

Abstract

Thiophene S,S-dioxides are underutilized tools for the de novo construction of benzene rings in organic synthesis. We report a collective synthesis of nine illudalane sesquiterpenes using bicyclic thiophene S,S-dioxides as generalized precursors to the indane core of the natural products. Exploiting furans as unusual dienophiles in this inverse electron demand Diels–Alder cascade, this concise and convergent approach enables the synthesis of these targets in as little as five steps. Theoretical studies rationalize the reactivity of thiophene S,S-dioxides with both electron-poor and electron-rich dienophiles and reveal reaction pathways involving either nonpolar pericyclic or bifurcating ambimodal cycloadditions. Overall, this work demonstrates the wider potential of thiophene S,S-dioxides as convenient and flexible precursors to polysubstituted arenes.
Introduction
Polysubstituted benzene rings are challenging synthetic targets due to the difficulty of regioselective introduction of different functional groups onto the aromatic core. Such motifs are commonly found in natural products such as the illudalane sesquiterpenoids1 (Figure 1a), pharmaceuticals,2 and organic materials.3 In the context of the illudalane natural products, the functionalization of pre-formed arenes offers one approach (Figure 1b, path a), but this can be challenging due to issues of regioselectivity compounded by steric considerations, resulting in lengthy synthetic routes that are specific to a single target.4 An alternative strategy involves the de novo construction of the benzene ring, simultaneously installing all required substituents; for the illudalanes, this has almost exclusively involved fully intramolecular (path b),5 or two-component (path c) metal-catalyzed alkyne cyclizations.6
Figure 1.

(a) Illudalane natural products. (b) Previous approaches and the strategy employed in this work. (c) Intramolecular thiophene S,S-dioxide/ynamide cycloaddition in the formal synthesis of dictyodendrins B, C, and E.8
A different approach to benzene synthesis involves the Diels–Alder cycloaddition/retro-cycloaddition of dienes equipped with “leaving groups” such as N2, nitriles, and CO2.7 Single-atom variants such as SO2 or CO (i.e., cheletropic extrusions) are also possible but have been surprisingly little used in target-oriented synthesis. In the case of SO2 extrusion, the majority of research has exploited the ability of sulfolenes (2,5-dihydrothiophenes) to reveal dienes by the loss of SO2, which subsequently engage with a dienophile.9 Far less studied are thiophene S,S-dioxides: to the best of our knowledge, only a single use of these motifs in natural product synthesis exists in an elegant formal synthesis of dictyodendrins B, C, and E by Kabuki and Yamaguchi, involving intramolecular cycloaddition of an electron-rich ynamide with a thiophene S,S-dioxide (Figure 1c).8,10 Simple intermolecular cycloadditions with alkynes, alkenes, and furans are known in a methodological context but have mainly employed (poly)halogenated or symmetric thiophenes.11
We questioned whether thiophene S,S-dioxides could offer an efficient and general entry to the illudalane sesquiterpenoid natural products and specifically if intermolecular cycloaddition cascades of these substrates, which are unprecedented in natural product synthesis, could be employed in this context (Figure 1b, path d). Here, we describe concise syntheses of nine members of the illudalane natural product family, where variation of the orientation of a bicyclic thiophene S,S- dioxide (i.e., a 2,3- or 3,4-fused bicyclic framework) brings strategic flexibility in the positioning of the arene substituents. We also report theoretical studies into the nature of thiophene S,S-dioxide cycloaddition reactions: studies using both electron-poor and electron-rich dienophiles revealed a balance of reactivity pathways from classical nonpolar Diels–Alder to bifurcating “ambimodal” cycloadditions.
Results and Discussion
Our studies commenced with the preparation of the 2,3-fused bicyclic thiophene S,S-dioxide 1 (Scheme 1). Thiophene 2 was first constructed using a Fiesselmann synthesis,12 whereby the reaction of commercially available 3,3-dimethylcyclopentanone 3 with POCl3/DMF led to intermediate β-chloroenal 4, which was directly reacted with methyl thioglycolate to yield 2. Oxidation of 2 to the targeted S,S-dioxide 1 was initially found to be challenging due to the electron-withdrawing methyl ester, with oxidants such as m-CPBA or Oxone proving unsuccessful. However, the use of in situ-generated trifluoroacetic peracid, as described by Nenajdenko and co-workers,13 afforded 1 in good yield (73%).
Scheme 1. 2,3-Fused Bicyclic Thiophene S,S-Dioxide 1 as a Precursor to Five Illudalane Natural Products.

With 1 in hand, the key intermolecular cycloaddition/SO2 extrusion was investigated. Efforts to engage 1 with various alkynes were unsuccessful, but to our delight, the use of furan 5 as dienophile (at 100 °C in toluene) constructed the illudalane benzenoid core 6 in good yield. This reaction presumably proceeds via the initial formation of the (4 + 2) adduct 7, which aromatizes via cheletropic extrusion of SO2 and ring-opening elimination of the furan; however, the path for the formation of 7 could also conceivably involve a stepwise sequence of 1,6-Michael addition followed by cyclization of an intermediate zwitterion (i.e., the extreme of a concerted asynchronous cycloaddition pathway, vide infra). Notwithstanding the reaction pathway, the illudalane core was thus constructed in just four steps from commercial materials.
