 g; P < 0.0001).
Skeletal muscle improvements in ZSF1‐obese rats after MyoMed‐205 treatment
After the 12 week feeding intervention, muscle weights, normalized to tibia length, indicated no difference for SO after 12 weeks of treatment. In contrast, EDL weights were significantly increased in the obese‐MyoMed‐205 group by treatment (Figure 2A,B). In TA, the development of HFpEF was associated with muscle atrophy (Figure 2C). Feeding with MyoMed‐205 attenuated both, TA weight and myofibre cross sectional area (CSA) loss in obese rats (Figure 2C,D).
Figure 2.
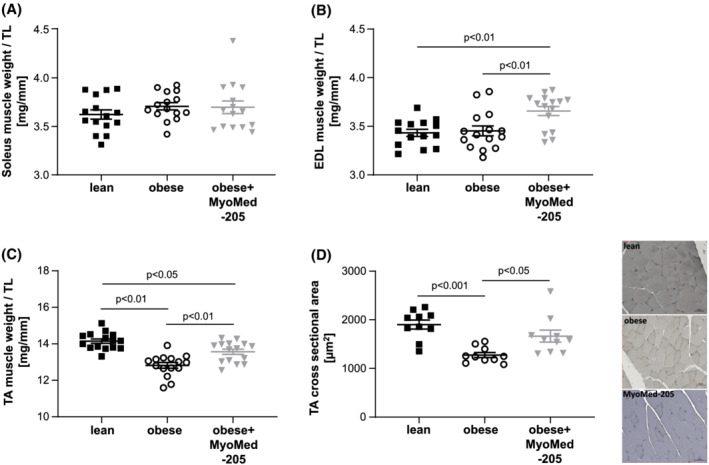
MyoMed‐205 increased muscle weight and cross sectional area in TA muscle. Muscle wet weight normalized to tibia length was measured in the soleus (A), EDL (B), and TA (C) muscle of ZSF1‐lean (lean), ZSF1 obese (obese), and ZSF1‐obese rats treated with MyoMed‐205 (obese + MyoMed‐205) (n = 14–15 per group). In addition, cross sectional area (CSA) was quantified in the TA muscle of each group (n = 10 per group) (D). Representative histological images (nuclei stained with haemalaun) for CSA are depicted. Results are expressed as mean ± SEM.
Evaluating muscle function at week 32, explanted EDL and SO muscles from obese rats had significantly reduced absolute muscle forces when compared with lean controls (Figure 3A,B): maximum absolute force was impaired in ZSF1‐obese by 8% for EDL (Figure 3C) and 17% for SO (Figure 3D). In contrast, obese animals treated with MyoMed‐205 until week 32 Sol and EDL had maximal absolute forces that were not significantly different from lean healthy controls (Figure 3C,D). Next, we normalized forces to CSA to determine if fibre loss was causative or rather myofibrils were impaired per se in their force generation. Determination of this specific muscle force indicated significantly lower specific muscle forces in the EDL (Figure 3E) and SO muscles in obese animals (Figure 3F). Maximal specific force was reduced by 16% in EDL (Figure 3G) and 14% in the SO fibres (Figure 3H). Treatment with MyoMed‐205 significantly improved maximal specific force in EDL (Figure 3G). In SO, the force increase was small and not significant after normalization to CSA (Figure 3H). With respect to muscle fibre fatigue, we detected earlier fatigue of Sol and EDL fibres from ZSF1‐obese animals compared with ZSF1‐lean healthy controls (Figure S4). MyoMed‐205 had no effect on muscle fatigability.
Figure 3.
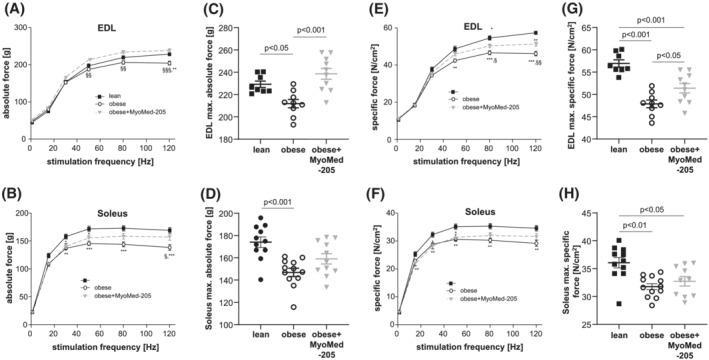
MyoMed‐205 improved muscle function of EDL and soleus muscle. Absolute (A–D) and specific (E–H) muscle force were measured in vitro from ZSF1‐lean (lean, black squares), ZSF1 obese (obese, open circles), and ZSF1‐obese rats treated with MyoMed‐205 (obese + MyoMed‐205, grey triangles) in the EDL and soleus muscle. Results are expressed as mean ± SEM (n = 9–11 per group). *P < 0.05, **P < 0.01, ***P < 0.001 vs. lean; § P < 0.05, §§ P < 0.01, §§§ P < 0.001 vs. obese + MyoMed‐205.
These MyoMed‐205 induced muscle changes were independent of changes in muscle fat content: the triglyceride content of the TA muscle did not change with MyoMed‐205 treatment (lean: 0.71 ± 0.15; obese: 2.25 ± 0.41*, obese + MyoMed‐205: 2.29 ± 0.57* nmol/mg protein; *P < 0.05 vs. lean).
Molecular signatures underlying SKM myopathy in ZSF1‐obese rats and their attenuation by MyoMed‐205
As shown earlier, the development of HFpEF in the ZSF1‐obese animals was associated with muscle atrophy, most pronounced in the TA muscle, and this was partially reversed by MyoMed‐205 (Figure 2). An important mechanism inducing muscle atrophy is the activation of the ubiquitin proteasome system which is promoted by specific ubiquitin E3‐ligases including MuRF1, MafBx and Trim72 (also known as Mitsugumin‐53). Therefore, we analysed the protein synthesis of MuRF1 (Figure 4A), MafBx (Figure 4B), and Trim72 (Figure 4C) in TA muscle. This revealed a significant up‐regulation of MuRF1 and Trim72 in obese animals, which was blunted after MyoMed‐205 treatment. Consistent with MuRF1 and Trim72 elevation, total ubiquitinated proteins were also up‐regulated in ZSF1‐obese rats. Again, this effect was partially reversed by MyoMed‐205 (Figure 4D). For MafBx, no difference was seen between the three groups.
Figure 4.
