

Abstract
Brilacidin (PMX-30063), a non-peptide defensin-mimetic small molecule, inhibits SARS-CoV-2 viral infection but the anti-viral mechanism is not defined. Here we determined its effect on the specific step of the viral life cycle. Brilacidin blocked SARS-CoV-2 infection but had no effect after viral entry. Brilacidin inhibited pseudotyped SARS-CoV-2 viruses expressing spike proteins from the P.1 Brazil strain and the B.1.1.7 UK strain. Brilacidin affected viral attachment in hACE2-dependent and independent manners depending on the concentrations. The inhibitory effect on viral entry was not mediated through blocking the binding of either the spike receptor-binding domain or the spike S1 protein to hACE2 proteins. Taken together, brilacidin inhibits SARS-CoV-2 infection by blocking viral entry and is active against SARS-CoV-2 variants.
Keywords: SARS-CoV-2, Brilacidin, Viral Entry
Introduction
Severe acute respiratory syndrome-related coronavirus (SARS-CoV-2), an enveloped, non-segmented, positive-sense RNA virus, causes coronavirus disease 19 (COVID-19) [1,2]. While several effective vaccines against SARS-CoV-2 virus have been developed, global inequalities of vaccine access and challenges of anti-vaccine aggression, in part, have contributed to the difficulty of halting spread of the virus [3–5]. Newly emerged SARS-CoV-2 variants that are more transmissible remain major challenges for combating COVID-19 [6]. Thus, effective agents with broad-spectrum anti-viral activity to prevent the virus spread are urgently needed.
Defensins are a major class of anti-microbial peptides active against a broad spectrum of microbes including various enveloped and non-enveloped viruses [7]. We have recently shown that human α-defensins, such as human neutrophil peptides 1–3 (HNP1–3) and human defensin 5 (HD5), inhibit SARS-CoV-2 infection by blocking viral entry [8]. Despite their potent anti-viral activities, large scale production of defensins (~35 amino acids) remains the main challenge impeding their use in clinical applications. The development of synthetic molecules with the properties of defensins would be an approach to overcoming the obstacles to developing defensins as therapeutics.
Brilacidin (PMX-30063) is a synthetic, non-peptide, defensin-mimetic with an amphiphilic structure (Figure 1A). Brilacidin exhibits broad-spectrum anti-microbial activities and has demonstrated efficacy in treating Acute Bacterial Skin and Skin Structure Infections (ABSSSI) in Phase 2 clinical trials as an intravenous agent [9–11]. In vitro, brilacidin at 0.5 – 1 µM has potent bactericidal activities against Gram-positive and Gram-negative bacteria, with minimal propensity for the development of resistance [9,10,12,13]. Similar to the lipopeptidic drug daptomycin, brilacidin appears to cause membrane depolarization in Staphylococcus aureus (Gram-positive) [9]. In a Phase 2b clinical trial, brilacidin showed comparable efficacy as a single-dose compared to a 7-day regimen of daptomycin [14].
Figure 1:
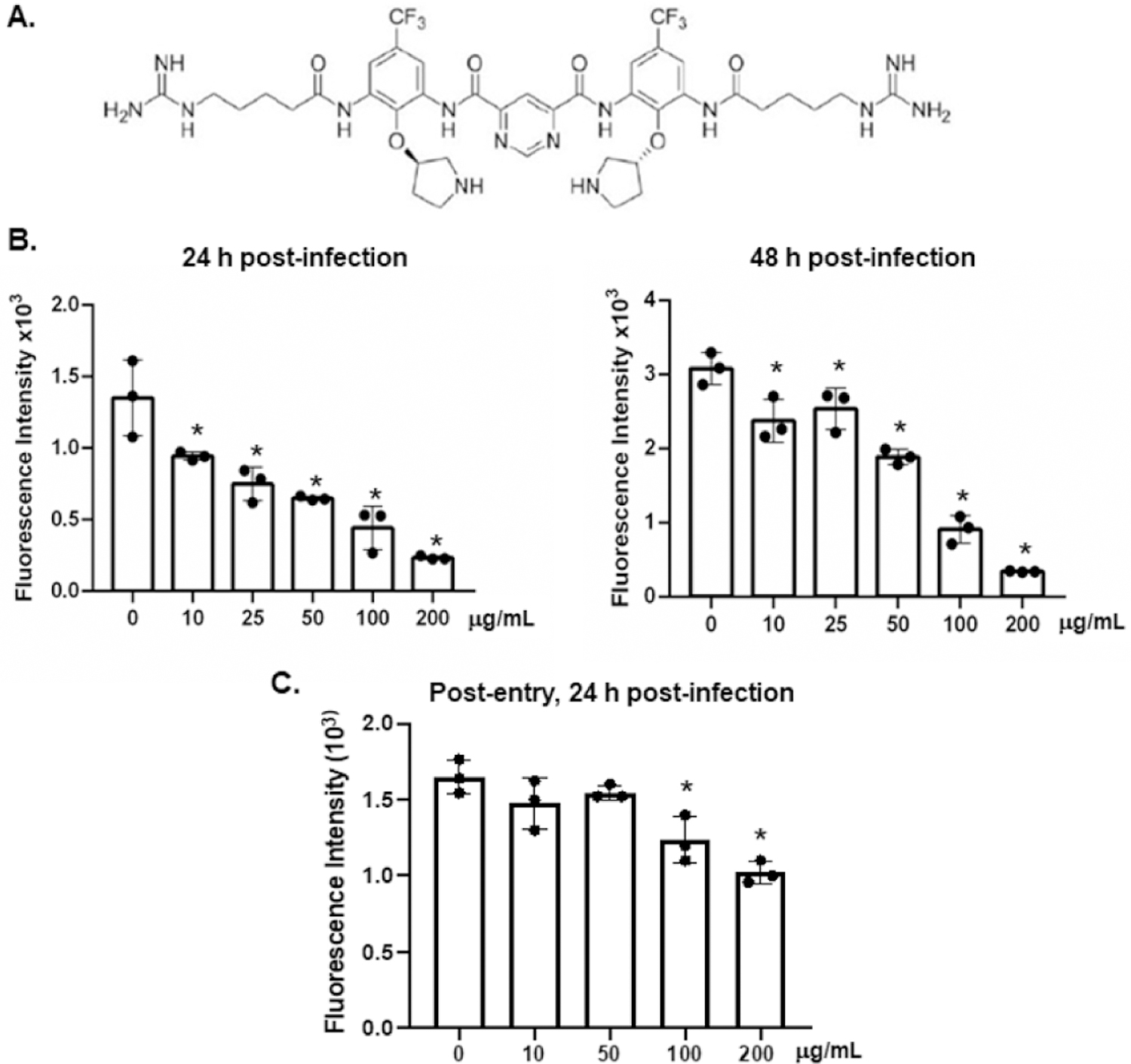
Brilacidin inhibits infection by replication-competent SARS-CoV-2 viruses. (A) The structure of brilacidin tetrahydrochloride. (B) Replication-competent SARS-CoV-2 viruses expressing mNeonGreen were incubated (MOI of 5) with brilacidin at different concentrations at 37°C for 1h and then added to Vero E6 cells for 2h. Infected cells were then cultured in FluoroBrite medium with 10% FBS in the presence of the inhibitor for 24 or 48h. (C) Vero E6 cells were exposed to replication-competent SARS-CoV-2 viruses expressing mNeonGreen for 2h. Infected cells were washed with PBS and then treated with brilacidin at different concentrations for 24h. Fluorescence from productive viral infections was measured using a Biotek Cytation 5. Differences between brilacidin-treated viruses and non-treated control viruses were calculated by one-way ANOVA; *p < 0.05. Data represent mean ± SD of three replicates.