The presence of the two carbonyl functionalities in 6 facilitates direct access to a number of illudalane natural products. Reduction of 6 with LiAlH4 afforded riparol B, the selective benzylic oxidation of which gave granulolactone in high yield. Mindful of the previously reported conversion of granulolactone to echinolactone A by Zhang and co-workers,6c in which the efficiency of indane oxidation was hampered by poor differentiation between C1 and C3 (using CrO3/AcOH), more selective conditions were explored. Pleasingly, we found that the use of the heterogeneous co-oxidant system KMnO4/MnO214 conferred respectable selectivity for oxidation at C1 (3:1), giving echinolactone A in 59% yield; reduction with NaBH4 afforded radulactone.6c
Thiophene dioxide 1 could also be deployed in the synthesis of alcyopterosin O (Scheme 1b). Subjection of 1 to the cycloaddition cascade with 2-methoxyfuran afforded the bicyclic tetrasubstituted arene 8; reduction of both esters (9) followed by selective hydrogenolysis of the benzylic alcohol gave 10. To introduce the required hydroxymethyl group onto the benzene ring, the remaining alcohol was then utilized in a hydroxyl-directed ortho C–H alkenylation;15 pleasingly, this afforded enoate 11 in a 58% yield, which was advanced to the natural product in a further four steps (65%).
Although alcyopterosin O could be accessed using this route, we questioned whether an alternative approach could be devised with a lower step count. Comparison of alcyopterosin O with riparol B reveals the structural source of the problem—namely, the transposition of the arene methyl and hydroxymethyl substituents between the two targets, which necessitated multiple post-cycloaddition manipulations. A different approach involves “rotating” the orientation of the thiophene relative to its fused cyclopentane ring to enable the direct introduction of all arene substituents with suitable degrees of oxygenation. Thiophene S,S-dioxide 12 (Scheme 2) was therefore targeted, the synthesis of which again commenced with 3,3-dimethylcyclopentanone 3. Treatment of 3 with carbon disulfide under basic conditions, followed by the addition of ethyl bromoacetate, led to thiophene 13. Reduction of the exocyclic C–S bond using PdCl2/Et3SiH, followed by oxidation to the bicyclic thiophene S,S-dioxide 12, proceeded in high yield (74% over two steps). The key (4 + 2) cycloaddition/SO2 extrusion step again proceeded smoothly using furan 5, giving 2,2-dimethylindane core 14 with the methyl group “transposed” to the C6 position. Reduction of the two carboxyl functionalities completed the synthesis of alcyopterosin O in a much improved five steps from 3.
Scheme 2. 3,4-Fused Bicyclic Thiophene S,S-Dioxide 12 as a Precursor to Alcyopterosins O, A, B, C, and H.

Once again, this initial natural product served as a gateway to other members of the illudalane family. Hydrogenolysis of the benzylic alcohol (15) followed by chlorination gave alcyopterosin A. Alternatively, tosylation of 15 and reaction with NaNO3 afforded alcyopterosin B, which could be converted to alcyopterosin C by oxidation with KMnO4/MnO2, albeit this time with poor regioselectivity (1:1) owing to the lack of an electron-withdrawing group on the arene. Finally, reduction of the indanone gave alcyopterosin H.
The facile reaction of furans with the thiophene S,S-dioxides led us to question the nature of the cycloaddition process. The ability of thiophene S,S-dioxides to undergo genuine (4 + 2) cycloadditions was first confirmed by the successful reaction of S,S-dioxide 16 with cyclopentenone and dimethylacetylene dicarboxylate (DMAD; Figure 2a). Computational examination of these reactions led to the identification of pericyclic transition states (e.g., Figure 2a; TS1 for reaction with cyclopentenone, ΔG‡ = 28.5 kcal mol–1).
Figure 2.
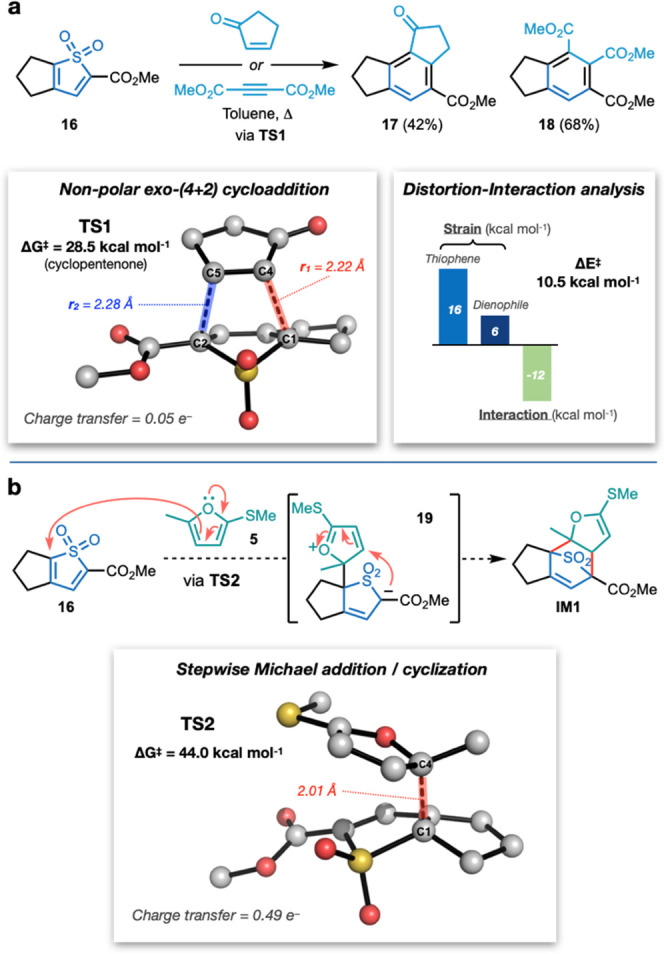
(a) Reaction of thiophene S,S-dioxide 16 with electron-deficient dienophiles, calculated TS1 for nonpolar Diels–Alder reaction of 16 with cyclopentenone, and distortion–interaction analysis. (b) Possible stepwise Michael addition/cyclization mechanism and calculated TS2 for the reaction of 16 with furan 5. Calculations at the CPCM(toluene)-DLPNO-CCSD(T)/def2-TZVPP//CPCM(toluene)-M06-2X/def2-TZVPP level of theory at 373.15 K/1 M. Hydrogen atoms are omitted for clarity.