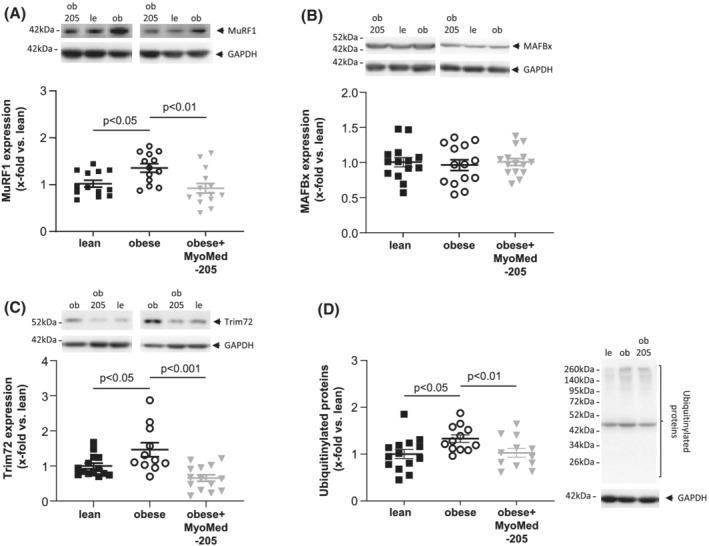
Impact of MyoMed‐205 on protein synthesis of atrophy related proteins in TA muscle. Protein synthesis of atrophy related proteins (A–D) was quantified by western blot analysis in TA muscle homogenates obtained from ZSF1‐lean (lean), ZSF1 obese (obese), and ZSF1‐obese rats treated with MyoMed‐205 (obese + MyoMed‐205). As atrophy related proteins MuRF1 (A), MAFBx (B), Trim72 (C), and ubiquitinylated proteins (D) were measured. Results are expressed as mean ± SEM (n = 10–14 per group). Representative western blots are depicted.
Unbiased proteome analysis of TA muscle revealed that from a total of 3179 protein fragments detected, 81 were significantly different in their abundance between the ZSF1‐lean and ZSF1‐obese groups (P < 0.01, Benjamini–Hochberg corrected). When comparing TA proteomes of ZSF‐1 obese treated and non‐treated groups, 31 proteins were differently expressed (P < 0.05) (the results from the proteome analysis are depicted in Table S4 ). Finally, eight proteins were independently identified as being differentially expressed in both data sets (Figure 5A; and not different between lean and obese + MyoMed‐205). Of these eight proteins, five are involved in mitochondrial dynamics, autophagy, or energy metabolism. For Ndufb5, Ndufb6, Fis1, and glutathione synthase (GSS), we confirmed their differential expression by WBs or RT‐PCR in the respective TA muscles (Figure 5B–E). In previous investigations, subunits of the mitochondrial NADH dehydrogenase were noted in MuRF1 Y2H screens. 31 Taken together the alterations in the HFpEF proteomes indicated mitochondrial stress, and MyoMed‐205 treatment attenuated the effects on mitochondrial metabolism.
Figure 5.
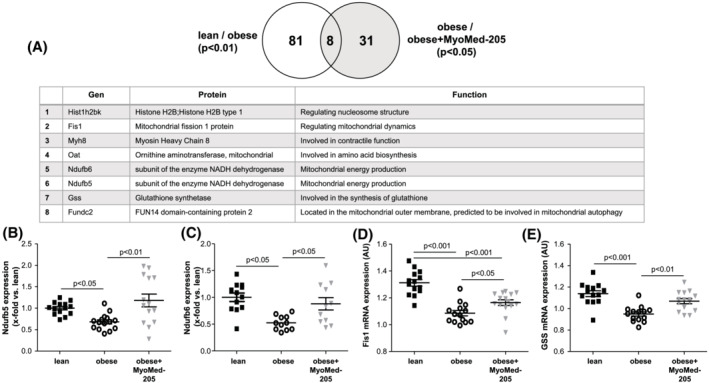
Proteome analysis of TA muscle. The proteomes expressed in TA muscle samples were analysed to identify differentially expressed proteins in the different groups (n = 5 per group). The set of 81 differentially expressed proteins between lean and obese and of 31 proteins of obese versus obese + MyoMed‐205 share a group of eight proteins that are differentially expressed in both sets and exhibit no difference between lean and obese + MyoMed‐205 (A). The eight proteins are listed in the table. Four out of these eight proteins were further investigated by western blot analysis (B,C) or RT‐PCR (D,E) in the TA muscle of ZSF1‐lean (lean), ZSF1 obese (obese) and ZSF1‐obese rats treated with MyoMed‐205 (obese + MyoMed‐205). Results are expressed as mean ± SEM (n = 11–15 per group).
Impact of MyoMed‐205 on components of the mitochondrial respiratory chain
Based on the proteome results and in consideration that a connection between the mitochondrial network and its energy supply and muscle atrophy has been described, 32 we assessed next the synthesis and/or activity of components of the mitochondrial respiratory chain. When quantifying total mitochondrial content via the synthesis of porin (Figure 6A) or the ratio of mitochondrial/nuclear DNA (Figure S5), we detected no significant difference between the three groups. Assessment of mitochondrial complex I activity revealed a significant up‐regulation in MyoMed‐205 treated animals when compared with untreated obese animals (Figure 6B). Quantifying the protein synthesis of mitochondrial complex I (Figure 6C), complex II (Figure 6D), complex III (Figure 6E), complex IV (Figure 6F), and complex V (Figure 6G), an up‐regulation was seen in obese animals fed with MyoMed‐205. In addition, CS enzyme activity was significantly up‐regulated in obese‐MyoMed‐205 TA muscle when compared with untreated obese rats (Figure 6I). Conversely, uncoupling protein 3 (UCP3) was significantly down‐regulated after MyoMed‐205 feeding with no change between lean and obese (Figure 6J).
Figure 6.
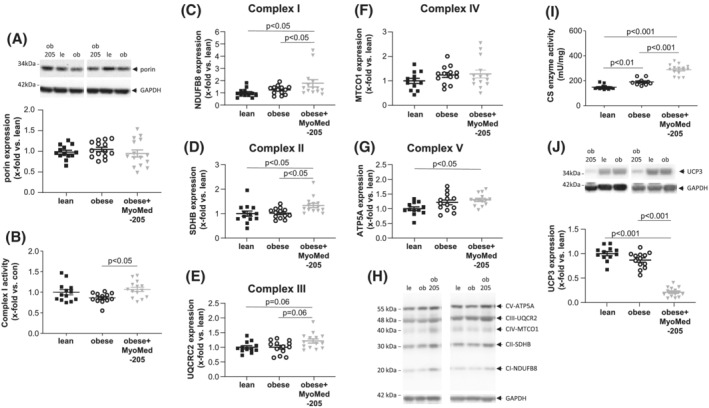
Impact of MyoMed‐205 on mitochondrial protein synthesis and enzyme activity in TA muscle. Protein synthesis of mitochondrial proteins or enzyme activities (A–E) were quantified by western blot analysis or specific enzyme assays in TA muscle homogenates obtained from ZSF1‐lean (lean), ZSF1 obese (obese) and ZSF1‐obese rats treated with MyoMed‐205 (obese + MyoMed‐205). Porin (A), mitochondrial complex I (C), mitochondrial complex II (D), mitochondrial complex III (E), mitochondrial complex IV (F), and mitochondrial complex V (G) synthesis were assessed by western blot and mitochondrial complex I enzyme activity was determined (B). Moreover, the enzymatic activity of citrate synthase (CS) in TA (I) and the protein synthesis of UCP3 was quantified (J). Results are expressed as mean ± SEM (n = 10–14 per group). A representative western blot shows the detection of mitochondrial complex I–V (H).