Anti-SARS-CoV-2 activity of brilacidin has recently been demonstrated in vitro using a multiple-round infection assay with replication-competent viruses [15]. Cells were pre-treated with brilacidin followed by SARS-CoV-2 infection of Vero E6 and Calu-3 cells, and brilacidin was present during the viral attachment and infection [15]. After confirmed limited cytotoxic activity, the 50 percent and 90 percent inhibitory concentrations (IC50 and IC90) of brilacidin against SARS-CoV-2 viruses were 0.565 and 2.63 µM, respectively [15]. Brilacidin inhibits various human coronaviruses possibly through interacting with heparan sulfate proteoglycans for viral attachment [16]. In development under U.S. FDA Fast Track designation, the efficacy of brilacidin in treating moderate-to-severe COVID-19 in hospitalized patients via intravenous delivery is being assessed in a Phase 2 randomized, blinded, placebo-controlled clinical trial (NCT04784897) [17]. To gain a better understanding of the anti-SARS-CoV-2 properties of brilacidin, we determined the step of viral infection affected by brilacidin and its impact on SARS-CoV-2 variants.
Materials and Methods
Reagents
The construct for full-length SARS-CoV-2-Wuhan-Hu-1 surface (spike) proteins (GenBank accession number QHD43416) [18] was codon optimized, synthesized, and subcloned into the pcDNA3.1(+) vector (Thermo Fisher Scientific, Waltham, MA, USA) as described [19]. Plasmids encoding spike proteins of B.1.1.7 and P.1 variants were kindly provided by Dennis Burton (The Scripps Research Institute, La Jolla, CA, USA). Brilacidin, as brilacidin tetrahydrochloride (mwt 1082.7), was provided by Innovation Pharmaceuticals, Inc (Wakefield, MA, USA). HEK293T-hACE2 cells and HeLa-hACE2 cells were (kindly provided by Hyeryun Choe at The Scripps Research Institute, Jupiter, Florida, USA) [20] and by Dennis Burton (The Scripps Research Institute, La Jolla, CA, USA) [21]. HEK293T, Caco-2, HeLa, and Vero E6 cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). Tissue culture media and fetal bovine serum (FBS) were from Sigma-Aldrich.
Cell culture and virus infection
All cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated FBS. Replication-competent SARS-CoV-2 expressing mNeonGreen, kindly provided by Pei-Yong Shi at the University of Texas Medical Branch, Galveston, TX, USA, was propagated in Vero E6 cells as described previously [22]. Experiments were performed in a biosafety level 3 laboratory with personal protection equipment including powered air-purifying respirators (Breathe Easy, 3M), Tyvek suits, aprons, sleeves, booties, and double gloves. Virus titers were determined by plaque assays in Vero E6 cells as described previously [23]. For the infection assay, Vero E6 cells were seeded at 1.5 x 104 cells/well in black 96-well glass plates (Greiner, Monroe, NC, USA), and cultured overnight. Cells were exposed to viruses with or without brilacidin treatment in 30 μl at a multiplicity of infection (MOI) of 5 for 1h followed by the addition of 100 μl FluoroBrite medium containing 2% FBS in the presence of various concentrations of brilacidin. Fluorescence from productive viral infections was monitored at 24 and 48h after infection using a Biotek Cytation 5.
Replication-defective HIV-1 luciferase-expressing reporter viruses pseudotyped with SARS-CoV-2 spike proteins were produced as described previously [19]. Briefly, HEK293T cells were transfected (Lipofectamine 3000; Thermo Fisher Scientific, Waltham, MA, USA) with a plasmid encoding the envelope-deficient HIV-1 NL4–3 virus with the luciferase reporter gene (pNL4–3.Luc.R+E-from N. Landau, New York University) were co-transfected with a pcDNA3.1 plasmid expressing the SARS-CoV-2 spike proteins. The supernatant was collected 48 h after transfection and filtered. Virus stocks were analyzed for HIV-1 p24 antigen by the AlphaLISA HIV p24 kit (PerkinElmer, Waltham, MA, USA). Serum-free (SF) SARS-CoV-2 pseudotyped viruses were produced by changing media to DMEM without serum 24h after transfection. After an additional 24h incubation, SF viruses were collected. Virus stocks contained approximately 200 ng/ml of HIV p24 proteins.
For infection assays, 5 x 104 cells/well in a 48-well plate were cultured overnight. Pseudotyped SARS-CoV-2 viruses were incubated at 37°C for 1h with brilacidin. The brilacidin-virus mixture (100 ml) was then added to cells. After 1 – 2h viral attachment, medium with 10% FBS (500 ml) was added, and the infected cells were cultured for 48 – 72h. Brilacidin was not added back during the course of infection. Luciferase activity (relative light units; RLU) was then measured using Luciferase Substrate Buffer (Promega Inc, Madison, WI, USA) on a 2300 EnSpire Multilabel Plate Reader (PerkinElmer, Waltham, MA). Results are reported as the average percent of control calculated using the formula: (RLU of treated cells/RLUs of untreated cells) x 100.
Cytotoxicity assay
HeLa-hACE2 and HEK293T-hACE2 cells were plated at 1 x 104 cells/well in 96-well plates and treated with various concentrations of brilacidin for 24 hours. Cell viability was analyzed using MTS-based CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacture’s instruction.
Viral attachment assay
HeLa-hACE2 cells seeded at 5 x 104 per well in 48-well plates were cultured overnight. Cells were incubated with pseudotyped viruses with or without defensin treatment at 4°C for 2h. Cells were washed with cold PBS three times and lysed with 100 ml of 1% Triton X-100. Cell-associated HIV p24 was determined by AlphaLISA HIV p24 kit (PerkinElmer).