Domingo et al. have classified Diels–Alder (DA) reactions as “nonpolar” or “polar”.16 Nonpolar DA reactions are characterized by relatively high activation energies and proceed through highly synchronous pathways in which the transition state (TS) does not involve significant charge transfer between the fragments (CT < 0.1 e–). Polar DA reactions exhibit lower activation energies and are typified by asynchronous bond formation with significant charge transfer between the two fragments at the TS (CT > 0.1 e–). The reactions of 16 with the electron-deficient dienophiles cyclopentenone or DMAD can thus be interpreted as nonpolar DA reactions due to limited charge transfer (Figure 2a and Table S6, 0.05 e– and 0.03 e–, respectively) and only slight asynchronicity (r2 – r1 = 0.05 and 0.08 Å, respectively).17 Distortion–interaction analysis18 of TS1 revealed a modest distortion of the dienophile compared to that of the diene and an interaction energy of 12 kcal mol–1.
Attempts to identify a pericyclic (4 + 2) TS for the reaction of 16 with furan 5 were unsuccessful; however, this reaction could also proceed by a 1,6-Michael addition/cyclization via zwitterion 19 (Figure 2b). A transition state was found for this Michael addition (TS2), but displayed a significant activation barrier of 44.0 kcal mol–1. The magnitude of this barrier appears to be inconsistent with the observed reactivity.
The concerted cycloaddition pathway was then explored in greater depth by constructing a More O’Ferrall–Jencks plot19 for the reaction of 16 with 5 as a function of the forming C1–C4 and C2–C5 bonds (Figure 3). This revealed a third possibility: a highly asynchronous, ambimodal pathway that proceeds via TS3 (ΔG‡ = 27.7 kcal mol–1). This transition state features partial bond formation between the diene and dienophile at three distinct positions—one advanced (r1, C1–C4, 2.00 Å) and two less advanced (r2, C3–C6, 2.67 Å; and r3, C2–C5, 2.93 Å), with significant charge transfer observed (CT = 0.37 e–)—and is favored over the Michael addition pathway (TS2) by 16 kcal mol–1. Characteristic of TS3 is a bifurcation of the potential energy surface via a valley ridge inflection into two distinct intermediates: IM1 (the “expected” inverse electron demand cycloadduct, which is then consumed by the cheletropic extrusion of SO2) and IM2 (a normal electron demand cycloadduct with the furan acting as the 4π component).20 These intermediates are formally connected via a Cope rearrangement (TS4; Figure 3).
Figure 3.

More O’Ferrall–Jencks plot for the reaction between 16 and 5 as a function of the forming C1–C4 (red), C2–C5 (green) bond distances calculated at the CPCM(toluene)-M06-2X/def2-SVP level of theory. Minima and TSs were further optimized at the CPCM(toluene)-DLPNO-CCSD(T)/def2-TZVPP//CPCM(toluene)-M06-2X/def2-TZVPP level of theory at 373.15 K/1 M. Gibbs free energies are reported in kcal mol–1 relative to the starting materials.
Such cycloaddition transition states were first envisioned by Woodward and Katz during their investigations on the dimerization of cyclopentadiene and were named “two-stage-one-step” processes.21 Caramella later revisited this phenomenon computationally and clarified the bifurcating nature of the transition state, as distinct from the Cope rearrangement pathway that connects the products.22 A number of other cycloadditions have since been reported that proceed through “bipericyclic” ambimodal pathways, in which two distinct products are formed from the bifurcation point.23
It is notable that the less-advanced bond formations in TS3 (C3–C6, 2.67 Å and C2–C5, 2.93 Å) are highly asymmetric (Δr = 0.26 Å). This suggests a product distribution favoring IM2 over IM1,24 and analysis using Goodman’s ValleyRidge algorithm showed that IM2 is indeed expected as the dynamic product.25 However, as this process is reversible (ΔGrev0 = 0.7 kcal mol–1, ΔGrev‡ = 28.4 kcal mol–1), the “required” cycloadduct IM1 can be formed by recrossing from IM2 via the starting materials.24 While the abovementioned Cope rearrangement pathway could also operate, the barrier is too high to contribute significantly to the rearrangement (TS4, 35.2 kcal mol–1). Following this conversion of IM2 to IM1, the exergonic extrusion of SO2 to form the corresponding diene product was calculated to proceed with a barrier of 16.2 kcal mol–1 (ΔG0 = −15.1 kcal mol–1; see Table S4).26
Distortion–interaction analysis revealed a higher distortion of the dienophile in TS3 compared to the pericyclic transition state TS1 (15 kcal mol–1 vs 6 kcal mol–1), which is offset by a significant increase in interaction energy (−21 kcal mol–1 vs −12 kcal mol–1 for TS1). This additional stabilization can be explained by electron donation from the developing C1–C4 σ bond into the C5–C7 π* orbital of the furan (Figure 4a; as evidenced by the NBO analysis, 18.2 kcal mol–1), accompanied by the expected donation into the developing C1–C4 σ* orbital from the furan π system (17.5 kcal mol–1) and oxygen lone pair (16.8 kcal mol–1). Several smaller C–H and S=O reciprocal hyperconjugation effects with the developing C1–C4 bond further stabilize TS3 (Figure 4b). NBO population analysis also illustrates the asynchronicity of the process, with the forming C1–C4 bond (r1) being significantly populated (1.71 e–), while minimal population is observed for C3–C6 (r2) and C2–C5 (r3).26
Figure 4.
