

Abstract
Mutations in BRCA1 and BARD1 predispose carriers to breast and ovarian cancers. The BRCA1 and BARD1 proteins form a heterodimeric complex (BRCA1/BARD1) that regulates many biological processes including transcription and DNA double-stranded break repair. These functions are mediated by BRCA1/BARD1’s only known enzymatic function as an E3 ubiquitin ligase and its role as a central hub for many large protein complexes. But the mechanisms by which BRCA1/BARD1 interfaces with chromatin, where it exerts its major functions, have remained unknown. Here, we review recent advancements in structural and cellular biology that have provided critical insights into how BRCA1/BARD1 serves as both a nucleosome reader and writer to facilitate transcriptional regulation and DNA repair by homologous recombination.
Keywords: ubiquitin ligase, DNA damage repair, homologous recombination, transcriptional regulation, chromatin regulation
BRCA1/BARD1 performs chromatin-associated functions
The breast-cancer susceptibility gene BRCA1 was linked to heritable breast and ovarian cancers over thirty years ago [1–4]. More recently, mutations in the BARD1 gene were also linked to breast cancer [5–7]. The BRCA1 and BARD1 proteins associate to form a large obligate heterodimeric complex (BRCA1/BARD1) that localizes to the nucleus, where it acts as a central regulator of DNA-centric activity throughout the cell cycle. Well-established roles for BRCA1/BARD1 include transcriptional regulation (see Glossary) and promotion of DNA double-stranded break (DSB) repair by homologous recombination (HR) [8–10].
Formation of the BRCA1/BARD1 heterodimer is essential for its nuclear function. The two proteins associate via interactions governed by their N-terminal RING domains [11] (Figure 1). This region constitutes the sole known enzymatic activity of BRCA1/BARD1 which functions as a RING-type E3 ubiquitin (Ub) ligase [12,13]. Both proteins also contain folded C-terminal domains (BRCT for BRCA1; Ank-BRCT for BARD1) and expansive intervening stretches of intrinsically disordered regions (IDR). Sites throughout both subunits of the heterodimer serve as hubs for many protein interactions, including several extremely large complexes involved in transcriptional regulation and DNA damage repair [14,15] (a relevant subset of these are indicated in Figure 1). For example, BRCA1 has been shown to interact with a variety of transcription factors, including the core transcriptional machinery [16]. BRCA1/BARD1 is also recruited to DNA damage sites, forming so-called ‘foci’ with repair factors until the breaks are fixed [17–20]. What remains to be determined, however, is a detailed understanding of the molecular logic that governs the recruitment and function of BRCA1/BARD1 on chromatin, where it exerts its major functions.
Figure 1. Domain structure of the BRCA1/BARD1 heterodimer.
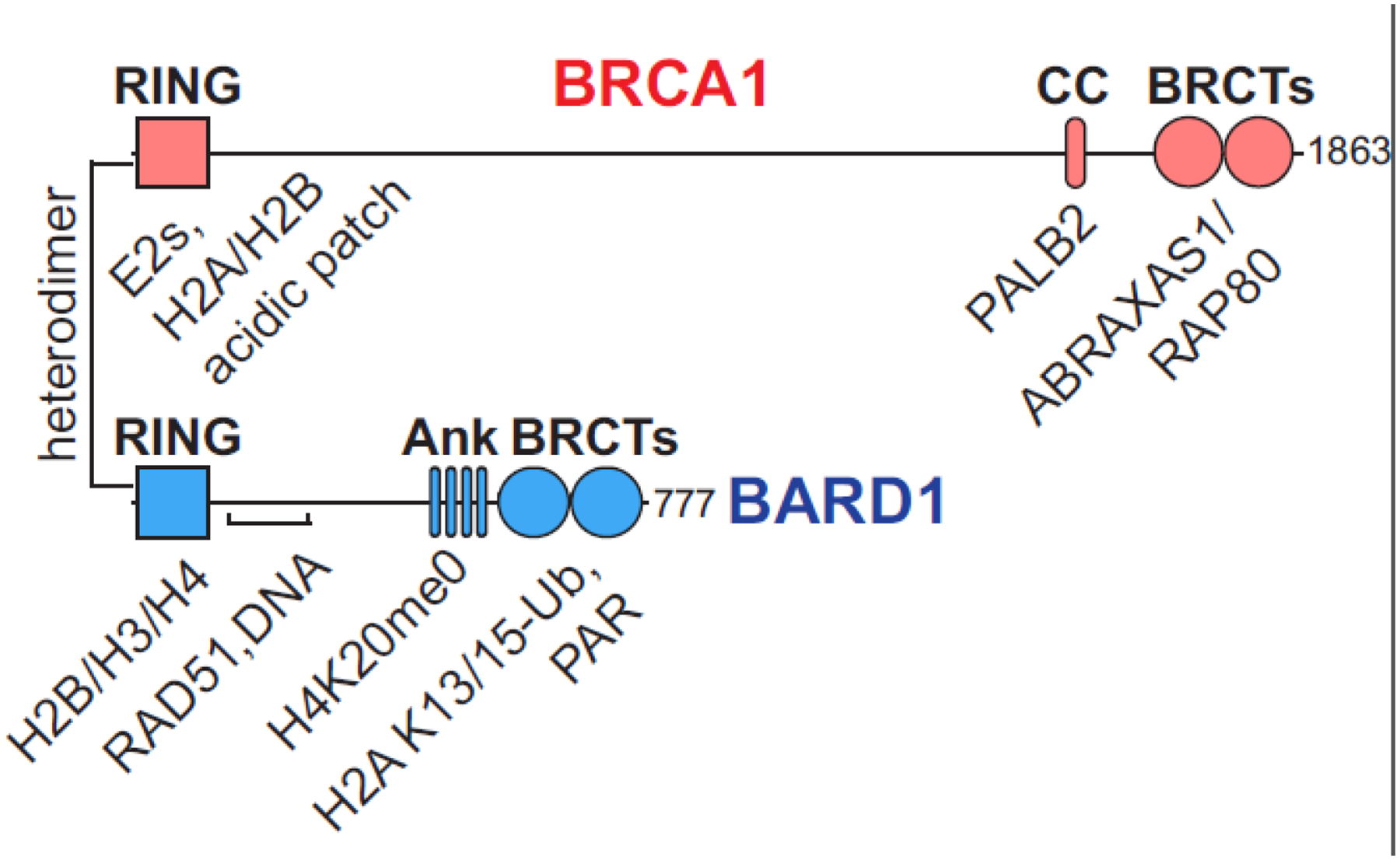
Folded domains are represented by shapes, with the intervening sequences composed of intrinsically disordered regions (RING, really interesting new gene; BRCT, BRCA1 C-terminal; Ank, ankyrin repeat domain; CC, coiled-coil). A subset of interacting proteins and nucleosome regions relevant to this review are labelled under the BRCA1/BARD1 domains to which they bind.