Activity of mitochondrial complex I and SDH in TA muscle of MuRF1 knockout animals
To investigate if the effect of MyoMed‐205 on mitochondrial metabolism, especially on mitochondrial complex I and SDH, is an ‘off‐target’ effect of this compound or possibly connected to its MuRF1 knock‐down activity, we next determined the mitochondrial complex I and SDH activities in the TA muscle of MuRF1‐KO mice. As depicted in Figure S6, the activity of mitochondrial complex I and SDH was significantly higher in the TA muscle of the MuRF1‐KO animals when compared with C57/BL6 animals (Figure S6A,B). This increase in activity was independent of mitochondrial content, because the synthesis of porin (marker for mitochondrial content) was similar in both groups (Figure S6C). In conclusion, our data suggest that MuRF1 activity/synthesis is negatively correlated to the activity of key markers (i.e. complex I and SDH) for mitochondrial oxidative phosphorylation.
Myocardial effects of Myomed‐205 on left ventricular compliance and protein synthesis
During ageing from 20 to 32 weeks, obese ZSF1 rats progressively developed diastolic dysfunction (increase in E/e′ and LVEDP), whereas systolic function (LVEF) remained preserved (Figure S7A,B). Treatment with MyoMed‐205 for 12 weeks significantly improved diastolic functions (see LVEDP and E/e′ in Figure S7). In particular, during the last 4 weeks, compound treatment did not only slow down the development of diastolic heart failure but even reversed progression towards diastolic heart failure. With regard to myocardial hypertrophy markers (heart weight, LVAW, and LVPW, Table 1), MyoMed‐205 treated obese animals showed a slight but significant reduction compared with untreated obese animals. In addition, MyoMed‐205 treatment reduced left ventricular myocardial stiffness (measured as stiffness factor beta) (Table 1).
Table 1.
Animal characteristics at 32 weeks of age
| Parameter | Control (n = 15) | HFpEF (n = 15) | HFpEF + MyoMed‐205 (n = 15) |
|---|---|---|---|
| Body weight (g) | 265 ± 4 | 559 ± 9*** | 558 ± 5*** |
| Tibia length (TL, mm) | 38.2 ± 0.1 | 38.1 ± 0.1 | 38.0 ± 0.1 |
| Heart weight/TL (mg/mm) | 23.54 ± 0.31 | 35.44 ± 0.50*** | 34.04 ± 0.35*** , § |
| Lung weight (wet/dry) | 4.72 ± 0.07 | 4.18 ± 0.05*** | 4.08 ± 0.03*** |
| Serum glucose (mg/dL) | 324 ± 20 | 572 ± 22*** | 505 ± 16*** |
| NT proBNP (pg/mL) | 96.9 ± 9.4 | 209.0 ± 38.2* | 162.8 ± 25.4 |
| Echocardiography | |||
| LV mass (mg) | 890 ± 11 | 1274 ± 20*** | 1224 ± 17*** |
| LVEF (%) | 79.0 ± 1.1 | 74.3 ± 1.2** | 79.6 ± 0.9 §§ |
| LVFS (%) | 52 ± 1.7 | 54 ± 1.3 | 45 ± 2.1 |
| LVSV (μL) | 441 ± 28 | 511 ± 17* | 489 ± 19 |
| LVEDV (μL) | 555 ± 34 | 691 ± 25*** | 605 ± 24 §§ |
| E/e′ | 21.6 ± 0.4 | 27.9 ± 0.7*** | 23.4 ± 0.6 §§§ |
| E/A | 1.2 ± 0.1 | 1.4 ± 0.1** | 1.2 ± 0.06 §§ |
| LAA (mm2) | 17 ± 1.1 | 21 ± 0.8** | 22.36 ± 0.6*** |
| LVAW (mm) | 1.76 ± 0.06 | 2.03 ± 0.07** | 2.08 ± 0.06** |
| LVPW (mm) | 1.66 ± 0.09 | 2.02 ± 0.09* | 2.15 ± 0.09** |
| LVID | 6.76 ± 0.11 | 7.97 ± 0.19*** | 7.47 ± 0.18** |
| Aortic valve area (mm2) | 5.64 ± 0.57 | 3.29 ± 0.21*** | 6.05 ± 0.64 §§§ |
| Aortic valve peak pressure (mmHg) | 13.39 ± 2.52 | 28.57 ± 3.39** | 19.82 ± 1.93 § |
| Invasive haemodynamic measurements | |||
| Ascending aorta | |||
| SAP (mmHg) | 125 ± 6 | 180 ± 4*** | 177 ± 6** |
| DAP (mmHg) | 95 ± 5 | 113 ± 3** | 115 ± 4** |
| MAP (mmHg) | 105 ± 6 | 136 ± 3*** | 136 ± 5*** |
| Left ventricle (LV) | |||
| LVEDP (mmHg) | 15 ± 1 | 22 ± 1*** | 16 ± 1 §§ |
| LVESP (mmHg) | 113 ± 8 | 165 ± 5*** | 159 ± 9*** |
| LVEDV (μL) | 434 ± 29 | 622 ± 47** | 507 ± 16 |
| LVESV (μL) | 134 ± 17 | 291 ± 31*** | 166 ± 13 §§ |
| dP/dt max (mmHg/s) | 5791 ± 310 | 9864 ± 411*** | 9885 ± 439*** |
| dP/dt min (mmHg/s) | −6055 ± 411 | −8148 ± 323*** | −8051 ± 416* |
| dV/dt max (μL/s) | 14 972 ± 1752 | 15 263 ± 926 | 12 407 ± 1598 |
| dV/dt min (μL/s) | −12 495 ± 1344 | −13 666 ± 1874 | −13 296 ± 1211 |
| LV stiffness constant betaw | 0.28 ± 0.05 | 2.54 ± 0.67* | 0.70 ± 0.14 § |
| Slope LV‐Ees (mmHg/μL) | 0.11 ± 0.02 | 0.25 ± 0.06* | 0.30 ± 0.05** |
P < 0.05.
P < 0.01.
P < 0.001 vs. control.
P < 0.05.
P < 0.01.
P < 0.001 vs. HFpEF.