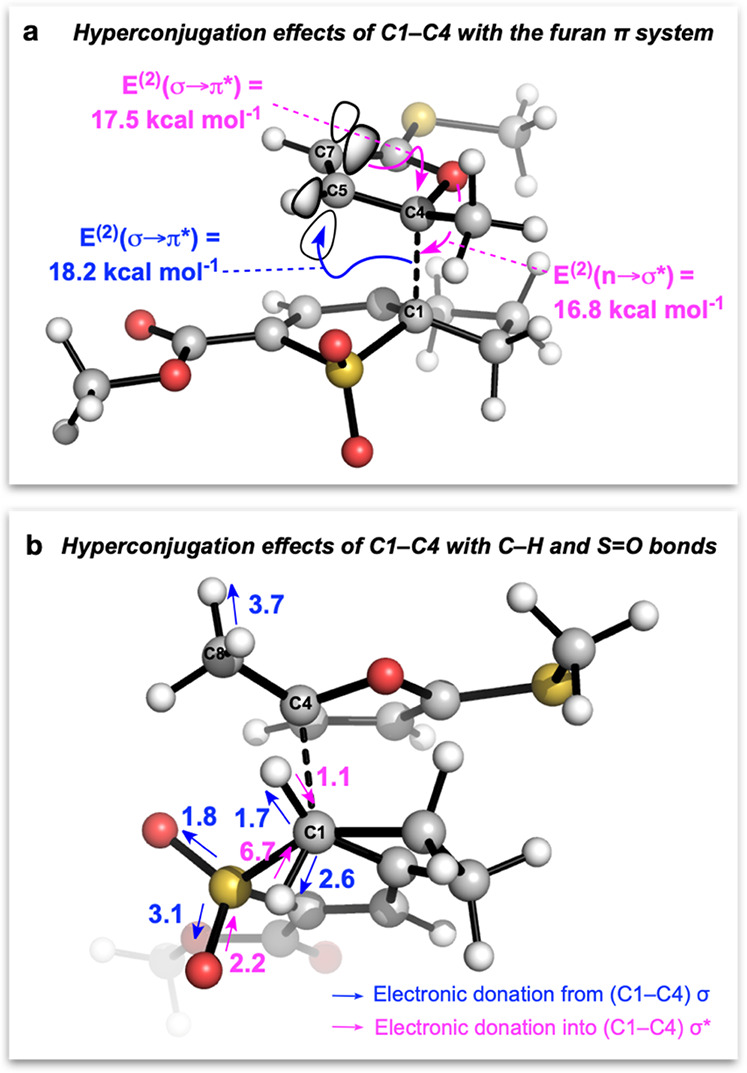
NBO second-order perturbation energy E(2) analysis of TS3 (Table S3). (a) Major contributions to stabilization and (b) minor hyperconjugation effects. Electron donation is from (blue) or into (pink) the developing C1–C4 bond. Calculations at the CPCM(toluene)-M06-2X/def2-TZVPP//CPCM (toluene)-M06-2X/def2-SVP level of theory at 373.15 K/1 M; all values are in kcal mol–1.
The reactions of 16 with electron-deficient dienophiles are typical of nonpolar cycloadditions (Figure 5a, left, and TS1). Comparison of TS3 with other ambimodal transition states27 reveals that it is moderately asynchronous with respect to r1 and r2 (Figure 5a, right) and highly asynchronous with respect to r2 and r3 (Figure 5b). These characteristics are similar to the reaction of diphenylketene with cyclopentadiene reported by Singleton,23e with the “asymmetry of ambimodality” (r3 – r2; Figure 5b) lying toward the upper end of such transition states characterized to date. Indeed, comparison with other ambimodal processes appears to suggest a qualitative inverse correlation between the “degree” and “asymmetry” of ambimodality, with the dimerization of cyclopentadiene reported by Caramella22a lying at the opposite end of this scale. It is interesting to consider whether these characteristics are critical or coincidental in enabling the reaction of 16 with an (aromatic) furan.
Figure 5.

Context of thiophene S,S-dioxide cycloadditions in ambimodal processes (geometries taken from refs (16, 34)). (a) Degree of asynchronicity of Diels–Alder cycloadditions and degree of ambimodality of bifurcating cycloadditions, with respect to the difference in forming bond lengths r1 and r2. (b) Asynchronicity of ambimodality with respect to the difference in forming bond lengths r2 and r3. r1 is the advanced bond formation. r1 ≪ r2 and r3, and r2 ≤ r3.
Conclusions
In summary, thiophene S,S-dioxides can undergo (4 + 2) cycloadditions via several reactivity channels. In the context of the illudalane natural products, reactions with furans proceed via ambimodal transition states that bifurcate reversibly to discrete cycloadducts, while electron-deficient dienophiles react through classical nonpolar pericyclic pathways. This multifaceted intermolecular reactivity profile enables the synthesis of the illudalanes in as little as five steps and underlines the utility of thiophene S,S-dioxides as powerful synthetic tools for the concise assembly of polysubstituted aromatic rings.
Computational Methods
All calculations were carried out using the ORCA suite of programs (version 4.2.1).28 Geometries were initially obtained via autodE using standard settings, with GFN2-XTB for conformational sampling and PBE-D3BJ/def2-SVP for geometry optimization.29 Dispersion corrections were considered using Grimme’s D3 empirical method with Becke–Johnson damping (D3BJ).30 The obtained geometries were then optimized at the CPCM(toluene)-M06-2X/def2-TZVPP level of theory. To support the computational validity of the M06-2X functional used herein, TS3 was also optimized at the CPCM(toluene)-SCS-MP2/def2-TZVPP level of theory. Significant differences in the asynchronicity of the ambimodal TS3 were found between two other DFT functionals and SCS-MP2; M06-2X was therefore used as it showed satisfactory agreement. A final CPCM(toluene)-DLPNO-CCSD(T)/def2-TZVPP single point calculation was performed on each structure to obtain reliable electronic energies (see the Supporting Information).31
Vibrational frequencies were computed at the optimization level of theory to confirm whether the structures correspond to minima or transition states. Grimme’s quasiRRHO approach was used to calculate free energies at 373.15 K.32 A standard state correction from 1 atm to 1 M was applied by adding RT ln(1/24.5) = 2.37 kcal mol–1 (T = 373.15 K) to the calculated free energy of each species. For calculating thermodynamic data, the python-script OTherm.py was used, with ω0 = 100 cm–1 replacing harmonic oscillators with free rotors below ω0.32,33
The 2D surface (More O’Ferrall–Jencks plot) in Figure 3 was generated in the space defined by the forming C1–C4 and C2–C5 bond distances using a grid of 0.1 Å. The area around stationary points TS3 and TS4 was sampled with a smaller grid size of 0.05 Å. These points were interpolated using the cubic spline function scipy.interp2d with matplotlib.py.