Based on several recent studies, a new paradigm has emerged for BRCA1/BARD1 recruitment and enzymatic activity at nucleosomes, the basic organizing units of chromatin. In support of its chromatin “writer” function, the BRCA1/BARD1 RING domains bind nucleosomes directly, facilitating mono-ubiquitylation of non-canonical sites in the unstructured C-terminal tail of H2A. In support of its chromatin “reader” function, the BARD1 C-terminal domains bind to H2A K13/15-ubiquitylated nucleosomes that are unmethylated at H4K20 (H4K20me0). These marks are hallmarks of damaged chromatin in the S/G2 phase of the cell cycle when a newly replicated sister chromatid is present to be used as a template for HR. Presently, the connection between the two activities remains to be fully defined. Here, we discuss recent advances in BRCA1/BARD1 structural and cellular biology and their implications in transcriptional regulation and DNA DSB repair.
BRCA1/BARD1 is an H2A-specific Ub ligase
In its capacity as a RING-type E3 Ub ligase, BRCA1/BARD1 facilitates the direct transfer of the small signaling protein ubiquitin (Ub) from an E2 Ub-conjugating enzyme to substrate proteins. The action of BRCA1/BARD1 as a Ub ligase is two-fold; it elicits a reactive conformation of an E2~Ub conjugate and simultaneously binds a substrate for Ub transfer [21,22]. While several BRCA1/BARD1 Ub ligase substrates have been described, few have provided compelling links to its major functions in transcriptional regulation and DNA DSB repair [23]. Moreover, whether the ligase function of BRCA1/BARD1 contributes to its role as a tumor suppressor has been the subject of controversy [24–27]. Early genetic and biochemical studies revealed that highly penetrant cancer-predisposing mutations concentrated in the BRCA1 RING domain cause inactivation of E3 Ub ligase function, but also disrupt heterodimerization with BARD1 – a critical interaction for tumor suppression [12].
Providing a direct link to chromatin regulation, nucleosomal histone H2A was identified as a target for BRCA1/BARD1 Ub ligase function in vivo. BRCA1/BARD1-mediated H2A ubiquitylation was shown to promote heterochromatin-mediated silencing of α-satellite DNA regions in a murine model [28] (see Figure 3A). These genomic regions are normally transcriptionally repressed, and the loss of BRCA1 or ectopic over-expression of α-satellite RNAs both cause genomic instability – a hallmark of BRCA1 deficiency. Expression of α-satellite RNAs was subsequently shown to induce breast cancer in mice, potentially linking BRCA1/BARD1 H2A-Ub ligase function to cancer phenotypes [29].
Figure 3. BRCA1/BARD1-dependent H2A-Ub in transcriptional regulation.

(A) Proposed model for heterochromatin-mediated silencing of α-satellite DNA regions by BRCA1/BARD1-dependent H2A-Ub. (B) Proposed model for regulation of estrogen metabolism through modulation of CYP450 expression levels in breast epithelial cells (MCF10a) by BRCA1/BARD1-dependent H2A-Ub.
BRCA1/BARD1 transfers mono-ubiquitin to a region with three closely spaced sites on the unstructured extreme C-terminal tail of canonical histone H2A in nucleosomes (K125/127/129, referred to here as K127-Ub) [30]. Importantly, these lysine sites differ from those targeted by other RING Ub ligases (H2A K13/15, referred to as K15-Ub, by RNF168; K119 by RING1B/BMI1; and H2B K120 by RNF20/40), discussed later in this review. The unique lysine sites suggest that BRCA1/BARD1-dependent ubiquitylation is a novel histone post-translational modification that is functionally non-redundant with marks deposited by other RING Ub ligases. Nucleosomes harboring H2A K127-Ub are likely recognized by specific chromatin-associated factors, as has been observed for other Ub-nucleosome signals [31–34]. It is also possible that BRCA1/BARD1-dependent H2A-Ub may directly influence chromatin structure, as has been reported for chromatin fibers containing H2A K15-Ub and H2A K120-Ub [35,36].
Specialized H2A isoforms regulate transcriptional processes and DNA DSB repair, and may also be substrates for BRCA1/BARD1 [37]. Though the folded histone core sequences are highly conserved, isoforms contain variable sequences in the H2A C-terminal tail where BRCA1/BARD1 directs its Ub signal. Nucleosomes containing the isoform H2A.X/γH2A.X are a molecular marker of DNA damage and repair and form functional complexes with BRCA1/BARD1 [38]. Additionally, residues within the H2A.X C-terminal tail have been identified as the targets of BRCA1/BARD1 in vitro [39]. MacroH2A1 was also identified as a substrate for BRCA1/BARD1 in a screen for Ub ligase substrates [40]; MacroH2A isoforms have been shown to regulate DNA DSB repair pathway choice in cancer cells and are associated with transcriptionally repressive chromatin [41–43]. Primary human fibroblasts expressing mutant macroH2A1 lacking the BRCA1/BARD1 ubiquitylation sites were deficient in cellular senescence and exhibited increased growth rates [40]. Other H2A isoforms H2A.J, H2A.V, and H2A.Z all have lysine residues in their C-terminal tails predicted to be efficiently ubiquitylated by BRCA1/BARD1 [44,45]. Ubiquitylation of H2A isoforms by BRCA1/BARD1 may mediate distinct functional outcomes in different nuclear processes.