Next, we studied the molecular signatures in LV tissues of treated and non‐treated ZSF1 obese rats to gain insights on the molecular mechanisms associated with HFpEF and its potential attenuation by MyoMed‐205 feeding. Previously suggested mechanisms responsible for causing a loss of myocardial compliance in HFpEF are fibrosis including up‐regulation of metalloproteinase‐2 (MMP2), titin filament stiffening by perturbed phosphorylation, 33 and increased advanced glycation end‐products (AGEs). 34 Consistent with this, LV tissue of obese animals presented with increased fibrosis and elevated MMP2 activity (Figure 7A,B). Titin was overall hypo‐phosphorylated compared with ZSF1‐lean (Figure 6C) and cGMP levels were lower in LV by 22%, but this did not reach statistical significance (lean: 1 ± 0.07 vs. obese: 0.78 ± 0.07 x‐fold vs. lean; P = 0.07). Feeding with MyoMed‐205 attenuated and in part reversed these alterations: perivascular fibrosis in the obese animals fed with MyoMed‐205 for 12 weeks was significantly lower when compared with lean and untreated obese animals (Figure 7A). Consistent with this, MMP2 activity was significantly decreased in the LV after feeding MyoMed‐205 when compared with control and untreated obese ZSF1 rats (Figure 7B). Feeding with MyoMed‐205 almost normalized the phosphorylation status of titin (Figure 7C) and increased cGMP levels in LV (obese: 0.78 ± 0.07 vs. obese + MyoMed‐205: 1.39 ± 0.11 x‐fold vs. lean; P < 0.001). AGEs were significantly lower in LV of MyoMed‐205 fed animals with no difference between lean and obese (Figure 7D).
Figure 7.
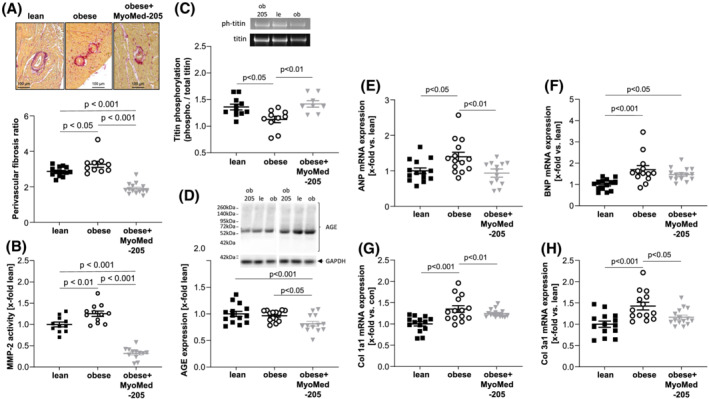
Impact of MyoMed‐205 on molecular parameters in the myocardium. The myocardium from ZSF1‐lean (lean), ZSF1 obese (obese), and ZSF1‐obese rats treated with MyoMed‐205 (obese + MyoMed‐205) was used to quantify the extent of perivascular fibrosis (A), the activity of matrix metalloproteinase 2 (MMP2) (B), the ratio of phosphorylated titin/total titin (C) and the synthesis of advanced glycation end products (AGE) modified proteins (D). Moreover, the mRNA expression of atrial natriuretic peptide (ANP) (E), B‐type natriuretic peptide (BNP) (F), collagen‐1a1 (Col1a1) (G), and collagen‐3a1 (Col3a1) (H) were quantified. Results are expressed as mean ± SEM (n = 10–14 per group). Representative western blots are depicted.
Finally, consistent with changes in fibrosis and extracellular matrix composition, mRNA expression analyses showed that ANP (Figure 7E), BNP (Figure 7F), collagen‐1 (Figure 7G), and collagen‐3 (Figure 7H) transcripts were significantly augmented in LV of obese animals. Feeding with MyoMed‐205 reduced the transcription of ANP, Col 1a1 and Col 3a1 and no significant difference to lean healthy control animals was detected (Figure 7E–H).
Endothelial function and the impact of MyoMed‐205
Measurement of maximal tension of carotid artery segments of ZSF1‐obese animals revealed a significant lower tension calculated per artery length when compared with ZSF1‐lean animals (Figure S8A). No beneficial impact of MyoMed‐205 feeding on tension development was observed (Figure S8A). We observed a significant impaired vasodilation in the obese animals compared with lean control animals when analysing endothelial‐dependent (Figure S8B) and endothelial‐independent (Figure S8C) vasodilation. Feeding of MyoMed‐205 to ZSF1‐obese animals for 12 weeks did not improve endothelial‐dependent (Figure S8B) or endothelial‐independent vasodilation (Figure S8C).
Discussion
Chronic heart failure types, although diverse in their mechanistic basis and pathophysiology, are in general accompanied during HF progression with an increasing SKM atrophy and muscle dysfunction. This SKM dysfunction further exacerbates quality of life and overall prognosis of patients. We recently reported that the targeting of MuRF1 by small molecules is a potential strategy to attenuate SKM dysfunction in HF using murine models with HF development following lung fibrosis and right ventricular hypertension, 22 or after post‐infarct left ventricular failure. 21 The aim of the present study was to test if MuRF1 attenuation might also diminish SKM atrophy in a HFpEF model, because diastolic heart failure differs regarding its pathophysiological basis due to preserved cardiac muscle contractility.
MyoMed‐205 and skeletal muscle
The effects after MyoMed‐205 feeding on SKM are consistent with our results from the previous murine cardiac cachexia interventions 21 , 22 : compound‐feeding attenuated muscle weight loss in TA and also protected its CSA. Future single myofibril studies will need to address differential effects of MyoMed‐205 on fibre‐types because previous studies on MURF1‐KO mice noted a preferential protection of SKM fast fibres. 35 Also, the SKM myocellular changes that we detected by western blot in TA were similar to our previous cardiac cachexia studies: MuRF1 and also TRIM72 were down‐regulated in TA from MyoMed‐205 fed ZSF1 rats. Consistent with this, total muscle protein ubiquitination was reduced. Compound feeding augmented the activity of mitochondrial oxidative phosphorylation enzymes in SKM extracts similar to our previous cardiac cachexia studies. Proteome analysis further demonstrated compound effects on mitochondrial metabolism as possible basis for the beneficial effects of MyoMed‐205 treatment: more than half of the proteins differentially expressed in SKM are mitochondrial proteins. Subunits of the mitochondrial NADH dehydrogenase were previously noted in MuRF1 Y2H screens. 31 Interestingly, studies on MuRF1 knockout animals also implicate MuRF1 in the negative regulation of mitochondrial oxidative phosphorylation. Therefore, the stimulative effects of MyoMed‐205 on SKM mitochondrial energy production are consistent with its role as MuRF1 inhibitor. The crosstalk between MuRF1 and mitochondria is further supported by proteome data, where besides structural proteins, also mitochondrial proteins involved in energy metabolism or maintaining mitochondrial function were detected: Timm9, Ndufa3, and Mrps5 in the HFrEF model 21 ; Smdt1, Cox3, Cpt2, Qil1, and Lyrm4 in the tumour cachexia model 23 ; Fis1, Oat, Ndufb6, Ndufb5, and Fundc2 in the present HFpEF model.