Acknowledgments
K.H.(K.)P. thanks the ASAN Foundation for a scholarship. N.F. thanks the Studienstiftung des Deutschen Volkes e.V. for a scholarship. E.A.A. thanks the EPSRC for support (EP/S013172/1). The authors acknowledge the use of the Cirrus U.K. National Tier-2 HPC Service at EPCC (http://www.cirrus.ac.uk) funded by the University of Edinburgh and EPSRC (EP/P020267/1). The authors thank Hanwen Zhang for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c03304.
Author Contributions
† K.H.(K.)P. and N.F. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Carbone M.; Núñez-Pons L.; Castelluccio F.; Avila C.; Gavagnin M. Illudalane Sesquiterpenoids of the Alcyopterosin Series from the Antarctic Marine Soft Coral Alcyonium grandis. J. Nat. Prod. 2009, 72, 1357–1360. 10.1021/np900162t. [DOI] [PubMed] [Google Scholar]; b Fraga B. M. Natural sesquiterpenoids. Nat. Prod. Rep. 2007, 24, 1350–1381. 10.1039/b706427f. [DOI] [PubMed] [Google Scholar]; c Fraga B. M. Natural sesquiterpenoids. Nat. Prod. Rep. 2013, 30, 1226–1264. 10.1039/c3np70047j. [DOI] [PubMed] [Google Scholar]; d Kokubun T.; Scott-Brown A.; Kite G. C.; Simmonds M. S. J. Protoilludane, Illudane, Illudalane, and Norilludane Sesquiterpenoids from Granulobasidium vellereum. J. Nat. Prod. 2016, 79, 1698–1701. 10.1021/acs.jnatprod.6b00325. [DOI] [PubMed] [Google Scholar]
- a Taylor R. D.; MacCoss M.; Lawson A. D. G. Rings in Drugs. J. Med. Chem. 2014, 57, 5845–5859. 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]; b Zhuang C.; Zhang W.; Sheng C.; Zhang W.; Xing C.; Miao Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang L.; Liang R.; Hang C.; Wang H.; Sun L.; Xu L.; Liu D.; Zhang Z.; Zhang X.; Chang F.; Zhao S.; Huang W. A facile solution-phase synthetic approach for constructing phenol-based porous organic cages and covalent organic frameworks. Green Chem. 2020, 22, 2498–2504. 10.1039/C9GC04033A. [DOI] [Google Scholar]; b Li M.; Liu Y.; Duan R.; Wei X.; Yi Y.; Wang Y.; Chen C.-F. Aromatic-Imide-Based Thermally Activated Delayed Fluorescence Materials for Highly Efficient Organic Light-Emitting Diodes. Angew. Chem., Int. Ed. 2017, 56, 8818–8822. 10.1002/anie.201704435. [DOI] [PubMed] [Google Scholar]
- a Kumari A.; Suresh M.; Singh R. B. Total synthesis of the proposed structure of Anti-TMV active tabasesquiterpene A. Tetrahedron 2021, 92, 132282 10.1016/j.tet.2021.132282. [DOI] [Google Scholar]; b Suresh M.; Kumari A.; Singh R. B. Total synthesis of (±)–4,8,14-trihydroxyilludala-2,6,8-triene. Tetrahedron 2019, 75, 3605–3608. 10.1016/j.tet.2019.05.031. [DOI] [Google Scholar]; c Suresh M.; Kumari A.; Das D.; Singh R. B. Total Synthesis of Onitin. J. Nat. Prod. 2018, 81, 2111–2114. 10.1021/acs.jnatprod.8b00335. [DOI] [PubMed] [Google Scholar]; d Suresh M.; Kumar N.; Veeraraghavaiah G.; Hazra S.; Singh R. B. Total Synthesis of Coprinol. J. Nat. Prod. 2016, 79, 2740–2743. 10.1021/acs.jnatprod.6b00277. [DOI] [PubMed] [Google Scholar]
- a Xun M.-M.; Bai Y.; Wang Y.; Hu Z.; Fu K.; Ma W.; Yuan C. Synthesis of Four Illudalane Sesquiterpenes Utilizing a One-Pot Diels–Alder/Oxidative Aromatization Sequence. Org. Lett. 2019, 21, 6879–6883. 10.1021/acs.orglett.9b02511. [DOI] [PubMed] [Google Scholar]; b Kramer N. J.; Hoang T. T.; Dudley G. B. Reaction Discovery Using Neopentylene-Tethered Coupling Partners: Cycloisomerization/Oxidation of Electron-Deficient Dienynes. Org. Lett. 2017, 19, 4636–4639. 10.1021/acs.orglett.7b02261. [DOI] [PubMed] [Google Scholar]; c Hoang T. T.; Birepinte M.; Kramer N. J.; Dudley G. B. Six-Step Synthesis of Alcyopterosin A, a Bioactive Illudalane Sesquiterpene with a gem-Dimethylcyclopentane Ring. Org. Lett. 2016, 18, 3470–3473. 10.1021/acs.orglett.6b01665. [DOI] [PubMed] [Google Scholar]; d Welsch T.; Tran H.-A.; Witulski B. Total Syntheses of the Marine Illudalanes Alcyopterosin I, L, M, N, and C. Org. Lett. 2010, 12, 5644–5647. 10.1021/ol102432q. [DOI] [PubMed] [Google Scholar]; e Witulski B.; Zimmermann A.; Gowans N. D. First total synthesis of the marine illudalane sesquiterpenoid alcyopterosin E. Chem. Commun. 2002, 2984–2985. 10.1039/b209573d. [DOI] [PubMed] [Google Scholar]