Structural basis for site-specific H2A ubiquitylation
The H2A targeting specificity of BRCA1/BARD1 is fully encoded within the RING domains of BRCA1 and BARD1, implying that the RING/RING Ub ligase domain binds a nucleosome substrate and an E2~Ub conjugate simultaneously [30]. Although RINGs are canonically employed as E2~Ub binding units, the BARD1 RING has no E2-binding activity [46]. Cancer-predisposing mutations in the BARD1 RING domain specifically abrogate nucleosome binding and H2A ubiquitylation, revealing a substrate-binding function for this RING domain in the context of its nucleosomal activity [5]. While most RING-type Ub ligases employ auxiliary substrate binding domains or even adaptor proteins [47,48], direct substrate binding by RING domains appears to be a common feature among those that mono-ubiquitylate nucleosomes [39,44,49–51].
In addition to BRCA1/BARD1, two other RING-type Ub ligases, RNF168 and RING1B/BMI1, are known to mono-ubiquitylate distinct H2A lysine residues. RNF168-mediated H2A K15-Ub serves as a signal in the DNA DSB repair cascade that recruits repair factors to DSB sites [20]. RING1B/BMI1 is a component of the Polycomb repressive 1 (PRC1) complex that is a critical regulator of transcription and responsible for the majority of H2A-Ub, namely H2A K119-Ub, in many cell-types [52,53]. Like BRCA1/BARD1, H2A targeting-specificity for these ligases is encoded within their RING domains [50,54].
A series of recent structural studies has revealed the basis for site-specific ubiquitylation of H2A by RNF168, RING1B/BMI1, and BRCA1/BARD1 (Figure 2A). All three ligases exploit a basic arginine anchor residue to bind to the same region of the H2A/H2B acidic patch – a surface used by many (if not most) chromatin-binding factors [55]. For the monomeric RING RNF168, additional interactions orient the E2 (UBE2D) towards the N-terminal region of H2A where K15 is located [49] (Figure 2A, left). The E2~Ub active site is oriented directly above its H2A target lysines, revealing how they are selectively ubiquitylated. In the RING1B/BMI1/UBE2D/nucleosome complex, the E2-binding RING1B contacts the acidic patch via a canonical arginine anchor [50] (Figure 2A, middle). In addition, a loop at the base of the non-E2-binding BMI1 RING domain forms interactions with the H3 α1-L1 elbow. Together, these interactions orient the RING1B E2-binding interface towards the nucleosome DNA entry/exit and position the E2~Ub active site directly over H2A K119 for Ub transfer. The RING1B-bound E2 also forms interactions with nucleosomal DNA, contributing to its affinity for the nucleosome substrate.
Figure 2. Structural basis for site-specific ubiquitylation of nucleosomal H2A.
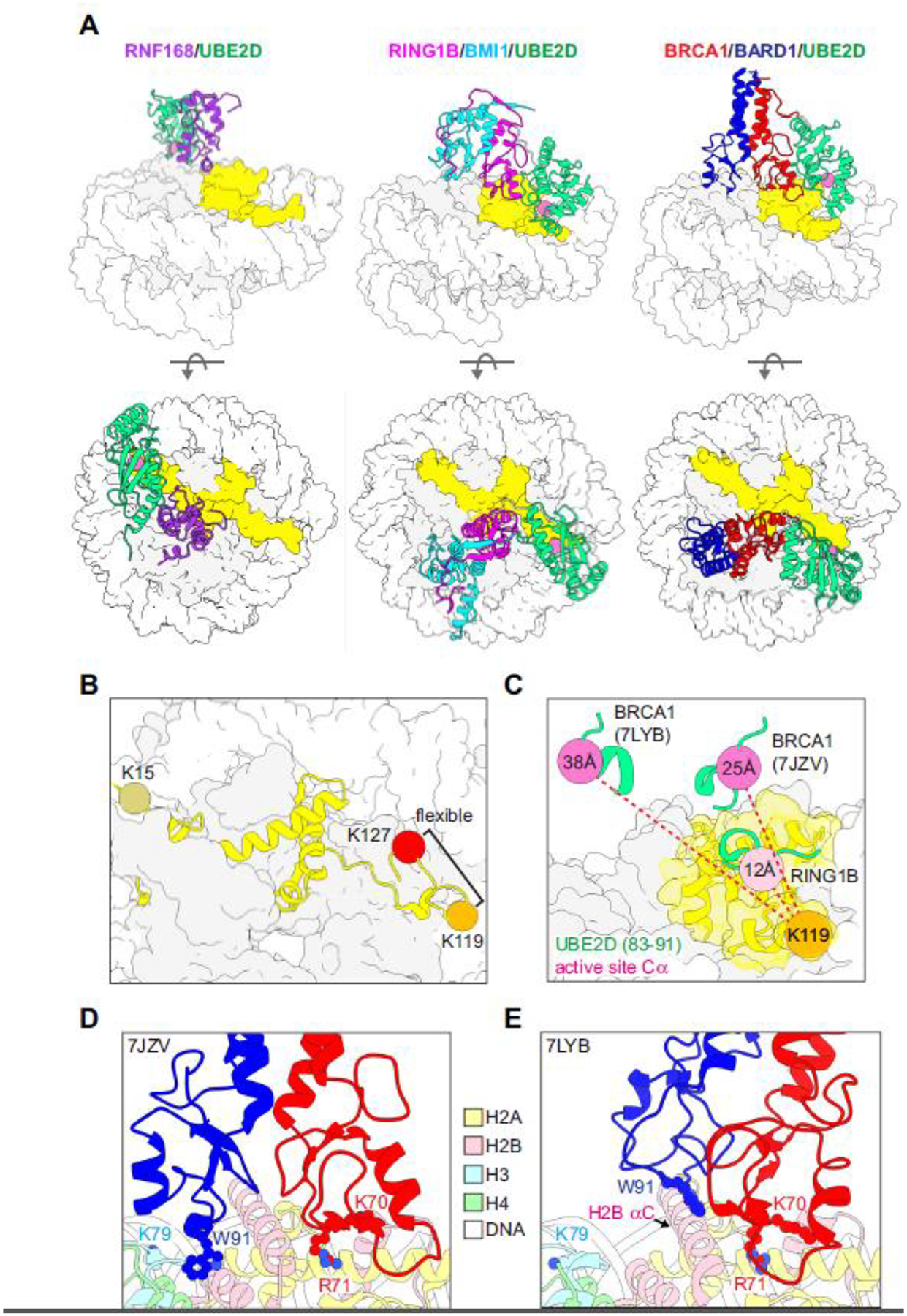
(A) Structural models of RING/E2/nucleosome complexes (PDB-Dev: 00000028, PDB: 4R8P, 7JZV). The active site Cα of the E2 (UBE2D, green) in each model is shown as a pink sphere and H2A is yellow. (B) Locations of representative target lysine residues for H2A-modifying Ub ligases on a nucleosome (PDB: 1KX5). The C-terminal tail of H2A beyond K119 is unstructured and not observed in x-ray or cryo-EM structures but is modelled in PDB 1KX5. (C) Positions of the E2 active site (Cα, pink spheres) from the indicated RING/E2/nucleosome structural models (PDB: 4R8P, 7JZV, 7LYB) relative to H2A K119 in the RING1B/BMI1/nucleosome complex (PDB: 4R8P, orange sphere). The Cα-Cα distances from H2A K119 to the respective E2 active sites in the E3/E2/nucleosome complexes (dashed red lines) are reported inside the active site spheres. For the measurements, models were aligned by H2B in the nucleosome. (D, E) BRCA1/BARD1 RING-histone interactions from two published structural models (PDB: 7JZV and 7LYB). The models are aligned by H2B on the E3/E2-bound face of the the nucleosome (see H2B αC helix) and shown side-by-side instead of overlaid for clarity. The largest difference between the two structures is in the positioning of the BARD1 RING domain. Critical histone-binding BRCA1/BARD1 sidechains and the H3 K79 sidechain in the H3 α1-L1 elbow are shown as sticks. Histone colors are indicated in the legend, and BRCA1 is red and BARD1 is blue.