SARS-COV-2 RBD/S1 protein and hACE2 binding assay
Greiner Microlon 200 plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated with recombinant SARS-CoV-2 S1 proteins or receptor-binding domain (RBD) proteins at 100 ng/well in 0.1M bicarbonate buffer pH 9.6 at 4°C for 24h. The plates were blocked with 2% non-fat dry milk in PBS (w/v) for 30 minutes at 37°C, washed with PBS+0.05% tween20, and treated with different concentrations of brilacidin diluted in 2% non-fat dry milk in PBS (w/v) for 1h at 37°C. Biotinylated hACE2 proteins (0.5 μg/mL) were added to the samples and incubated at 37°C for 1h. After washing, the bound hACE2 protein was detected by incubating with alkaline phosphatase-conjugated streptavidin at 37°C for 1h. Plates were washed, and the binding of hACE2 protein to recombinant S1 or RBD proteins was detected by adding 1 mg/mL of ρ-nitrophenol in DEA buffer at pH 9.8 and measuring the resulting signal at 405 nm using a Tecan Infinite F50 Absorbance Microplate Reader (Tecan Life Sciences, Männedorf, Switzerland).
Statistical analysis
Statistical comparisons were performed using one-way ANOVA; p < 0.05 was considered significant. Prism 8 (GraphPad Software, LLC, San Diego, CA, USA) was used.
Results
Brilacidin inhibits replication-competent SARS-CoV-2 virus infection when added before viral entry
We first determined the effect of brilacidin on infection by replication-competent SARS-CoV-2 (Wuhan strain) viruses expressing mNeonGreen. This assay system allows us to detect infection by continuous measurement of fluorescence over time and has an advantage of assessing anti-viral effect within 24h to avoid drug-induced cytotoxicity after pro-longed incubation. Viruses were incubated with brilacidin at different concentrations for 1h before infection of Vero E6 cells. Brilacidin was present during the infection. The fluorescent signal from viral infection was measured at 24 and 48h after infection. In agreement with a previous report using WA and Italian strains of SARS-CoV-2 [24], brilacidin blocked replication competent viral infection in a dose-dependent manner with an IC50 of 28.5 µM at 24h post-infection (Figure 1B). The effect of brilacidin (9.2 – 46 µM, 10 – 50 μg/ml) on viral infection diminished at 48h post-infection (Figure 1B). Brilacidin at 100 and 200 μg/ml inhibited SARS-CoV-2 viruses by 70 – 89% at 24 or 48h.
To determine whether brilacidin inhibited SARS-CoV-2 infection after viral entry in this replication component virus system, cells were exposed to viruses for 2h and then washed with PBS to remove unbound viruses. Infected cells were treated with brilacidin for 24h before measuring the fluorescent signal. Brilacidin at 10 – 50 μg/ml had no effect on viral infection when added after viral entry. Brilacidin at high concentrations (100 – 200 μg/ml) exhibited a weak anti-viral effect on infected cells (Figure 1C). Because the inhibitor at 100 μg/ml had a slight inhibitory effect on cell proliferation (Figure 2) [24], the anti-viral activity of brilacidin at high concentrations might be associated with brilacidin-associated cytotoxicity. Our subsequent infection experiments were conducted using non-cytotoxic concentrations (50 μg/ml).
Figure 2:
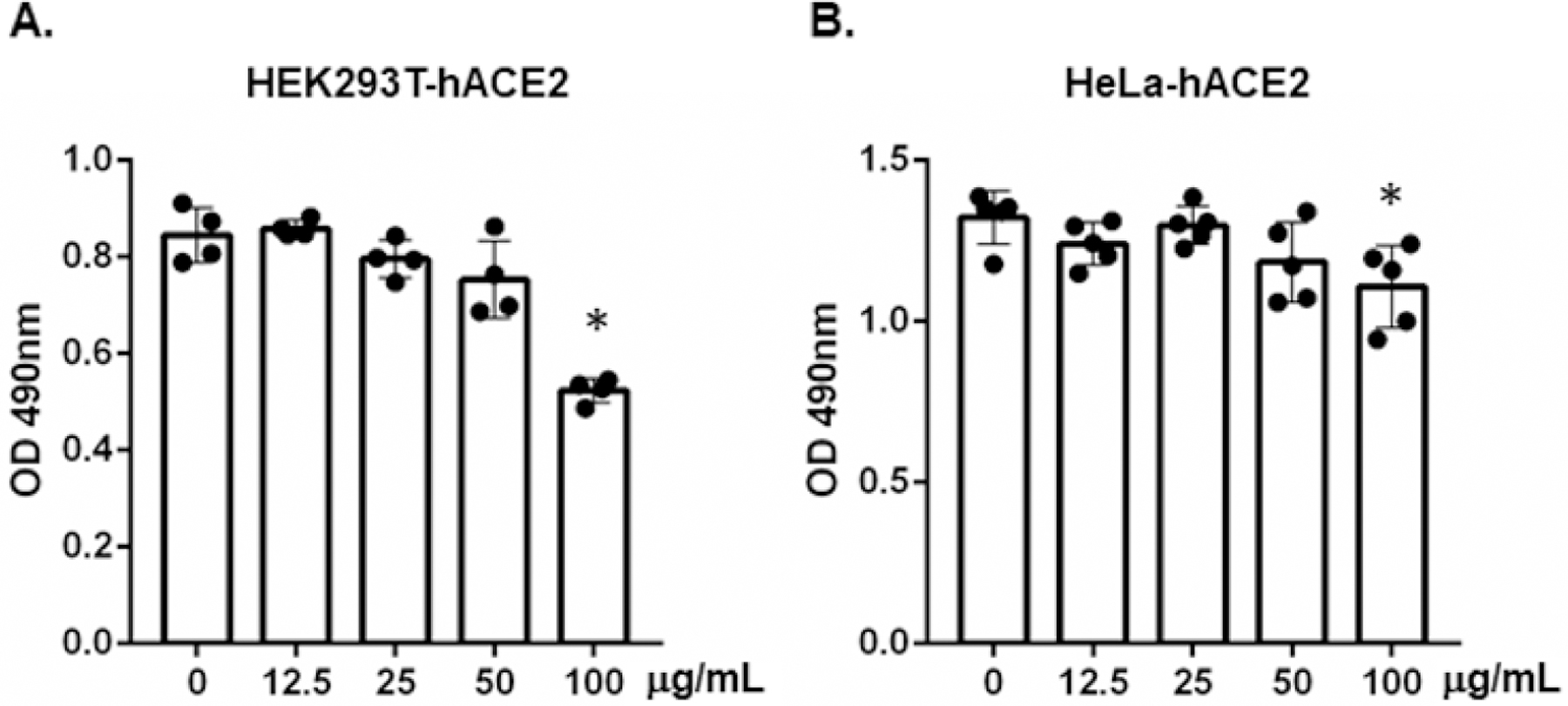
The effect of brilacidin on cell viability. HEK293T-hACE2 cells (A) and HeLa-hACE2 cells (B) were treated with the indicated concentrations of brilacidin for 24h, and cell viability was determined by MTS-based CellTiter 96® AQueous One Solution Cell Proliferation Assay. Data represent mean ± SD of three or four replicates, and were analyzed by one-way ANOVA; *p < 0.05.