- a Tavakoli A.; Dudley G. B. Synthesis of 4,4-Dimethyl-1,6-heptadiyne and Alcyopterosin O. Org. Lett. 2020, 22, 8947–8951. 10.1021/acs.orglett.0c03356. [DOI] [PubMed] [Google Scholar]; b Gaston R.; Geldenhuys W. J.; Dudley G. B. Synthesis of Illudinine from Dimedone and Identification of Activity as a Monoamine Oxidase Inhibitor. J. Org. Chem. 2020, 85, 13429–13437. 10.1021/acs.joc.0c01301. [DOI] [PubMed] [Google Scholar]; c Zeng Z.; Zhao Y.; Zhang Y. Divergent total syntheses of five illudalane sesquiterpenes and assignment of the absolute configuration. Chem. Commun. 2019, 55, 4250–4253. 10.1039/C9CC00933G. [DOI] [PubMed] [Google Scholar]; d Tanaka R.; Nakano Y.; Suzuki D.; Urabe H.; Sato F. Selective Preparation of Benzyltitanium Compounds by the Metalative Reppe Reaction. Its Application to the First Synthesis of Alcyopterosin A. J. Am. Chem. Soc. 2002, 124, 9682–9683. 10.1021/ja027008o. [DOI] [PubMed] [Google Scholar]
- Ichihara A. Retro-Diels-Alder Strategy in Natural Product Synthesis. Synthesis 1987, 1987, 207–222. 10.1055/s-1987-27894. [DOI] [Google Scholar]
- Kabuki A.; Yamaguchi J. Formal Syntheses of Dictyodendrins B, C, and E by a Multi-substituted Indole Synthesis. Synthesis 2022, 10.1055/a-1786-9881. [DOI] [Google Scholar]
- a Nicolaou K. C.; Barnette W. E.; Ma P. A remarkably simple, highly efficient, and stereoselective synthesis of steroids and other polycyclic systems. Total synthesis of estra-1,3,5(10)-trien-17-one via intramolecular capture of o-quinodimethanes generated by cheletropic elimination of sulfur dioxide. J. Org. Chem. 1980, 45, 1463–1470. [Google Scholar]; b Nicolaou K. C.; Vassilikogiannakis G.; Mägerlein W.; Kranich R. Total Synthesis of Colombiasin A. Angew. Chem., Int. Ed. 2001, 40, 2482–2486. . [DOI] [PubMed] [Google Scholar]; c Mejorado L. H.; Pettus T. R. R. Total Synthesis of (+)-Rishirilide B: Development and Application of General Processes for Enantioselective Oxidative Dearomatization of Resorcinol Derivatives. J. Am. Chem. Soc. 2006, 128, 15625–15631. 10.1021/ja062987w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Haut F.-L.; Habiger C.; Speck K.; Wurst K.; Mayer P.; Korber J. N.; Müller T.; Magauer T. Synthetic Entry to Polyfunctionalized Molecules through the [3+2]-Cycloaddition of Thiocarbonyl Ylides. J. Am. Chem. Soc. 2019, 141, 13352–13357. 10.1021/jacs.9b07729. [DOI] [PubMed] [Google Scholar]; e Prilezhaeva E. N. Sulfones and sulfoxides in the total synthesis of biologically active natural compounds. Russ. Chem. Rev. 2000, 69, 367–408. 10.1070/RC2000v069n05ABEH000561. [DOI] [Google Scholar]
- a Tanaka S.; Asako T.; Ota E.; Yamaguchi J. Synthesis of a Pentaarylcarbazole: Installation of Different Aryl Groups on a Benzenoid Moiety. Chem. Lett. 2020, 49, 918–920. 10.1246/cl.200302. [DOI] [Google Scholar]; b Asako T.; Suzuki S.; Itami K.; Muto K.; Yamaguchi J. Synthesis of a Heptaarylisoquinoline: Unusual Disconnection for Constructing Isoquinoline Frameworks. Chem. Lett. 2018, 47, 968–970. 10.1246/cl.180429. [DOI] [Google Scholar]; c Suzuki S.; Asako T.; Itami K.; Yamaguchi J. Modular synthesis of heptaarylindole. Org. Biomol. Chem. 2018, 16, 3771–3776. 10.1039/C8OB00993G. [DOI] [PubMed] [Google Scholar]; d Suzuki S.; Itami K.; Yamaguchi J. Synthesis of Octaaryl Naphthalenes and Anthracenes with Different Substituents. Angew. Chem., Int. Ed. 2017, 56, 15010–15013. 10.1002/anie.201709332. [DOI] [PubMed] [Google Scholar]; e Suzuki S.; Segawa Y.; Itami K.; Yamaguchi J. Synthesis and characterization of hexaarylbenzenes with five or six different substituents enabled by programmed synthesis. Nat. Chem. 2015, 7, 227–233. 10.1038/nchem.2174. [DOI] [PubMed] [Google Scholar]
- a Nakayama J.; Yamaoka S.; Nakanishi T.; Hoshino M. 3,4-Di-tert-butylthiophene 1,1-dioxide, a convenient precursor of o-Di-tert-butylbenzene and its derivatives. J. Am. Chem. Soc. 1988, 110, 6598–6599. 10.1021/ja00227a069. [DOI] [Google Scholar]; b Raasch M. S. Addition-rearrangement reactions of halogenated thiophene dioxides with furans. J. Org. Chem. 1980, 45, 867–870. 10.1021/jo01293a020. [DOI] [Google Scholar]; c Moiseev A. M.; Balenkova E. S.; Nenajdenko V. G. Reactions of electron-withdrawing thiophene 1,1-dioxides with furans. A novel reaction pathway. Russ. Chem. Bull. 2006, 55, 712–717. 10.1007/s11172-006-0318-8. [DOI] [Google Scholar]; d Dmowski W.; Manko V. A.; Nowak I. 3-Chloro-4-fluorothiophene-1,1-dioxide. A new synthetically useful fluorodiene. J. Fluorine Chem. 1998, 88, 143–151. 10.1016/S0022-1139(98)00109-2. [DOI] [Google Scholar]