RNF168 and RING1B/BMI1 ubiquitylate H2A lysine residues within ordered regions of the nucleosome. In contrast, BRCA1/BARD1 mediates the transfer of Ub to lysines in a flexible disordered region of the C-terminal tail fewer than ten residues away from the target of RING1B/BMI1, calling to question how this is orchestrated (Figure 2B). Two studies reporting cryo-EM structures of BRCA1/BARD1/UBE2D/nucleosome complexes have elucidated the RING-mediated specificity determinants of site-specific H2A ubiquitylation [39,44] (Figure 2A, right, 2D, 2E). The structures show that the E2-binding BRCA1 subunit utilizes a canonical arginine-anchor to interact with the H2A/H2B acidic patch akin to RING1B. However, the non-E2-binding BARD1 RING domain forms interactions with histones that are distinct from its counterpart, BMI1. These differences in non-E2 binding RING-histone interactions lead to a unique “stance” of the BRCA1/BARD1 RING heterodimer on the nucleosome compared to RING1B/BMI1. This stance positions the E2 bound by the BRCA1 RING away from the ordered histone surface where H2A K119 is located, providing a rationale for why this E3 ligase does not efficiently ubiquitylate the RING1B/BMI1 target lysine (K119, Figure 2C). Instead, the flexible H2A C-terminal tail has sufficient mobility in the complex to sample the BRCA1-bound E2 active site for Ub transfer to H2A K127 [39,44]. Unlike the RING1B-bound E2, the BRCA1-bound E2 does not interact with nucleosomal DNA.
Notably, the BARD1 RING-histone interactions differ between the two published structures (Figure 2D, 2E). In Witus et al., the BARD1 RING binds close to the H3 α1-L1 elbow, inserting its W91 sidechain in a cleft formed by H2B and H4 [44]. This interaction is consistent with the requirement for a nucleosome substrate as opposed to an H2A/H2B dimer for BRCA1/BARD1-dependent H2A ubiquitylation activity (unpublished observation). In Hu et al, the BARD1 RING domain interacts with the H2B αC helix, again using the W91 sidechain [39]. These interactions are not located near the H3 α1-L1 elbow region as observed in the Witus et al. structure. Despite these differences, the E2 is positioned away from the histone surface in both complexes, consistent with H2A K127 being selectively ubiquitylated (Figure 2C).
The source of differences in the two RING orientations in the BRCA1/BARD1 structures is unclear. Variability analysis of the Hu et al. cryo-EM map indicates a large amount of structural heterogeneity of the RING domains on the nucleosome, accounting for the lower resolution in this region compared to the histone core observed in both maps [39]. To overcome the modest affinity of the complexes, each group used a genetic fusion strategy, but the details of the constructs employed differ. Importantly, although both groups utilized a truncated minimal RING/RING heterodimer for structural studies, the full-length BRCA1/BARD1 complex exhibits stronger binding and increased activity for a nucleosome substrate, suggesting that other regions of the ligase promote functional interactions with mono-nucleosomes [39,44]. A complete understanding of relevant regions in the full-length complex that facilitate BRCA1/BARD1-dependent H2A-Ub will be important to understand its activity and the effects of patient mutations outside the RING domains.