The anti-SARS-CoV-2 effect of brilacidin in a single-cycle infection assay
Replication-competent SARS-CoV-2 viruses induce cytopathic effects on cells. Thus, we determined the step of the viral life cycle affected by brilacidin using a non-pathogenic, replication-defective HIV pseudotyped viruses expressing SARS-CoV-2 spike proteins [25]. A single-cycle infection also allowed us to clearly distinguish pre- and post-entry steps since synchronization of infection by replication competent viruses is more challenging. Viruses were pretreated with brilacidin at non-cytotoxic concentrations at 37°C for 1h. The mixture of virus-brilacidin was added to HEK293T-hACE2 and HeLa-hACE2 cells for 2h. HEK293T-hACE2 cells had a higher abundance of hACE2 and TMPRSS receptors than did HeLa-hACE2 cells. Infected cells were cultured for 2 days before measuring luciferase activity. We found that brilacidin pre-treatment blocked infection by pseudotyped SARS-CoV-2 viruses in HEK293T-hACE2 cells in a dose-dependent manner. Brilacidin at 0.1 μg/mL suppressed viral infection by 28.7%, although the inhibition was not statistically significant. At 1 and 12.5 μg/mL, brilacidin significantly inhibited viral infection by 40% and 42%, respectively. Infection was suppressed by 64% and 73% when viruses were pre-treated with brilacidin at 25 and 50 μg/mL, respectively (Figure 3A). The IC50 from 4 experiments was 4.15 μg/ml (3.82 µM). In HeLa-hACE2 cells, infection was inhibited by 45% and 55% when viruses were pre-treated with brilacidin at 25 and 50 μg/mL, respectively (Figure 3B). Brilacidin also suppressed infection of Caco-2 cells, which expressed endogenous ACE2 (Figure 3C) to a similar degree.
Figure 3:
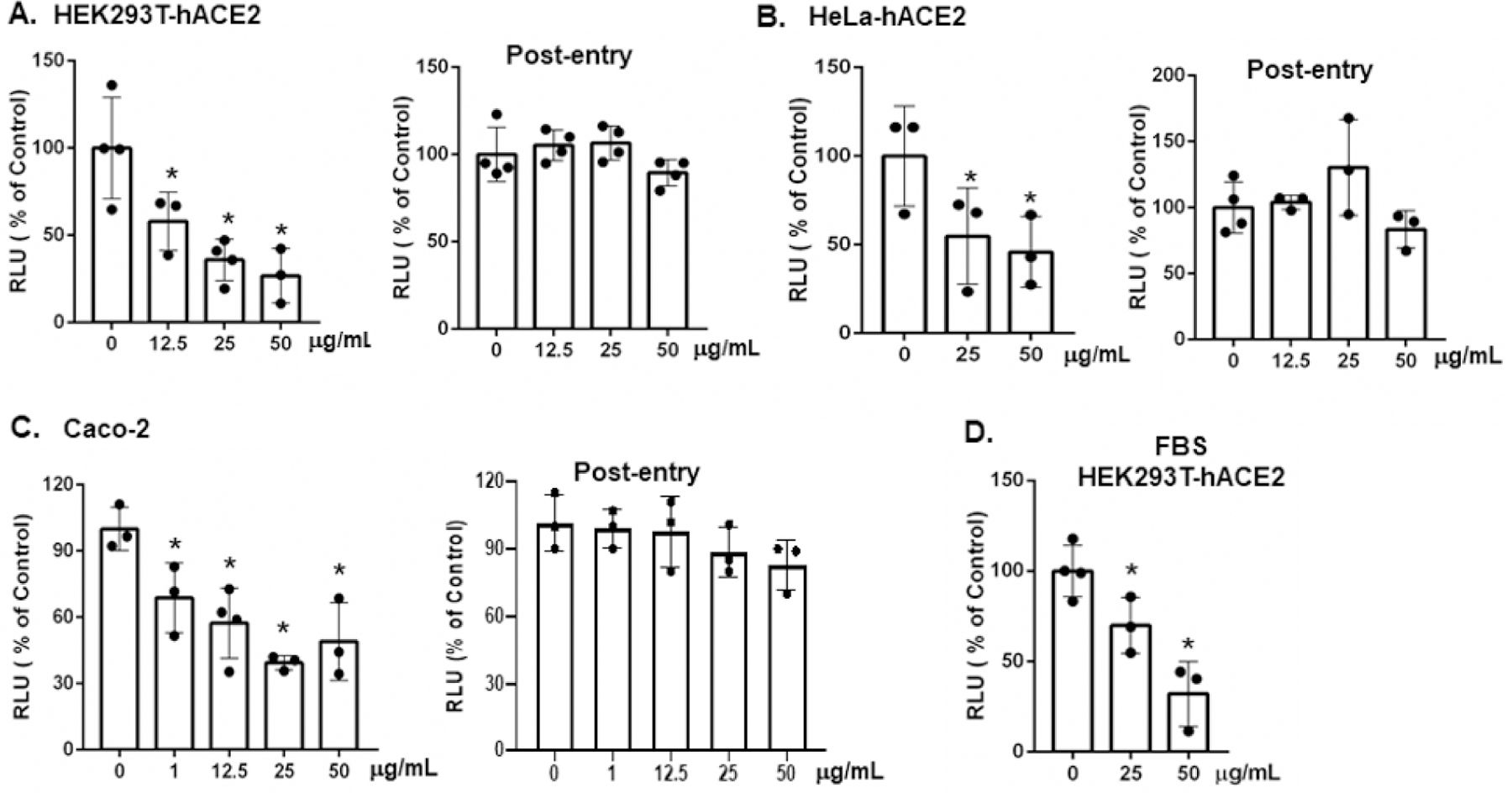
Brilacidin inhibits infection by pseudotyped viruses expressing SARS-CoV-2 spike proteins. HIV pseudotyped luciferase reporter viruses expressing SARS-CoV-2 spike proteins were treated with the indicated concentrations of brilacidin at 37°C for 1h before infection of HEK293T-hACE2 cells (A), HeLa-hACE2 cells (B) or (C) CaCo-2 cells as described in Methods. For post-entry experiments, cells were infected by pseudotyped SARS-CoV-2 viruses for 2h. After washing off unbound viruses, infected cells were treated with the indicated concentrations of brilacidin for 3 days. (D) Pseudotyped SARS-CoV-2 viruses were treated with brilacidin in the presence of 10% FBS at 37°C for 1h before infection of HEK293T-hACE2 cells. Data represents means ± SD of three or four replicates and were analyzed by one-way ANOVA; *p < 0.05.