- Wang X.; Barbosa J.; Blomgren P.; Bremer M. C.; Chen J.; Crawford J. J.; Deng W.; Dong L.; Eigenbrot C.; Gallion S.; Hau J.; Hu H.; Johnson A. R.; Katewa A.; Kropf J. E.; Lee S. H.; Liu L.; Lubach J. W.; Macaluso J.; Maciejewski P.; Mitchell S. A.; Ortwine D. F.; DiPaolo J.; Reif K.; Scheerens H.; Schmitt A.; Wong H.; Xiong J.-M.; Xu J.; Zhao Z.; Zhou F.; Currie K. S.; Young W. B. Discovery of Potent and Selective Tricyclic Inhibitors of Bruton’s Tyrosine Kinase with Improved Druglike Properties. ACS Med. Chem. Lett. 2017, 8, 608–613. 10.1021/acsmedchemlett.7b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenajdenko V. G.; Moiseev A. M.; Balenkova E. S. A novel method for the oxidation of thiophenes. Synthesis of thiophene 1,1-dioxides containing electron-withdrawing substituents. Russ. Chem. Bull. 2004, 53, 2241–2247. 10.1007/s11172-005-0107-9. [DOI] [Google Scholar]
- Shaabani A.; Mirzaei P.; Naderi S.; Lee D. G. Green oxidations. The use of potassium permanganate supported on manganese dioxide. Tetrahedron 2004, 60, 11415–11420. 10.1016/j.tet.2004.09.087. [DOI] [Google Scholar]
- Li L.; Liu Q.; Chen J.; Huang Y. Alcohol-Directed ortho-C–H Alkenylation. Synlett 2019, 30, 1366–1370. 10.1055/s-0037-1611538. [DOI] [Google Scholar]
- Domingo L. R.; Saez J. A. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. 10.1039/b909611f. [DOI] [PubMed] [Google Scholar]
- Domingo L. R.; Chamorro E.; Perez P. Understanding the mechanism of non-polar Diels-Alder reactions. A comparative ELF analysis of concerted and stepwise diradical mechanisms. Org. Biomol. Chem. 2010, 8, 5495–5504. 10.1039/c0ob00563k. [DOI] [PubMed] [Google Scholar]
- Bickelhaupt F. M.; Houk K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Ferrall R. A. M. Relationships between E2 and E1cB mechanisms of β-elimination. J. Chem. Soc. B 1970, 0, 274–277. 10.1039/J29700000274. [DOI] [Google Scholar]; b Jencks W. P. General acid-base catalysis of complex reactions in water. Chem. Rev. 1972, 72, 705–718. 10.1021/cr60280a004. [DOI] [Google Scholar]
- Hare S. R.; Tantillo D. J. Post-transition state bifurcations gain momentum – current state of the field. Pure Appl. Chem. 2017, 89, 679–698. 10.1515/pac-2017-0104. [DOI] [Google Scholar]
- a Woodward R. B.; Katz T. J. The mechanism of the Diels-Alder reaction. Tetrahedron 1959, 5, 70–89. 10.1016/0040-4020(59)80072-7. [DOI] [Google Scholar]; b Dewar M. J. S. Mechanism of the diels-alder reaction. Tetrahedron Lett. 1959, 1, 16–18. 10.1016/S0040-4039(01)82720-9. [DOI] [Google Scholar]; c Hoffmann R.; Woodward R. B. Selection Rules for Concerted Cycloaddition Reactions. J. Am. Chem. Soc. 1965, 87, 2046–2048. 10.1021/ja01087a034. [DOI] [Google Scholar]; d Houk K. N.; Liu F.; Yang Z.; Seeman J. I. Evolution of the Diels-Alder Reaction Mechanism since the 1930s: Woodward, Houk with Woodward, and the Influence of Computational Chemistry on Understanding Cycloadditions. Angew. Chem., Int. Ed. 2021, 60, 12660–12681. 10.1002/anie.202001654. [DOI] [PubMed] [Google Scholar]
- a Caramella P.; Quadrelli P.; Toma L. An unexpected bispericyclic transition structure leading to 4+2 and 2+4 cycloadducts in the endo dimerization of cyclopentadiene. J. Am. Chem. Soc. 2002, 124, 1130–1131. 10.1021/ja016622h. [DOI] [PubMed] [Google Scholar]; b Toma L.; Romano S.; Quadrelli P.; Caramella P. Merging of 4+2 and 2+4 cycloaddition paths in the regiospecific dimerization of methacrolein. A case of concerted crypto-diradical cycloaddition. Tetrahedron Lett. 2001, 42, 5077–5080. 10.1016/S0040-4039(01)00891-7. [DOI] [Google Scholar]; The term “bispericyclic” was proposed to describe bifurcating pathways that lead to the same reaction product, as opposed to “bipericyclic” where different products arise from a single transition state (see ref (23)g).