Therefore, in summary, our rodent intervention studies with MyoMed‐205 or ID#704946 suggest a potential to attenuate SKM myopathy in the three mechanistically different heart failure types studied so far [right ventricular failure, 22 left ventricular post MI failure, 21 and diastolic heart failure (present study)]. The unexpected range of drug effects makes future studies mandatory to determine how MyoMed‐205 attenuated muscle atrophy and improved mitochondrial oxidative phosphorylation: this may involve possible off‐target effects and/or MuRF1‐mediated regulatory crosstalk between mitochondrial energy production and muscle trophicity, including perhaps titin filament stiffness regulation by phosphorylation and ROS mediated modifications. The modulation of mitochondrial enzymes activities like mitochondrial complex I might reflect a MuRF1‐specific role and not merely an off‐target effect of MyoMed‐205, because the same effects were seen in MuRF1 knockout animals (Figure S6). Another unexpected finding is the significant down‐regulation of UCP3 protein in the TA muscle of obese rats treated with MyoMed‐205. We speculate that a reduced UCP3 synthesis could hypothetically enhance the coupling between the TCA cycle and mitochondrial ATP synthesis, enhancing energetic efficiency.
MyoMed‐205 and myocardial effects
In any treatment strategy of SKM myopathy in a HF setting, potential harmful effects on the cardiac function must be critically assessed and weighted with benefits. Based on previous studies, a potential cardio‐toxicity when targeting MuRF1,2 needs to be considered: knockout of MuRF1 in mice makes their hearts highly vulnerable to left ventricular hypertrophy after aortic constriction induced pressure overload, 36 whereas overexpression of MuRF1 antagonizes cardiac myocyte hypertrophic response to agonists. 37 Consistent with this, patients with mutations in MuRF1 gene suffer from hypertrophic cardiomyopathy, and the overexpression of this mutant MuRF1 protein in mouse hearts was associated with cardiac hypertrophy. 38 Finally, a recent study suggested that the interaction of MuRF1 and titin is essential as increased binding of MuRF1 to titin due to mutations is associated with hypertrophic cardiomyopathy. 39 Therefore, we assessed both, endothelial and myocardial effects of MyoMed‐205 feeding. We could not detect major MyoMed‐205 effects on vascular functions. With regard to the myocardium neither echocardiographic nor haemodynamic measurements nor heart mass indicated signs of cardiotoxicity after 12 week compound intervention to ZSF1 animals or healthy control animals. Instead, we surprisingly detected a marked improvement of diastolic functions after MyoMed‐205 intervention as indicated by E/e′, LVEDP, stiffness factor beta, and LVEF in the HFpEF cohort. In addition, we noted that MyoMed‐205 did not increase lung wet to dry ratios, consistent with our earlier study in HFrEF. 21
Next, we studied the potential mechanism underlying improved ventricular compliance after MyoMed‐205 by analysing LV tissues of treated and non‐treated ZSF1‐obese rats: histology indicated a marked protection from myocardial fibrosis particularly in the perivascular sections. Consistent with this, after treatment WBs detected a reduced collagen expression and reduced MMP2 activity, respectively. MMP2 is an extracellular matrix‐directed metalloproteinase implicated in fibrosis in a number of pathological conditions including heart failure. 40 In conclusion, protection from myocardial fibrosis augmenting ECM‐based stiffness might be one mechanism that protects myocardial elasticity in ZSF1‐obese rats. Intracellularly, the elastic titin filament is the major source of passive stiffness in cardiomyocytes during physiological amounts of stretch (for a review, see Granzier et al. 41 ). Consequently, titin filament stiffening has been suggested to be a major mechanism underlying myofibrillar stiffening in HFpEF, 42 and mechanistically, this has been correlated with hypo‐phosphorylation of the titin filament. 12 When measuring the level of global titin phosphorylation, we detected a small but significant reduction in the LV of ZSF1‐obese animals compared with lean controls (Figure 4C) and a trend towards lower levels of cGMP. Future studies are required with site‐specific titin antibodies that can differentiate between PKB and PKG phosphorylation (linked to titin relaxation), or PKC phosphorylation (linked to titin stiffening) of titin spring elements. Nevertheless, based on the observed lower LV cGMP levels in of obese animals, we speculate that PKG activity might be reduced, thereby leading to hypo‐phosphorylation of titin. Finally, titin stiffening might also occur pathologically during metabolic stress by augmented Maillard reaction modifications of the titin filament, leading to advanced glycation end products (AGE). Consistent with this hypothesis, we noted a reduction of AGE‐modified proteins in ZSF1‐obese rats, and AGE species were reduced in the ZSF1‐obese MyoMed‐205 treated group. In summary, MyoMed‐205 protection of intracellular titin‐based elasticity by increased phosphorylation via the cGMP‐PKG axis is very speculative and future studies are required to determine the relative efficacies of MyoMed‐205 on extracellular and intracellular myocardial stiffness.
Future studies will also need to address if the improved myocardial function in the ZSF1‐obese treated animals is due to a direct effect of MyoMed‐205 on the myocardium or rather secondary after improved peripheral muscle function: in our earlier studies, 21 , 22 the related analogue ID#704946 affected muscle atrophy but had no impact on myocardial hypertrophy. In contrast, we note here a clear beneficial effect on diastolic function of MyoMed‐205. Possibly, this might be due to its augmented serum stability when compared with ID#704946 (Figure S1). Alternatively, improved SKM functions impacted secondarily on myocardial function: recent exercise training studies 43 reported that an improved exercise performance and better muscular function after exercise were closely correlated with an improved myocardial function.
Study limitations
The present study is the first report on beneficial effects of MyoMed‐205 on SKM atrophy/function and myocardial diastolic function in animals exhibiting HFpEF. However, the following limitations have to be considered.
First, in the present study, the concentration of MyoMed‐205 (0.1% w/w) was chosen based on previous studies using MyoMed‐205 23 or the original lead compound #704946 21 , 22 , 23 in mouse models of different cachectic diseases. To date, no data are available on testing different compound concentrations for their SKM and myocardial effects. The effects of MyoMed‐205 in the present study are comparable with the effects of ID#704946 in our earlier studies. 21 , 22 , 23 In our previous murine studies, muscle atrophy in TA was improved by 10–20%, whereas in the present study, CSAs in the TA increased by 26% after MyoMed‐205 treatment. Differences in SKM effects might result from different serum stabilities of MyoMed‐205 and ID#704946 that complicates direct comparisons (Figure S1).
Second, complete gene inactivation of MuRF1 family members in mice is cardiotoxic. 44 In the present study, rats were treated for 12 weeks with a small molecule interfering with MuRF1 target recognition. In the case of patients, longer treatment periods exceeding 1 year will be needed. Possible side effects when Myomed‐205 is given over a longer time period may preclude its application for treatment of HFpEF patients. Potentially, toxicology data of non‐rodent large animal models may preclude the launch of human trials and are urgently needed.