To determine whether brilacidin inhibited SARS-CoV-2 infection after viral infection, HEK293T-hACE2, HeLa-hACE2 or Caco-2 cells were infected with pseudotyped SARS-CoV-2 viruses at 37°C for 2h and then treated with brilacidin for 2 days before measuring luciferase activity in infected cells. We found that the protective effect of brilacidin was lost when the inhibitor was added after viral infection (Figure 3A–3C), indicating that brilacidin acts to block SARS-CoV-2 infection at the step of viral entry.
FBS interferes with the anti-viral effect of defensins when viral inhibition is mediated through the lectin properties of defensins [26–28]. Because brilacidin is a defensin-mimetic molecule, we examined the effect of FBS on brilacidin-mediated anti-SARS-CoV-2 activity and found that the presence of FBS at the pre-treatment of virus of the experiment diminished the anti-viral activity of brilacidin at 25 μg/mL to 30% inhibition compared to 64% inhibition in the samples without FBS (Figure 3D). However, FBS did not have a significant impact on anti-viral activity of brilacidin at 50 μg/mL (68% in the presence of FBS vs 73% inhibition in the absence of FBS).
Brilacidin inhibits infection by SARS-CoV-2 P.1 and B.1.1.7 variants
SARS-CoV-2 P.1 and B.1.1.7 variants from Brazil and the United Kingdom, respectively, are highly transmissible and increase the risk of death [29–32]. Anti-viral agents have more potential if they block infection across various SARS-CoV-2 variants. We determined the effect of brilacidin on pseudotyped viruses expressing spike proteins from the P.1 and B.1.1.7 variants. Pseudotyped viruses expressing SARS-CoV-2 spike proteins from the P.1 and B.1.1.7 variants were incubated with brilacidin at different concentrations for 1h and were then used to infect HeLa-hACE2 cells. Infection was determined by measuring luciferase activities at day 2 post-infection. Note that brilacidin was not added back during the infection. At 25 and 50 μg/mL, brilacidin suppressed infection by the P.1 variant by approximately 60% and 74%, respectively (Figure 4A). Brilacidin appeared to be less potent against the B.1.1.7 UK variant, as at 25 and 50 μg/mL, brilacidin blocked infection by the UK variant by 31% and 40%, respectively (Figure 4B).
Figure 4:
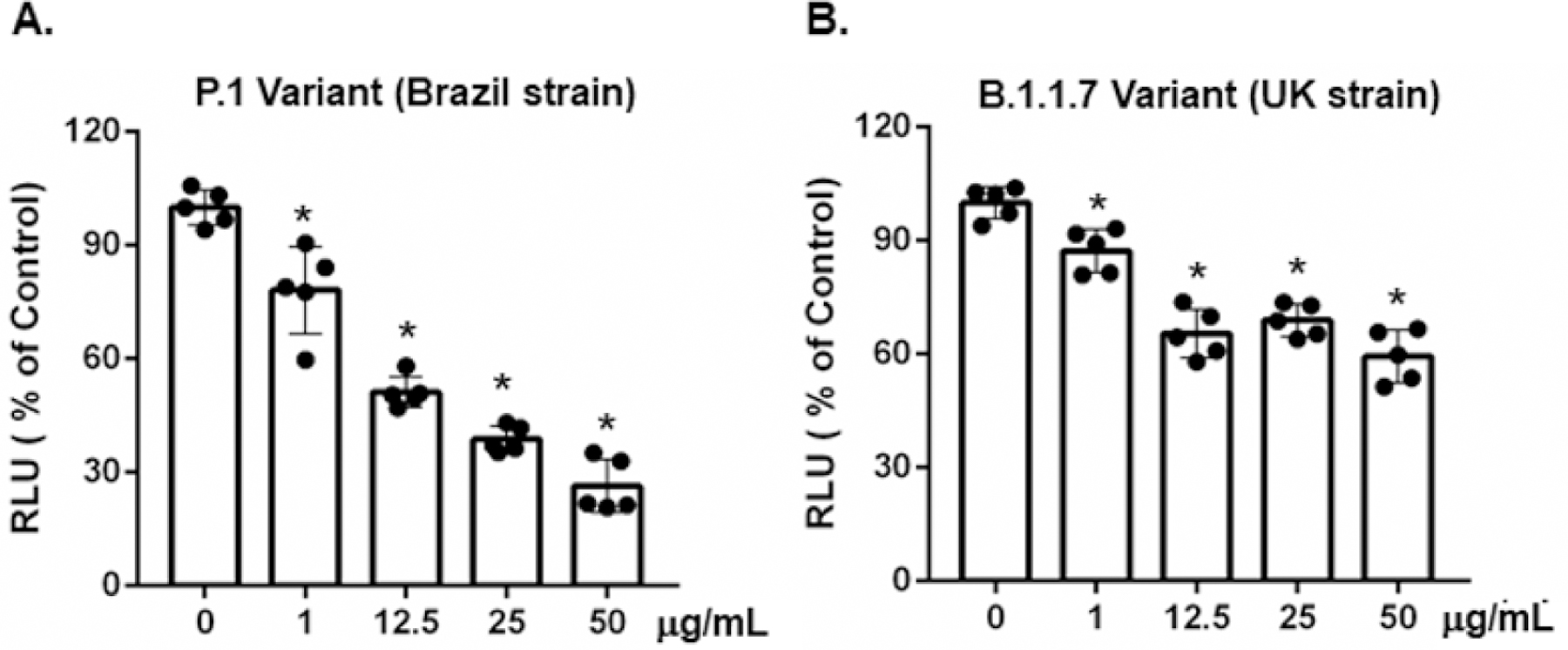
Brilacidin inhibits infection by pseudotyped SARS-CoV-2 variants including P.1 (Brazil strain) and B.1.1.7 (UK strain). Pseudotyped luciferase reporter viruses expressing spike protein from SARS-CoV-2 P.1 (A) or B1.1.7 (B) variants were incubated with various concentrations of brilacidin at 37°C for 1h before adding to HeLa-hACE2 cells for 2h. Cultures were continued, and luciferase activity was measured at 48h as described in Methods. Data represent mean ± SD of five replicates, and were analyzed by one-way ANOVA; *p < 0.05.
The effect of brilacidin on SARS-CoV-2 attachment and on the binding of spike receptor-binding domain (RBD) to hACE2
Viral attachment and the binding of spike proteins through RBD to hACE2 are critical steps for viral entry. To assess the effect of brilacidin on viral attachment, pseudotyped SARS-CoV-2 viruses with or without brilacidin pre-treatment were incubated with HeLa-hACE2 cells at 4°C for 2h. After washing off the unbound viruses, cells were lysed, and the level of cell-associated HIVp24 was determined. Because SARS-CoV-2 interacts with cellular glycosaminoglycan (heparan sulfate) that promotes the binding of viruses to hACE2 [33,34], HeLa cells (without hACE2 overexpression) were included to determine the effect of brilacidin on hACE2-independent viral attachment. Interestingly, in HeLa-hACE2 cells, brilacidin at 10 μg/ml inhibited viral attachment by 20% but did not have any effect on viral attachment to HeLa cells (Figure 5A). Brilacidin at 50 or 100 μg/ml promoted viral attachment to both HeLa-hACE2 and HeLa cells, suggesting the effect of brilacidin at the high concentrations on viral attachment was not dependent on hACE2.