- a Xu M.-M.; Yang L.; Tan K.; Chen X.; Lu Q.-T.; Houk K. N.; Cai Q. An enantioselective ambimodal cross-Diels–Alder reaction and applications in synthesis. Nat. Catal. 2021, 4, 892–900. 10.1038/s41929-021-00687-x. [DOI] [Google Scholar]; For selected examples of Diels-Alder reactions proceeding through ambimodal transition states, see:; b Zhang H.; Novak A. J. E.; Jamieson C. S.; Xue X.-S.; Chen S.; Trauner D.; Houk K. N. Computational Exploration of the Mechanism of Critical Steps in the Biomimetic Synthesis of Preuisolactone A, and Discovery of New Ambimodal (5 + 2)/(4 + 2) Cycloadditions. J. Am. Chem. Soc. 2021, 143, 6601–6608. 10.1021/jacs.1c01856. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang Z.; Jamieson C. S.; Zhao Y.-L.; Li D.; Ohashi M.; Houk K. N.; Tang Y. Enzyme-Catalyzed Inverse-Electron Demand Diels–Alder Reaction in the Biosynthesis of Antifungal Ilicicolin H. J. Am. Chem. Soc. 2019, 141, 5659–5663. 10.1021/jacs.9b02204. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pham H. V.; Houk K. N. Diels-Alder reactions of allene with benzene and butadiene: concerted, stepwise, and ambimodal transition states. J. Org. Chem. 2014, 79, 8968–8976. 10.1021/jo502041f. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ussing B. R.; Hang C.; Singleton D. A. Dynamic effects on the periselectivity, rate, isotope effects, and mechanism of cycloadditions of ketenes with cyclopentadiene. J. Am. Chem. Soc. 2006, 128, 7594–7607. 10.1021/ja0606024. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ess D. H.; Wheeler S. E.; Iafe R. G.; Xu L.; Celebi-Olcum N.; Houk K. N. Bifurcations on potential energy surfaces of organic reactions. Angew. Chem., Int. Ed. 2008, 47, 7592–7601. 10.1002/anie.200800918. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review, see:; g Houk K. N.; Xue X.-S.; Liu F.; Chen Y.; Chen X.; Jamieson C. Computations on Pericyclic Reactions Reveal the Richness of Ambimodal Transition States and Pericyclases. Isr. J. Chem. 2022, 62, e202100071 10.1002/ijch.202100071. [DOI] [Google Scholar]
- a Wang Z.; Hirschi J. S.; Singleton D. A. Recrossing and dynamic matching effects on selectivity in a Diels-Alder reaction. Angew. Chem., Int. Ed. 2009, 48, 9156–9159. 10.1002/anie.200903293. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Thomas J. B.; Waas J. R.; Harmata M.; Singleton D. A. Control elements in dynamically determined selectivity on a bifurcating surface. J. Am. Chem. Soc. 2008, 130, 14544–14555. 10.1021/ja802577v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lee S.; Goodman J. M. VRAI-selectivity: calculation of selectivity beyond transition state theory. Org. Biomol. Chem. 2021, 19, 3940–3947. 10.1039/D1OB00234A. [DOI] [PubMed] [Google Scholar]; b Lee S.; Goodman J. M. Rapid Route-Finding for Bifurcating Organic Reactions. J. Am. Chem. Soc. 2020, 142, 9210–9219. 10.1021/jacs.9b13449. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- Yang Z.; Dong X.; Yu Y.; Yu P.; Li Y.; Jamieson C.; Houk K. N. Relationships between Product Ratios in Ambimodal Pericyclic Reactions and Bond Lengths in Transition Structures. J. Am. Chem. Soc. 2018, 140, 3061–3067. 10.1021/jacs.7b13562. [DOI] [PubMed] [Google Scholar]
- a Neese F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]; b Neese F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327 10.1002/wcms.1327. [DOI] [Google Scholar]
- a Young T. A.; Silcock J. J.; Sterling A. J.; Duarte F. autodE: Automated Calculation of Reaction Energy Profiles- Application to Organic and Organometallic Reactions. Angew. Chem., Int. Ed. 2021, 60, 4266–4274. 10.1002/anie.202011941. [DOI] [PubMed] [Google Scholar]; b Bannwarth C.; Ehlert S.; Grimme S. GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]
- a Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]; b Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Liakos D. G.; Sparta M.; Kesharwani M. K.; Martin J. M.; Neese F. Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theory Comput. 2015, 11, 1525–1539. 10.1021/ct501129s. [DOI] [PubMed] [Google Scholar]
- Grimme S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem.–Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
- Young T.duartegroup/otherm, 2020https://github.com/duartegroup/otherm.
- a Ussing B. R.; Hang C.; Singleton D. A. Dynamic effects on the periselectivity, rate, isotope effects, and mechanism of cycloadditions of ketenes with cyclopentadiene. J. Am. Chem. Soc. 2006, 128, 7594–7607. 10.1021/ja0606024. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Toma L.; Romano S.; Quadrelli P.; Caramella P. Merging of 4+2 and 2+4 cycloaddition paths in the regiospecific dimerization of methacrolein. A case of concerted crypto-diradical cycloaddition. Tetrahedron Lett. 2001, 42, 5077–5080. 10.1016/S0040-4039(01)00891-7. [DOI] [Google Scholar]; c Caramella P.; Quadrelli P.; Toma L. An unexpected bispericyclic transition structure leading to 4+2 and 2+4 cycloadducts in the endo dimerization of cyclopentadiene. J. Am. Chem. Soc. 2002, 124, 1130–1131. 10.1021/ja016622h. [DOI] [PubMed] [Google Scholar]; d Patel A.; Chen Z.; Yang Z.; Gutierrez O.; Liu H. W.; Houk K. N.; Singleton D. A. Dynamically Complex [6+4] and [4+2] Cycloadditions in the Biosynthesis of Spinosyn A. J. Am. Chem. Soc. 2016, 138, 3631–3634. 10.1021/jacs.6b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.