Third, in the present study, we determined an improvement in the overall titin phosphorylation and a modulation of the extracellular matrix. The relative contributions of both modulations for increased myocyte stiffness still need to be determined. Furthermore, it will be of importance to identify the specific phosphorylation site in the titin molecule because phosphorylation at different sites in the titin molecule results in different titin stiffness—PKB and PKG phosphorylation is linked to titin relaxation whereas the PKC dependent phosphorylation in the PEVK region of the titin molecule is linked to titin stiffening.
Fourth, the importance of MyoMed‐205 induced modulation of the synthesis of components of the mitochondrial oxidative phosphorylation (complex I‐V) needs to be validated by metabolic readouts like direct measurements of oxidative phosphorylation using a Clark electrode.
Conclusions
MyoMed‐205 administered over 12 weeks improves myocardial diastolic function and skeletal muscle atrophy/function in an animal model of HFpEF. Molecular pathways affected by MyoMed‐205 in HFpEF include the UPS and mitochondrial oxidative phosphorylation for SKM (for a hypothetical model, see Figure S9) and posttranslational modification of titin and fibrosis in the myocardium. The observed effects on myocardial and SKM function are probably independent of modulating hypertension and glucose concentration (Table 1). This in accordance with an earlier study by our group were the application of Sacubitril/Valsartan to ZSF1‐obese animals lowered blood pressure but had no effect on SKM atrophy/function. 24 Also, in a separate mouse study, we found that MyoMed‐205 had no significant effects on basal glucose or oral glucose tolerance. 45 Future studies are warranted to characterize the molecular mechanisms responsible for the beneficial effects of MyoMed‐205 on skeletal and myocardial function and its pharmacology and to address the question if off‐target effects are playing an important role. Nevertheless, it seems that targeting MuRF1 may represent a new strategy to improve skeletal and myocardial function in HFpEF.
Funding
This work was supported by grant from the Foundation Leducq (network 13CVD04) and the European Union Horizon 2020 Research and Innovation Program (grant agreement no. 645648 ‘Muscle Stress Relief’). V.A. was supported by a grant from the DZHK (German Center for Cardiovascular Research).
Conflict of interest
Volker Adams reports a patent filing for MyoMed‐205, ID#704946 and further derivatives for its application to chronic muscle stress states (patent accession No WO2021023643A1).
Antje Schauer, Antje Augstein, Virginia Kirchhoff, Runa Draskowski, Anett Jannasch, Keita Goto, Gemma Lyall, Anita Männel, and Peggy Barthel have nothing to disclose. Norman Mangner reports personal fees from Edwards Lifesciences, Medtronic, Biotronik, Novartis, Sanofi Genzyme, AstraZeneca, Pfizer, Bayer, Abbott, Abiomed, and Boston Scientific outside the submitted work. Ephraim B. Winzer reports personal fees from Boehringer‐Ingelheim, CVRx, and Novartis outside the submitted work. Axel Linke reports grants from Novartis, personal fees from Medtronic, Abbott, Edwards Lifesciences, Boston Scientific, Astra Zeneca, Novartis, Pfizer, Abiomed, Bayer, Boehringer, and other from Picardia, Transverse Medical, and Claret Medical outside the submitted work. Siegfried Labeit reports a patent filing for MyoMed‐205, ID#704946 and further derivatives for its application to chronic muscle stress states (patent accession No WO2021023643A1).
Supporting information
Table S1 List of primary antibodies used
Table S2: List of PCR primers
Table S3: Animal characteristics at 20 weeks of age
Figure S1: Plasma stability for substance ID#704946 and MyoMed‐205 was tested in vitro. 1 μmol/L of test compound was incubated with 70 μl murine plasma for different time points at 37 °C with shaking at 100 rpm. After stopping the reaction with acetonitrile, the concentration of the test compound was determined by HPLC system coupled with tandem mass spectrometer. The structural formulas of the two compounds are depicted.
Figure S2: Skeletal muscle weight of the TA (A), soleus (B) and EDL (C) as well as muscle function of the soleus muscle (D,E) was measured in the myocardium of Sprague Dawley rats (con) and Sprague Dawley rats receiving MyoMed‐205 for 12 weeks (con‐MyoMed205). In addition heart weight (F), cardiac output (G) and left ventricular ejection fraction was quantified in both groups.
Figure S3: MMP2 activity was determined in left ventricular tissue homogenate of Sprague Dawley rats (con) and Sprague Dawley rats receiving MyoMed‐205 for 12 weeks (con‐MyoMed205) by zymography. The original zymography is shown on top and the quantification of MMP2 normalized to the mean value of the control animals is depicted below.
Figure S4: Skeletal muscle fatigue was assessed in the soleus (A) and EDL muscle (B) at an age of 32 weeks in ZSF1‐lean control animals (lean) and ZSF1‐obese animals developing HFpEF (obese) or being treated for 12 weeks with MyoMed‐205 (obese+MyoMed‐205). Soleus and EDL muscle fatigued significantly earlier in HFpEF when compared to controls. Only in the EDL muscle MyoMed‐205 improved fatigue marginal. * p < 0.05 vs. lean.
Figure S5: The ratio of mitochondrial DNA (mtDNA) over nuclear DNA (nDNA) was assessed in TA muscle of lean, obese and obese treated with MyoMed‐205 for 12 weeks. The ratio of 16 s RNA (A) or ND1 (B) over the geometrical mean of Myod1/Foxo3/Sirt1 as nDNA is depicted.
Figure S6: Expression and activity of mitochondrial proteins in TA muscle of WT and MuRF1‐KO animals. The enzyme activity of mitochondrial complex‐I (A) and SDH (B) were asses in the TA muscle of wildtype (WT) and MuRF1‐KO animals. In addition the protein expression of porin as marker for mitochondrial content was assessed by Western blot (C). Results are expressed as mean ± SEM (n = 7–11 per group).
Figure S7: Impact of MyoMed‐205 on functional parameters of the myocardium. The E/e′ ratio/A), the left ventricular end‐diastolic pressure (LVEDP) (B) and the left ventricular ejection fraction (LVEF) (C) was assessed in ZSF1‐lean (lean, filled square or black bars), ZSF1 obese (obese, open circles or white bars) and ZSF1‐obese rats treated with MyoMed‐205 (obese+MyoMed‐205, triangles or grey bars). Results are expressed as mean ± SEM (n = 10–14 per group). **p < 0.01, ***p < 0.001 vs. lean; §§§p < 0.001 vs. obese.
Figure S8: Assessment of endothelial function. Endothelial function was measured in vitro using carotid artery rings obtained from ZSF1‐lean (lean, filled square), ZSF1 obese (obese, open circles) and ZSF1‐obese rats treated with MyoMed‐205 (obese+MyoMed‐205, triangles). Tensions of the artery after titration with increasing concentration of phenylephrine were recorded (A) and endothelial dependent (B) and –independent (C) vasodilation were assessed. Results are expressed as mean ± SEM (n = 14–15 per group). *p < 0.05, **p < 0.01, ***p < 0.001 vs. lean.