Figure 5:
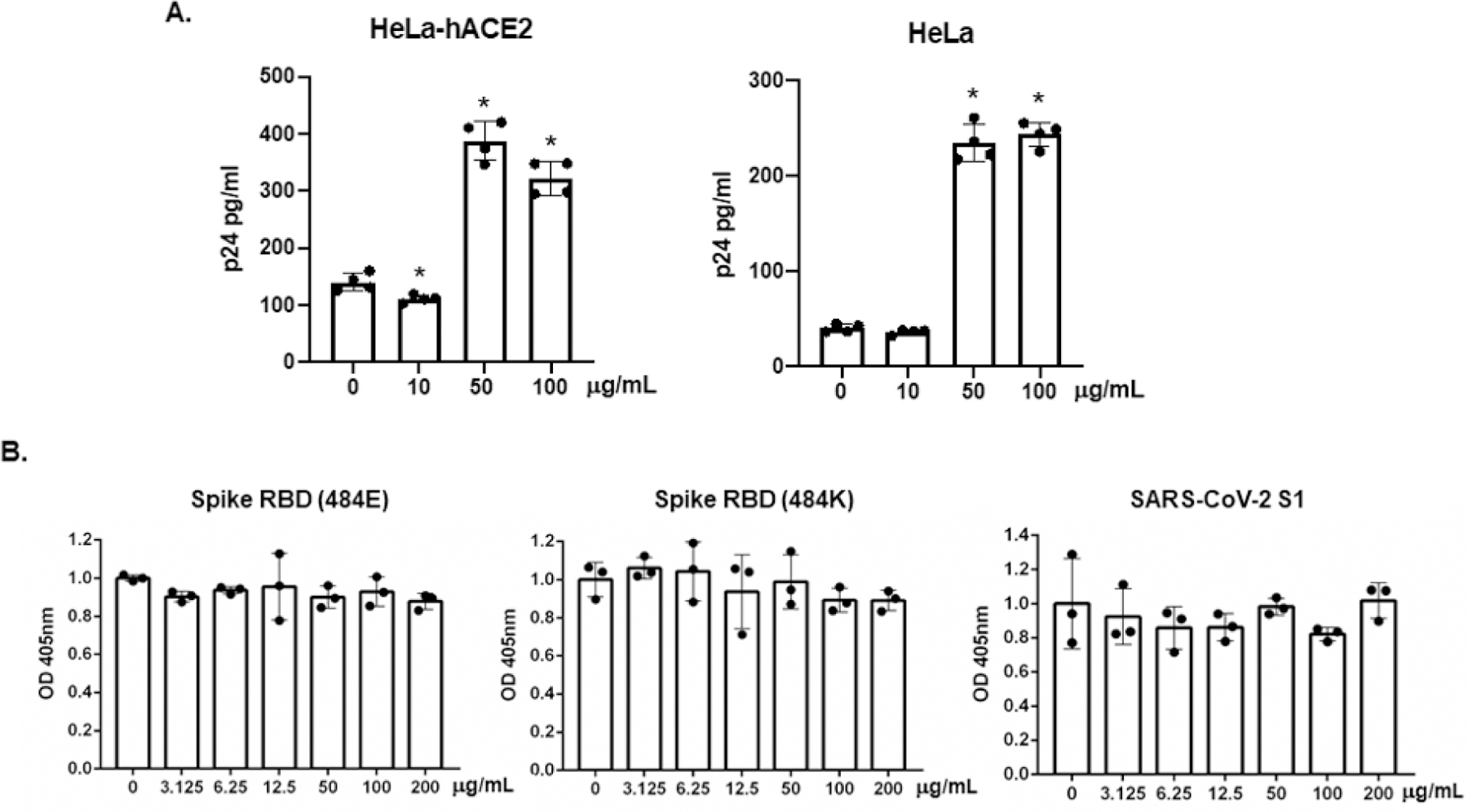
The effect of brilacidin on viral attachment and the binding of spike RBD or S1 proteins to hACE2. (A) Pseudotyped SARS-CoV-2 viruses were treated with brilacidin at different concentrations for 1h at 37°C. The virus-defensin mixture was added to HeLa-hACE2 cells, and cells were incubated at 4°C for 2h. After washing off unbound viruses, cells were lysed, and cell-associated HIVp24 was determined as described in the Methods. (B) Immobilized spike RBD (484E), RBD (484K), or S1 proteins on the plate were pre-treated with various concentrations of brilacidin for 1h followed by incubation with biotinylated hACE2 proteins at 37°C for 1h. After incubation, plates were washed, and the bound hACE2 proteins were detected by incubation with alkylate phosphatase (AP)-conjugated streptavidin followed by the AP colorimetric assay as described in Methods. Data are mean ± SD of triplicate samples.
We then examined the effect of brilacidin on the binding of spike RBD proteins to hACE2 proteins. Spike RBD (484E) from early circulating SARS-CoV-2 strains and RBD (484K) from P.1 and B.1.1.7 variants were used in the binding assay as mutations in SARS-CoV-2 RBD under selective pressure are involved in reduced efficacy of COVID-19 treatments and preventative measures. S1 proteins that contained the RBD region were also included. We found that brilacidin had no effect on the binding of hACE2 proteins to spike RBD (484E or 484K) or S1 proteins (Figure 5B).
Discussion
In this study, we show that brilacidin, a defensin-mimetic molecule, inhibited SARS-COV-2 infection in both multiple-round and single-cycle infection assays. Brilacidin suppressed infection by P.1 strain and B.1.1.7 strain, although the anti-viral activity was less potent against the B.1.1.7 UK strain. Brilacidin did not affect viral infection when added after attachment, indicating that brilacidin inhibited SARS-CoV-2 by blocking viral entry. Brilacidin affected viral attachment in hACE2 dependent and independent manners (Figure 6A). The inhibition of viral entry by brilacidin was not mediated through the binding of spike RBD to hACE2.