H2A K127-Ub in transcriptional regulation
BRCA1/BARD1 was first identified as a regulator of transcription, but this aspect of BRCA1/BARD1 activity is less well studied than its DNA damage repair function [56,57]. To date, BRCA1/BARD1-mediated H2A-Ub has primarily been associated with repressing gene expression, but studies are limited to relatively few genes identified through candidate-based approaches (Figure 3A, 3B).
For example, BRCA1/BARD1-mediated H2A-Ub has been implicated in the transcriptional regulation of estrogen metabolism, a process known to be disrupted in breast and ovarian cancers [58,59]. In non-cancerous breast epithelial cells (MCF10a), depletion of BRCA1 or BARD1 is associated with elevated transcript levels of certain estrogen-metabolizing cytochrome P450 (CYP450) genes [5] (Figure 3B). Several of these CYP450 proteins mediate the hydroxylation of estradiol, forming toxic intermediates that damage DNA and have been linked to hormone-related cancers [60]. CYP450 transcript levels were elevated in BARD1-deficient MCF10a cells and restored to near wild-type levels by ectopic expression of wild-type BARD1 or an H2A-Ub fusion gene [5]. In another study, BRCA1 deficiency led to increased levels of toxic estrogen metabolites that induced DNA breaks in MCF10a cells [61]. While not explicitly tested, it is likely that these effects are similarly linked to BRCA1/BARD1-directed H2A-Ub. Relevant to patient cancer phenotypes, both studies revealed substantial effects of BRCA1 or BARD1 haploinsufficiency in regulating transcription [5,61]. Together, the data indicate that transcriptional repression is a downstream effect of H2A ubiquitylation by BRCA1/BARD1. They also imply that estrogen-utilizing cells with BRCA1/BARD1-ligase deficiencies potentially experience over-exposure to DNA-damaging toxic estrogen metabolites, a compelling hypothesis that links BRCA1/BARD1 deficiency to tissue-specific cancers. Identification of a comprehensive set of genes regulated by BRCA1/BARD1-dependent H2A-Ub in relevant cell-types is an important goal.
BRCA1/BARD1 has also been shown to interact with other transcriptional regulators. These include the core transcriptional machinery, histone modifying enzymes, and a cadre of transcription factors [8]. Interactions with sequence-specific transcription factors (e.g., ERα, OCT1, ZBRK1, c-Myc, and others) may recruit the ligase to specific chromatin regions where it deposits H2A-Ub, thereby directly regulating transcription. It will be important to fully elucidate the role of BRCA1/BARD1 in modulating transcription via H2A-Ub, and how this process interfaces with the large and diverse array of transcriptional regulators with which it functions. It is also possible that specific histone modifications may influence BRCA1/BARD1’s presence and/or H2A-Ub enzymatic activity at genomic regions to regulate transcription of target genes.
Recruitment of BRCA1/BARD1 to damaged chromatin
Double-stranded DNA breaks are mainly repaired through two competing pathways depending on the cell cycle [10]. HR is facilitated by BRCA1/BARD1 in S/G2 phases, where broken DNA ends are resected to expose single-stranded DNA and a sister chromatid serves as a template to guide high-fidelity repair. Alternatively, an error-prone repair pathway called non-homologous end joining (NHEJ) promoted by the protein 53BP1 rapidly ligates broken DNA ends to avoid catastrophic chromosome breakages. It is well established that competition between BRCA1/BARD1 and 53BP1 determines pathway choice between HR and NHEJ and, hence, the maintenance of genome integrity following DNA DSBs [62–65]. Previously, two mechanisms have been implicated in the recruitment and retention of BRCA1/BARD1 at DSB sites. These include interactions between the BRCA1-A complex (BRCA1/ABRAXAS1/RAP80) and K63-linked Ub chains deposited on linker histone H1 and/or interactions between the BARD1 BRCT domains and poly (ADP-ribose) (PAR; Figure 1) [18,66,67]. Specifically lacking, however, is an explanation for BRCA1/BARD1’s ability to compete with the NHEJ factor 53BP1 for DSB site occupancy to promote HR in S/G2 phase of the cell cycle when a sister chromatid is present.
Recent studies have elucidated a mechanism for sensing the cell cycle through multivalent interactions between the BARD1 C-terminal domains (Ank-BRCTs; Figure 1) and specifically modified nucleosomes [68,69]. This is dependent on co-occurrence of the DNA DSB signal H2A K15-Ub deposited by RNF168 and the presence of H4K20me0, a marker of replicative chromatin and sister chromatid availability. Together, these marks serve to recruit the BRCA1/BARD1 complex to DNA DSBs to facilitate HR [69,70]. 53BP1 specifically binds to H2A K15-Ub and H4K20me2 nucleosomes, so this serves as a logic switch to determine whether BRCA1/BARD1 or 53BP1 is retained at DSB sites to license HR or NHEJ, respectively [32,71] (see figure 5A, left pathway). Notably, H4K20 methylation status is a potent marker of the cell-cycle, with around 80% of post-replicative chromatin containing H4K20me1/2/3 and newly synthesized histones in S/G2 largely devoid of this mark [72].
Figure 5. Nucleosome-based recruitment and activity of BRCA1/BARD1 in DNA DSB repair.