Figure S9: Hypothetical working model. The schematic drawing summarizes how MyoMed‐205 might protect muscle atrophy and muscle dysfunction in settings of metabolic stress situations. UPS = ubiquitin proteasome system
Table S4. Supporting information
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle. 46 We thank the DZHK partner site Mass Spectrometry Core Facility Lab Bad Nauheim, Germany, for expert proteome analysis and the Chemical Biology Core Facility at the EMBL Heidelberg, Germany, for expert chemical biology support.
Adams V., Schauer A., Augstein A., Kirchhoff V., Draskowski R., Jannasch A., Goto K., Lyall G., Männel A., Barthel P., Mangner N., Winzer E. B., Linke A., and Labeit S. (2022) Targeting MuRF1 by small molecules in a HFpEF rat model improves myocardial diastolic function and skeletal muscle contractility, Journal of Cachexia, Sarcopenia and Muscle, 13, 1565–1581, 10.1002/jcsm.12968
Contributor Information
Volker Adams, Email: volker.adams@tu-dresden.de.
Siegfried Labeit, Email: labeit@medma.de.
References
- 1. Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, et al. Outcome of heart failure with preserved ejection fraction in a population‐based study. New Engl J Med 2006;355:260–269. [DOI] [PubMed] [Google Scholar]
- 2. Adamczak DM, Oduah MT, Kiebalo T, Nartowicz S, Beben M, Pochylski M, et al. Heart failure with preserved ejection fraction—a concise review. Curr Cardiol Rep 2020;22:82–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmederer Z, Rolim N, Bowen TS, Linke A, Wisloff U, Adams V. Endothelial function is disturbed in a hypertensive diabetic animal model of HFpEF: moderate continuous vs. high intensity interval training. Int J Cardiol 2018;273:147–154. [DOI] [PubMed] [Google Scholar]
- 4. Akiyama E, Sugiyama S, Matsuzawa Y, Konishi M, Suzuki H, Nozaki T, et al. Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal left ventricular ejection fraction. J Am Coll Cardiol 2012;60:1778–1786. [DOI] [PubMed] [Google Scholar]
- 5. Arena R, Myers J, Aslam SS, Varughese EB, Peberdy MA. Peak VO2 and VE/VCO2 slope in patients with heart failure: a prognostic comparison. Am Heart J 2004;147:354–360. [DOI] [PubMed] [Google Scholar]
- 6. Haykowsky MJ, Brubaker PH, Morgan TM, Kritchevsky S, Eggebeen J, Kitzman DW. Impaired aerobic capacity and physical functional performance in older heart failure patients with preserved ejection fraction: role of lean body mass. J Gerontol A Biol Sci Med Sci 2013;68:968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haykowsky MJ, Kouba EJ, Brubaker PH, Nicklas BJ, Eggebeen J, Kitzman DW. Skeletal muscle composition and its relation to exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Cardiol 2014;113:1211–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 2019;139:1435–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bowen TS, Rolim NPL, Fischer T, Baekkerud FH, Medeiros A, Werner S, et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail 2015;17:263–272. [DOI] [PubMed] [Google Scholar]
- 10. Seiler M, Bowen TS, Rolim N, Dieterlen MT, Werner S, Hoshi T, et al. Skeletal muscle alterations are exacerbated in heart failure with reduced compared with preserved ejection fraction: mediated by circulating cytokines? Circ Heart Fail. 2016;9:e003027. [DOI] [PubMed] [Google Scholar]
- 11. Bowen TS, Herz C, Rolim NPL, Berre AMO, Halle M, Kricke A, et al. Effects of endurance training on detrimental structural, cellular, and functional alterations in skeletal muscles of heart failure with preserved ejection fraction. J Card Fail 2018;24:603–613. [DOI] [PubMed] [Google Scholar]
- 12. Hamdani N, Franssen C, Lourenco A, Falcao‐Pires I, Fontoura D, Leite S, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 2013;6:1239–1249. [DOI] [PubMed] [Google Scholar]
- 13. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos VE, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 14. Sandri M. Protein breakdown in muscle wasting: role of autophagy‐lysosome and ubiquitin‐proteasome. Int J Biochem Cell Biol 2013;45:2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scalabrin M, Adams V, Labeit S, Bowen TS. Emerging strategies targeting catabolic muscle stress relief. Int J Mol Sci 2020;21:4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen T, Bowen TS, Augstein A, Schauer A, Gasch A, Linke A, et al. Expression of MuRF1 or MuRF2 is essential for the induction of skeletal muscle atrophy and dysfunction in a murine pulmonary hypertension model. Skeletal Muscle 2020;10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eddins MJ, Marblestone JG, Suresh Kumar KG, Leach CA, Sterner DE, Mattern MR, et al. Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem Biophys 2011;60:113–118. [DOI] [PubMed] [Google Scholar]
- 18. Castillero E, Alamdari N, Lecker SH, Hasselgren PO. Suppression of atrogin‐1 and MuRF1 prevents dexamethasone‐induced atrophy of cultured myotubes. Metabolism 2013;62:1495–1502. [DOI] [PubMed] [Google Scholar]
- 19. Wintrich J, Kindermann I, Ukena C, Selejan S, Werner C, Maack C, et al. Therapeutic approaches in heart failure with preserved ejection fraction: past, present and future. Clin Res Cardiol 2020;109:1079–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. New Engl J Med 2021;385:1451–1461. [DOI] [PubMed] [Google Scholar]
- 21. Adams V, Bowen TS, Werner S, Barthel P, Amberger C, Konzer A, et al. Small‐molecule‐mediated chemical knock‐down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J Cachexia Sarcopenia Muscle 2019;10:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bowen TS, Adams V, Werner S, Fischer T, Vinke P, Brogger MN, et al. Small‐molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J Cachexia Sarcopenia Muscle 2017;8:939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adams V, Gußen V, Zozulya S, Cruz A, Moriscot A, Linke A, et al. Small‐molecule chemical knockdown of MuRF1 in melanoma bearing mice attenuates tumor cachexia associated myopathy. Cell 2020;9:2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schauer A, Adams V, Augstein A, Jannasch A, Draskowski R, Kirchhoff V, et al. Sacubitril/valsartan improves diastolic function but not skeletal muscle function in a rat model of HFpEF. Int J Mol Sci 2021;22:3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Männel A, et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail 2020;7:2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mangner N, Garbade J, Heyne E, van den Berg M, Winzer EB, Hommel J, et al. Molecular mechanisms of diaphragm myopathy in humans with severe heart failure. Circ Res 2021;128:706–719. [DOI] [PubMed] [Google Scholar]
- 27. Quiros PM, Goyal A, Jha P, Auwerx J. Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol 2017;7:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non‐ischemic heart failure. J Cardiol 2012;60:416–421. [DOI] [PubMed] [Google Scholar]