Figure 6:
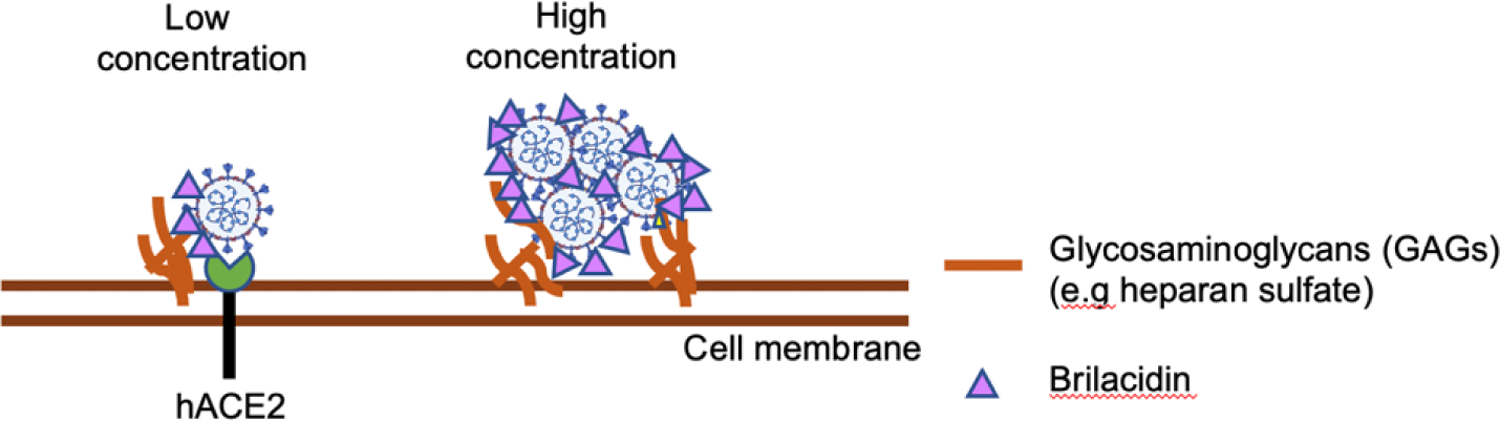
Potential mechanisms of anti-SARS-CoV-2 activities of brilacidin. Brilacidin inhibits SARS-CoV-2 viral entry. The inhibitory effect on viral entry is not involved in the binding of spike RBD to hACE2. Brilacidin at a lower concentration blocks viral attachment in a hACE2 dependent manner. Higher concentrations of brilacidin inhibit viral entry by aggregating viruses.
As a defensin-mimetic molecule, brilacidin shares similar anti-SARS-CoV-2 profiles with human α-defensins and RC101, a θ-defensin analog [8]. Both brilacidin and defensins inhibited the entry of SARS-CoV-2, but did not affect the binding of spike RBD to hACE2 proteins, and were less effective against the B.1.1.7 UK variant. Although higher concentrations of brilacidin (IC50, 3.82 µM) were needed to achieve anti-SARS-CoV-2 activity comparable to that of HNP1 (IC50, 290 nM) in the same single-cycle infection assay, brilacidin has the advantage that it is easily manufactured, unlike HNP1, which is difficult to produce in large quantity. In addition, brilacidin has been evaluated in clinical trials where it has shown effectiveness against a broad-spectrum of bacterial skin infections and exhibited robust anti-inflammatory properties [35]. Thus, brilacidin is a promising candidate against SARS-CoV-2 viruses.
In our study, the anti-SARS-CoV-2 activity of brilacidin was more potent against replication-defective pseudotyped viruses expressing SARS-CoV-2 spike proteins (IC50, 3.82 µM) compared to replication-competent SAR-CoV-2 viruses (at 24h post-infection; IC50, 28.5 µM), suggesting that the anti-viral activity of brilacidin is mediated through common features of these viruses, which are the viral membrane (envelope) and spike proteins. The spike proteins may play a more dominant role. Although spike RBD plays a critical role for viral entry, our data showed that RBD was not involved in brilacidin-mediated viral inhibition. Brilacidin may interact with other domains of spike proteins such as the N-terminal domain important for viral entry [36]. Brilacidin at low vs high concentrations may inhibit viruses via different mechanisms. In HeLa-hACE2 cells, brilacidin at 10 μg/ml inhibited viral attachment by 20% but this inhibitory effect was absent in HeLa cells. Interestingly, brilacidin at high concentrations promoted viral attachment in a hACE2 independent manner, suggesting that brilacidin may inhibit viral infection by aggregating viruses, similar to some anti-viral activities of defensins [37]. It has been suggested that anti-viral activity of brilacidin is involved in binding to heparan sulfate proteoglycans on cells [16]. Thus, the inhibitor at a lower concentration may interfere with heparan sulfate/hACE2-mediated viral attachment via other SARS-CoV-2 spike domains (e.g. SARS-CoV-2 spike N-terminal domain, another glycosaminoglycan binding site on cells [34]). At high concentrations, brilacidin may aggregate viruses and block the subsequent entry events. Note that brilacidin also has concentration-dependent sensitivity to FBS (Figure 4D). At a lower concentration, brilacidin was less potent against SARS-CoV-2 viruses possibly due to high abundant fetuin that has been shown to bind to defensins [28]. However, the anti-viral activity of brilacidin at 50 μg/ml was not affected FBS (Figure 4D), suggesting that differential anti-viral mechanisms were involved.
Other possible anti-viral mechanisms may involve the viral membrane. Brilacidin is known to induce membrane depolarization in Staphylococcus aureus [38]; thus, it may suppress infection by affecting the structural integrity of the viral membrane although this mechanism cannot fully explain the differential anti-viral effect of brilacidin on pseudotyped viruses expressing SARS-CoV-2 spike variants. Studies using viruses expressing spike proteins with specific mutations, different types of glycosaminoglycans, target cells with treatments by various types of heparinases, and electron microscopy imaging of viruses may shed light on specific molecular mechanisms of anti-viral activities of brilacidin.
The IC50 of brilacidin against replication-competent SARS-CoV-2 viruses was 0.565 µM in a previous report [24], whereas we found that the IC50 was 28.5 µM in our replication-competent virus infection assay. Several factors may contribute to the difference. Bakovic and colleagues pre-treated both viruses and cells with brilacidin, and the inhibitor was present during the infection [24]. In our assay, cells were not pre-treated with the inhibitor. Brilacidin was present during a one-hour preincubation with the virus and during viral attachment but not throughout the course of infection. Because brilacidin is known to modulate host cell functions [39], it may induce host restriction factors in cells during the pre-treatment of cells in addition to its direct effects on the virions in the published study [24]. Among other factors contributing to differences between these two reports is that brilacidin was dissolved in DMSO in the previous report [3], whereas we dissolved it in water (1 mg/ml) and then diluted in the media for appropriate concentrations. Different cell lines were also used when evaluating brilacidin. Finally, different virus preparation and titers (MOIs) may also contribute to the different potency; in this regard, we have previously shown that the viral titer can impact the potency of HNP1 [8]. Thus, higher concentrations of brilacidin were required to inhibit infection by viruses at high MOIs in our assays. These differences aside, our findings confirm the anti-SARS-CoV-2 activity of brilacidin.