(A) Schematic of BRCA1/BARD1 and 53BP1 recruitment pathways and retention at damaged chromatin. The left two pathways are reliant on RNF168-mediated H2A K15-Ub, while the right pathway (separated by dashed line) is reliant on RNF8-mediated K63-linked poly-Ub deposited on linker histone H1. The center pathway reliant on H2A K15-Ub and H4K20me0 requires the BRCA1 RING domain and ligase proficiency for DSB site retention and HR, indicated by the curved arrow with an asterisk. (B) Schematic of BRCA1/BARD1-dependent H2A-Ub activity in the DNA end-resection step of HR. BRCA1/BARD1 is recruited to DSB sites and ubiquitylates H2A K127 (1, top). HR is licensed through limited end-resection of broken DNA ends via association of BRCA1/BARD1 with CtIP and MRN (1 and 2, top and middle, light green arrow). SMARCAD1 is recruited via BRCA1/BARD1-dependent H2A K127-Ub, excludes the NHEJ factor 53BP1 from break sites, and performs ATP-dependent nucleosome repositioning to facilitate long-range DNA end-resection (2, middle). This is antagonized by the action of the deubiquitylating enzyme USP48, which specifically removes H2A K127-Ub to prevent over-resected DNA tracts and facilitate HR (3, bottom). Question marks in panels denote unknown interactions (A) or speculative complexes (B).
Structures of the BARD1 Ank-BRCT C-terminal region bound to nucleosomes with H2A K15-Ub and H4K20me0 have revealed a highly specific network of interactions that are centered around an arginine anchor in the BARD1 BRCTs and the H2A/H2B acidic patch [39,73] (Figure 4A, 4B). The BARD1 Ank and BRCT domains also form intramolecular interactions that are not detected outside of a nucleosomal context [74]. The Ank domain binds to the H4 tail, utilizing a conserved pocket observed in other HR proteins that bind to H4K20me0 [68] (Figure 4C). Mutation of binding pocket residues BARD1 E467A/N470A/D500A (called BARD1–3A) or introduction of H4K20me2 impairs nucleosome binding in vitro. In cells, BARD1–3A causes defects in HR by disrupting accumulation of BRCA1/BARD1 at DSBs, thereby limiting DNA end-resection [68]. Consequently, 53BP1 accumulates at DSB sites and NHEJ becomes the dominant repair pathway.
Figure 4. Recognition of damaged chromatin by BARD1.
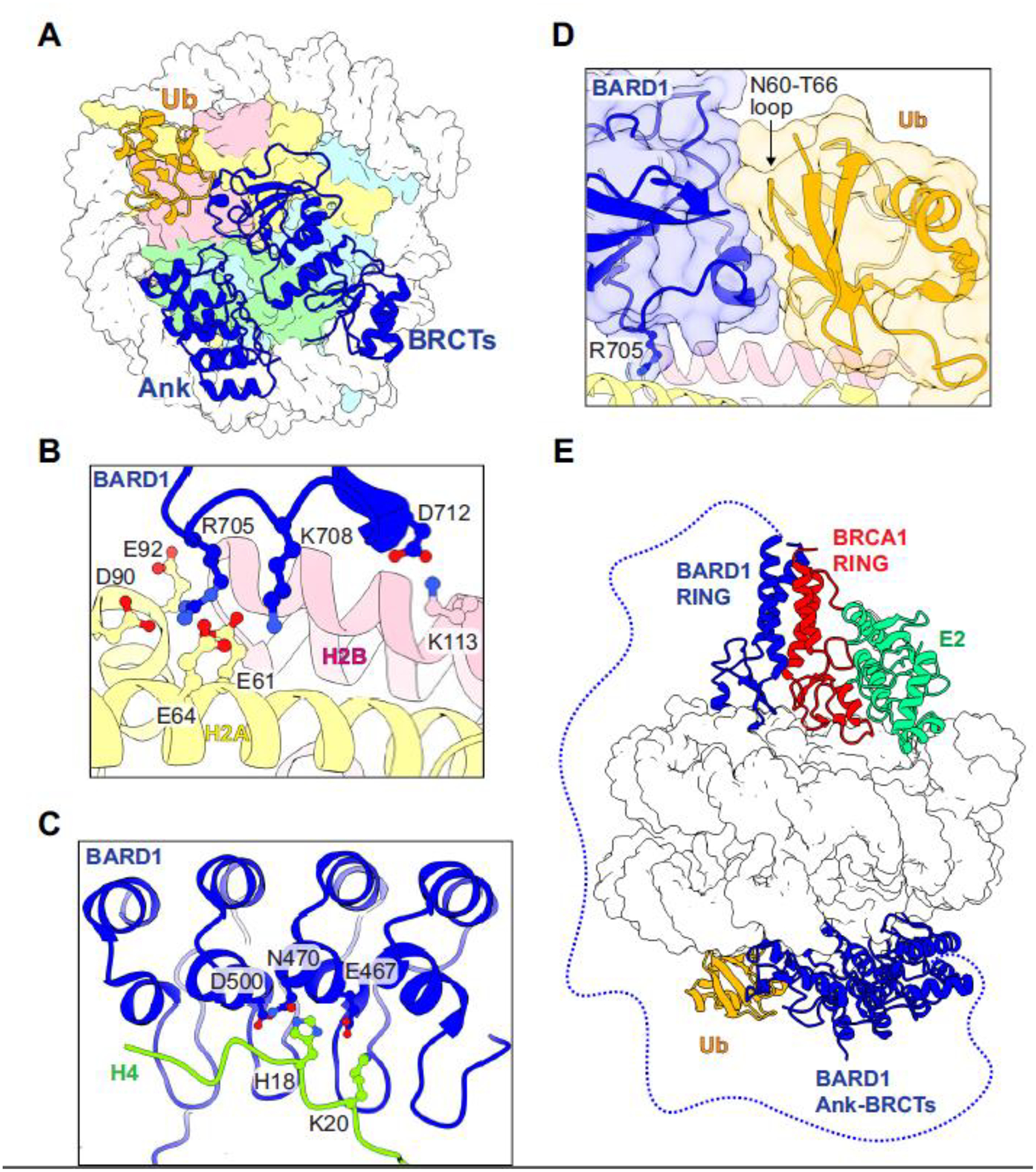
(A) Top-down view of the BARD1 Ank-BRCT domains (blue) bound to a nucleosome containing H2A K15-Ub and H4K20me0 (PDB: 7E8I). (B) Interactions between the BARD1 BRCT domain and the H2A/H2B acidic patch. (C) Interactions between the BARD1 Ank domain and the H4 N-terminal tail. Residue sidechains mutated in the “BARD1–3A” mutant are shown as blue sticks. (D) Interaction between the BARD1 BRCT Ub-directed recognition motif and H2A K15-Ub. The BARD1 “Arg anchor” sidechain interacting with the H2A/H2B acidic patch is shown in sticks. (E) Proposed model of binding for the RING heterodimer BRCA1/BARD1/E2 complex and the BARD1 Ank-BRCTs to both sides of the same nucleosome (overlay of PDB models 7JZV and 7E8I).