- 29. Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high‐molecular‐weight proteins. Electrophoresis 2003;24:1695–1702. [DOI] [PubMed] [Google Scholar]
- 30. Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J 2008;27:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Witt SH, Granzier H, Witt CC, Labeit S. Murf‐1 and Murf‐2 target a specific subset of myofibrillar proteins redundantly: towards understanding Murf‐dependent ubiquitination. J Mol Biol 2005;350:713–722. [DOI] [PubMed] [Google Scholar]
- 32. Romanello V, Sandri M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell Mol Life Sci 2020; Online ahead of print;78:1305–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinan AS, Lindsey ML. Titin phosphorylation. Circ Res 2009;105:611–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hartog JWL, van de Wal RM, Schalkwijk CG, Miyata T, Jaarsma W, Plokker HWT, et al. Advanced glycation end‐products, anti‐hypertensive treatment and diastolic function in patients with hypertension and diastolic dysfunction. Eur J Heart Fail 2010;12:397–403. [DOI] [PubMed] [Google Scholar]
- 35. Moriscot AS, Baptista IL, Bogomolovas J, Witt C, Hirner S, Granzier H, et al. MuRF1 is a muscle fiber‐type II associated factor and together with MuRF2 regulates type‐II fiber trophicity and maintenance. J Struct Biol 2010;170:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Willis MS, Schisler JC, Li L, Rodriguez JE, Hilliard EG, Charles PC, et al. Cardiac muscle ring finger‐1 increases susceptibility to heart failure in vivo. Circ Res 2009;105:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J, et al. Muscle ring finger protein‐1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J Cell Biol 2004;167:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, et al. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res 2012;111:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Higashikuse Y, Mittal N, Arimura T, Yoon SH, Oda M, Enomoto H, et al. Perturbation of the titin/MURF1 signaling complex is associated with hypertrophic cardiomyopathy in a fish model and in human patients. Dis Model Mech 2019;12:dmm041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Int J Cardiol 2001;79:277–286. [DOI] [PubMed] [Google Scholar]
- 41. Granzier H, Wu Y, Siegfried L, Titin LWM. Physiological function and role in cardiomyopathy and failure. Heart Fail Rev 2005;10:211–223. [DOI] [PubMed] [Google Scholar]
- 42. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 2015;131:1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edelmann F, Gelbrich G, Düngen HD, Fröhling S, Wachter R, Stahrenberg R, et al. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: results of the Ex‐DHF (exercise training in diastolic heart failure) pilot study. J Am Coll Cardiol 2011;58:1780–1791. [DOI] [PubMed] [Google Scholar]
- 44. Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest 2007;117:2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Labeit S, Hirner S, Bogomolovas J, Cruz A, Myrzabekova M, Moriscot A, et al. Regulation of glucose metabolism by MuRF1 and treatment of myopathy in diabetic mice with small molecules targeting MuRF1. Int J Mol Sci 2021;22:2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 List of primary antibodies used
Table S2: List of PCR primers
Table S3: Animal characteristics at 20 weeks of age
Figure S1: Plasma stability for substance ID#704946 and MyoMed‐205 was tested in vitro. 1 μmol/L of test compound was incubated with 70 μl murine plasma for different time points at 37 °C with shaking at 100 rpm. After stopping the reaction with acetonitrile, the concentration of the test compound was determined by HPLC system coupled with tandem mass spectrometer. The structural formulas of the two compounds are depicted.
Figure S2: Skeletal muscle weight of the TA (A), soleus (B) and EDL (C) as well as muscle function of the soleus muscle (D,E) was measured in the myocardium of Sprague Dawley rats (con) and Sprague Dawley rats receiving MyoMed‐205 for 12 weeks (con‐MyoMed205). In addition heart weight (F), cardiac output (G) and left ventricular ejection fraction was quantified in both groups.
Figure S3: MMP2 activity was determined in left ventricular tissue homogenate of Sprague Dawley rats (con) and Sprague Dawley rats receiving MyoMed‐205 for 12 weeks (con‐MyoMed205) by zymography. The original zymography is shown on top and the quantification of MMP2 normalized to the mean value of the control animals is depicted below.
Figure S4: Skeletal muscle fatigue was assessed in the soleus (A) and EDL muscle (B) at an age of 32 weeks in ZSF1‐lean control animals (lean) and ZSF1‐obese animals developing HFpEF (obese) or being treated for 12 weeks with MyoMed‐205 (obese+MyoMed‐205). Soleus and EDL muscle fatigued significantly earlier in HFpEF when compared to controls. Only in the EDL muscle MyoMed‐205 improved fatigue marginal. * p < 0.05 vs. lean.
Figure S5: The ratio of mitochondrial DNA (mtDNA) over nuclear DNA (nDNA) was assessed in TA muscle of lean, obese and obese treated with MyoMed‐205 for 12 weeks. The ratio of 16 s RNA (A) or ND1 (B) over the geometrical mean of Myod1/Foxo3/Sirt1 as nDNA is depicted.
Figure S6: Expression and activity of mitochondrial proteins in TA muscle of WT and MuRF1‐KO animals. The enzyme activity of mitochondrial complex‐I (A) and SDH (B) were asses in the TA muscle of wildtype (WT) and MuRF1‐KO animals. In addition the protein expression of porin as marker for mitochondrial content was assessed by Western blot (C). Results are expressed as mean ± SEM (n = 7–11 per group).
Figure S7: Impact of MyoMed‐205 on functional parameters of the myocardium. The E/e′ ratio/A), the left ventricular end‐diastolic pressure (LVEDP) (B) and the left ventricular ejection fraction (LVEF) (C) was assessed in ZSF1‐lean (lean, filled square or black bars), ZSF1 obese (obese, open circles or white bars) and ZSF1‐obese rats treated with MyoMed‐205 (obese+MyoMed‐205, triangles or grey bars). Results are expressed as mean ± SEM (n = 10–14 per group). **p < 0.01, ***p < 0.001 vs. lean; §§§p < 0.001 vs. obese.
Figure S8: Assessment of endothelial function. Endothelial function was measured in vitro using carotid artery rings obtained from ZSF1‐lean (lean, filled square), ZSF1 obese (obese, open circles) and ZSF1‐obese rats treated with MyoMed‐205 (obese+MyoMed‐205, triangles). Tensions of the artery after titration with increasing concentration of phenylephrine were recorded (A) and endothelial dependent (B) and –independent (C) vasodilation were assessed. Results are expressed as mean ± SEM (n = 14–15 per group). *p < 0.05, **p < 0.01, ***p < 0.001 vs. lean.
Figure S9: Hypothetical working model. The schematic drawing summarizes how MyoMed‐205 might protect muscle atrophy and muscle dysfunction in settings of metabolic stress situations. UPS = ubiquitin proteasome system
Table S4. Supporting information