Our results showed that brilacidin had no effect if it was added after viral infection. Additionally, the anti-viral activity of brilacidin against replication competent SARS-CoV-2 viruses was reduced at 48h post-infection, suggesting that increased viral loads from the multiple-rounds of infection overcame the anti-viral effect of brilacidin. Additionally, brilacidin at lower concentrations was sensitive to FBS in vitro, although it has shown efficacy in the presence of serum in human clinical trials for treatment of ABSSSI [9–11]. These results suggest that brilacidin possesses a unique potential as a preventative agent at the mucosa (e.g. in a nasal spray).
Conclusion
In summary, we showed that brilacidin, a synthetic, non-peptide defensin-mimetic molecule, inhibited SARS-COV-2 infection by blocking viral entry. Brilacidin suppressed the SARS-CoV-2 P.1 variant but was less effective against the B.1.1.7 UK variant. Because of its anti-microbial properties against a broad-spectrum of bacteria, combined with its immunomodulatory properties, brilacidin might be a promising candidate to be developed as a multi-functional agent to target both viral and bacterial infection and to mitigate pathogen-associated inflammation.
Acknowledgments
This work was supported by NIH grant NIH R01AI36948 to T.L.C.
Footnotes
Disclosure Statement
Authors declare that there is no conflict of interest except that J. A. H. and W. K. W. are employees of Innovation Pharmaceuticals, Inc.
Bibliography
- 1.Li Q, et al. “Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia”. New England Journal of Medicine 382.13 (2020): 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. https://coronavirus.jhu.edu/map.html .
- 3.Mathieu E, et al. “A global database of COVID-19 vaccinations”. Nature Human Behaviour 5.7 (2021): 947–953. [DOI] [PubMed] [Google Scholar]
- 4.Hotez P “COVID vaccines: time to confront anti-vax aggression”. Nature 592.7856 (2021): 661. [DOI] [PubMed] [Google Scholar]
- 5. https://covid.cdc.gov/covid-data-tracker/#vaccinations .
- 6.Harvey WT., et al. “SARS-CoV-2 variants, spike mutations and immune escape”. Nature Reviews Microbiology 19.7 (2021): 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang TL and Klotman ME. “Defensins: natural anti-HIV peptides”. AIDS Reviews 6.3 (2004): 161–168. [PubMed] [Google Scholar]
- 8.Xu C, et al. “Human Defensins Inhibit SARS-CoV-2 Infection by Blocking Viral Entry”. Viruses 13.7 (2021): 1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mensa B, et al. “Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16”. Antimicrobial Agents and Chemotherapy 58.9 (2014): 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kowalski RP., et al. “An Independent Evaluation of a Novel Peptide Mimetic, Brilacidin (PMX30063), for Ocular Anti-infective”. Journal of Ocular Pharmacology and Therapeutics 32.1 (2016): 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott RW and Tew GN. “Mimics of Host Defense Proteins; Strategies for Translation to Therapeutic Applications”. Current Topics in Medicinal Chemistry 17.5 (2017): 576–589. [DOI] [PubMed] [Google Scholar]
- 12.Tew GN., et al. “De novo design of biomimetic antimicrobial polymers”. Proceedings of the National Academy of Sciences of the United States of America 99.8 (2002): 5110–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi S, et al. “De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers”. Proceedings of the National Academy of Sciences of the United States of America 106.17 (2009): 6968–6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Institutes of Health: National Library of Medicine. ClinicalTrials.Gov. “Efficacy and Safety Study of Brilacidin to Treat Serious Skin Infections” (2021).
- 15.Bakovic A, et al. “Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture”. Viruses 13.2 (2021): 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Y, et al. “Brilacidin, a COVID-19 Drug Candidate, demonstrates broad-spectrum antiviral activity against human coronaviruses OC43, 229E and NL63 through targeting both the virus and the host cell”. bioRxiv (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Institutes of Health: National Library of Medicine. ClinicalTrials.Gov. “A Study to Evaluate the Efficacy and Safety of Brilacidin in Hospitalized Participants With COVID-19” (2021).
- 18.Wu F, et al. “A new coronavirus associated with human respiratory disease in China”. Nature 579.7798 (2020): 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu C, et al. “Human Immunodeficiency Viruses Pseudotyped with SARS-CoV-2 Spike Proteins Infect a Broad Spectrum of Human Cell Lines through Multiple Entry Mechanisms”. Viruses 13.6 (2021): 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore MJ., et al. “Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2”. Journal of Virology 78.19 (2004): 10628–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers TF., et al. “Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model”. Science 369.6506 (2020): 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie X, et al. “An Infectious cDNA Clone of SARS-CoV-2”. Cell Host and Microbe 27.5 (2020): 841–848.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu C, et al. “Differential Effects of Antiseptic Mouth Rinses on SARS-CoV-2 Infectivity In Vitro”. Pathogens 10.3 (2021): 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bakovic A, et al. “Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture”. Viruses 13.2 (2021): 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu F, et al. “A new coronavirus associated with human respiratory disease in China”. Nature 579.7798 (2020): 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang TL., et al. “Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity”. The Journal of Clinical Investigation 115.3 (2005): 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehrer RI., et al. “Multivalent binding of carbohydrates by the human {alpha}-defensin, HD5”. Journal of Immunology 183.1 (2009): 480–490. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, et al. “Retrocyclin, an antiretroviral theta-defensin, is a lectin”. Journal of Immunology 170.9 (2003): 4708–4716. [DOI] [PubMed] [Google Scholar]
- 29.Burki T “Understanding variants of SARS-CoV-2”. Lancet 397.10273 (2021): 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faria NR., et al. “Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil”. Science 372.6544 (2021): 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volz E Hill., et al. “Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity”. Cell 184.1 (2021): 64–75.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Washington NL., et al. “Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States”. Cell 184.10 (2021): 2587–2594. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clausen TM., et al. “SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2”. Cell 183.4 (2020): 1043–1057.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuurs ZP., et al. “Evidence of a putative glycosaminoglycan binding site on the glycosylated SARS-CoV-2 spike protein N-terminal domain”. Computational and Structural Biotechnology Journal 19 (2021): 2806–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pharmaceuticals., I. “Brilacidin, a Novel Anti-Inflammatory Drug Candidate: Shows Potential in Both Severe Oral Mucositis and Inflammatory Bowel Disease”. In Drug Discovery and Therapy World Congress; Boston, MA, USA: (2017). [Google Scholar]
- 36.Huang Y, et al. “Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for CO-VID-19”. Acta Pharmacologica Sinica 41 (2020): 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson SS., et al. “Antiviral mechanisms of human defensins”. Journal of Molecular Biology 425.24 (2013): 4965–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mensa B, et al. “Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16”. Antimicrobial Agents and Chemotherapy 58.9 (2014): 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nijnik A and Hancock R. “Host defence peptides: antimicrobial and immunomodulatory activity and potential applications for tack-ling antibiotic-resistant infections”. Emerging Health Threats Journal 2 (2009): e1. [DOI] [PMC free article] [PubMed] [Google Scholar]