Interactions between the BARD1 BRCT domain and Ub attached to H2A K15 involve conserved, charged residues on the second BARD1 BRCT repeat termed the BRCT domain ubiquitin-directed recruitment motif (BUDR) [39,69,73] (Figure 4D). The BARD1 BUDR motif interacts with an atypical surface of Ub that spans residues N60-T66 and is distinct from the highly utilized I44 or I36 patches recognized by scores of other proteins [75]. The Ub K63 sidechain is engaged with the BARD1 BRCT, blocking extension of K63-linked chains on H2A K15-Ub by RNF8 [39]. Like the BARD1–3A Ank mutation, substitution of conserved BUDR residues yields enhanced poly (ADP-ribose) polymerase inhibitor (PARPi) sensitivity and decreased recruitment to DSBs [69,73]. The putative PAR-binding pocket in BARD1 BRCT1 is unobstructed in the structure of the complex, possibly allowing for higher-order assemblies. Adding further complexity, phosphorylation of Ub-T12 in nucleosomal H2A K15-Ub decreases recognition by 53BP1 but allows for binding of BRCA1/BARD1 and other HR factors [76]. This feature is consistent with the different Ub surfaces recognized by the BARD1 BUDR and 53BP1 ubiquitin-directed recognition motif (UDR).
Finally, two redundant Ub-mediated pathways reliant on RNF168-mediated H2A K15-Ub or the BRCA1-A complex (ABRAXAS1/RAP80) serve to recruit BRCA1/BARD1 to damaged chromatin, adding an additional layer of complexity to the emerging picture [70,77] (Figure 5A). Of the two, only the RNF168-mediated pathway is dependent on BRCA1’s Ub ligase activity [77]. Simultaneous disruption of the BRCA1/ABRAXAS1/RAP80 interaction and BRCA1-E2 binding (BRCA1-I26A) caused complete loss of recruitment of BRCA1/BARD1 to ionizing radiation induced foci (IRIF). Furthermore, a nucleosome-binding mutant in the RING domain of BRCA1 (K70A/R71A) also displayed defective IRIF recruitment only upon co-depletion of RAP80 as observed for the BRCA1-E2 binding mutant, although with a smaller effect [77]. Together, the data suggest that BRCA1/BARD1 Ub ligase activity functions in the same pathway as RNF168 in HR and contributes to its own retention at DSB sites. It is unclear why BRCA1/BARD1 ligase function is required for its retention at DSB sites. Hypotheses that BRCA1/BARD1 Ub ligase activity serves to displace 53BP1 and potentially influence the activities of multiple BRCA1/BARD1 substrates (including but not limited to H2A) should be entertained. Importantly, this work may provide an explanation for the lack of HR phenotypes observed in other studies using BRCA1/BARD1 Ub ligase-deficient mutants discussed in the section below, in that the effects of the BRCA1/BARD1 mutants used may be masked by intact BRCA1-A complex interactions [24,68].
H2A K127-Ub in DNA double-stranded break repair
In addition to BARD1’s role as a nucleosome reader to recruit BRCA1/BARD1 to nucleosomes near DSB sites, BRCA1/BARD1-mediated ubiquitylation of nucleosomal H2A was found to promote HR after pathway commitment by facilitating long-range DNA end-resection [78] (Figure 5B). HeLa cells depleted of endogenous BARD1 by siRNA and expressing a BARD1 mutant reported to be a less effective ligase with certain E2 enzymes (BARD1-R99E) exhibited phenotypes consistent with HR deficiency, suggesting that the E3 ligase activity of BRCA1/BARD1 is important for HR. Notably, expression of an H2A-Ub genetic fusion presumed to mimic the natural product of BRCA1/BARD1-dependent ubiquitylation restored HR in BARD1-deficient cells, implying that the H2A-Ub signal promotes DNA end-resection. The H2A modification is required to recruit SMARCAD1, a SWI/SNF-like ATP-dependent chromatin remodeler that helps reposition nucleosomes and excludes 53BP1 from DSB sites to promote DNA end-resection (Figure 5B). In a cryo-EM structure of SMARCARD1 bound to a mono-nucleosome, its ATPase domains are bound in proximity to the H2A C-terminal region, but the flexible tail is not detected in the cryo-EM image [79]. Further investigation is needed to elucidate how H2A-Ub plays a role in SMARCARD1 recruitment or remodeling activity.
Further evidence in support of a role for BRCA1/BARD1 Ub ligase function in HR comes from studies involving de-ubiquitylation. Ubiquitylation is a dynamic and reversible modification that can be removed from substrates by deubiquitylating enzymes (DUBs). The DUB USP48 was shown to be recruited to sites of DNA damage in cells and to preferentially remove BRCA1/BARD1-mediated H2A-Ub marks (K127-Ub) over those deposited by RNF168 (K15-Ub) or RING1B/BMI1 (K119-Ub) in vitro [80]. USP48 acts in the same pathway as BRCA1 and SMARCAD1, and its depletion resulted in over-resected DNA ends, exclusion of 53BP1 from IRIF, decreased HR efficiency, and decreased cellular survival following PARPi treatment. Together, the findings indicate that USP48 antagonizes BRCA1/BARD1 Ub ligase-dependent activities in HR, creating a balance that regulates DNA end-resection tract length [78, 80] (Figure 5B). Additional support for BRCA1/BARD1’s Ub ligase activity in HR comes from studies in hTERT RPE-1 cells, where only concomitant deletion of the BRCA1 BRCT domain and introduction of an E2-binding deficient mutation in the BRCA1 RING domain caused PARPi survival defects [77]. However, whether this dependency on BRCA1/BARD1 Ub ligase function is due to H2A-Ub or other substrates remains to be determined.
Notably, the role of BRCA1/BARD1 as a Ub ligase in HR is not universally embraced, as several studies have arrived at differing observations and conclusions. Each study was performed in a different cell-type and used different methods to perturb or deplete BRCA1/BARD1 ligase functionality. For example, a study using an auxin inducible degron (AID) system to deplete endogenous BARD1 in HCT116 cells did not observe the same HR defects upon expression of BARD1 R99E (the mutant used in the study [78] described above) [68]. Additionally, in the BRCA1-deficient MDA-MB-436 cell line, several ligase-inactivating mutants did not yield PARPi survival defects and IRIF formation of HR factors was not dependent on Ub ligase function [70]. Lack of Ub ligase dependency in HR is also consistent with previous findings using a BRCA1 mutant that is defective in binding to and activity with the E2 enzyme UBE2D (BRCA1-I26A) [24,25]. Although the differences could be cell-type specific, each approach has intrinsic limitations, making it impossible to reach a unified understanding at present. Further investigation using separation-of-function mutants is warranted to clarify the requirements for BRCA1/BARD1 H2A-Ub activity in HR. Although challenging, it will be critical to examine BRCA1/BARD1 Ub ligase functions in HR in non-cancerous mammary and ovarian epithelial cells to ascertain cell-type specific dependencies. Current experimental limitations have also impeded characterization of BRCA1/BARD1-dependent H2A-Ub in cells and animals, underscoring the critical need for novel reagents and tools (Box 1).
Box 1. Current experimental limitations in understanding H2A K127-Ub.
An impediment to biological characterization of BRCA1/BARD1-dependent ubiquitylation of nucleosomal H2A is the lack of tools for robust detection and manipulation of this post-translation modification. Unlike the other H2A-Ub marks (K15-Ub and K119-Ub), no commercial antibodies are currently available. The lysine-rich extreme C-terminal H2A tail also precludes traditional mass spectrometry techniques that employ trypsin digestion, which generates peptide fragments that are likely too small to detect. Furthermore, the basal cellular amount of BRCA1/BARD1-mediated H2A-Ub appears to be extremely low compared to that of PRC1-mediated K119-Ub. Although expressing an H2A-Ub C-terminal genetic fusion to bypass ligase deficiency has proven to be an effective strategy, it is possible that overexpression of this fusion construct causes global perturbation of chromatin that could introduce artifacts. Together, these circumstances indicate a critical need for the development of reagents and tools capable of monitoring and manipulating BRCA1/BARD1-dependent H2A-Ub in cells.
Concluding Remarks
The BARD1 C-terminal Ank-BRCT “reader” domains and the N-terminal BRCA1/BARD1 RING “writer” domains bind to a fully overlapping surface on a nucleosome, precluding their simultaneous binding to one “face” of a nucleosome. Notably, a protein species containing only the BRCA1 RING domain but full-length BARD1 has increased H2A-Ub enzymatic activity for a mono-nucleosome substrate with pre-installed H2A K15-Ub [39]. This observation implies the possibility of a complex where the BRCA1/BARD1 RING domains and BARD1 Ank-BRCTs are simultaneously bound to opposite faces of one nucleosome through a wrapping mechanism, promoting its Ub ligase activity (i.e., the “writer” function; Figure 4E). It is also possible that such interactions may occur between neighboring nucleosomes as has been observed with other nucleosome-modifying enzymes [81], but this remains to be tested. In addition to H2A K15-Ub, a variety of other histone post-translational modifications (PTMs) may influence BRCA1/BARD1-dependent H2A-Ub activity as has been reported for the H2B K120-Ub-specific Ub ligase RNF20/40 [82]. A more complete understanding of BRCA1/BARD1 interactions with chromatin, the PTMs involved, and associated higher-order protein complexes will be critical to understand its many biological functions and to assess potentially pathogenic patient mutations of unknown significance (see outstanding questions).
OUTSTANDING QUESTIONS.
Full-length BRCA1/BARD1 binds more strongly and has increased activity for unmodified nucleosomes than the isolated RING/RING complex. How do regions outside the RING domains contribute to chromatin interactions?
In addition to H2A K15-Ub and K4K20me0, what combinations of histone PTMs regulate BRCA1/BARD1 recruitment to nucleosomes and H2A-Ub activity?
What are the patterns and dynamics of the H2A K127-Ub mark in different cell-types? This requires a method for nucleosome-Ub PTM detection that is currently lacking.
What is the full subset of genes regulated by BRCA1/BARD1-dependent H2A-Ub? How is BRCA1/BARD1 targeted to these genomic regions to deposit H2A-Ub? How does this differ between cell types?
What are the direct and indirect effects of BRCA1/BARD1-dependent H2A K127-Ub on chromatin? What factors other than SMARCAD1 and USP48 recognize and regulate this mark?
Studies have drawn conflicting conclusions regarding BRCA1/BARD1 Ub ligase participation in DNA DSB repair. What are the requirements for BRCA1/BARD1 Ub ligase activity in DNA DSB repair and how is H2A-Ub involved? Is BRCA1/BARD1-dependent H2A K127-Ub important for tumor suppressor function?
What is the interplay between BRCA1/BARD1 recruitment to damaged chromatin via its C-terminal domains and its Ub ligase activity in HR?
Supplementary Material
HIGHLIGHTS:
BRCA1/BARD1 binds directly to nucleosomes in chromatin via multiple interaction sites to regulate transcription and DNA double-stranded break repair.
BRCA1/BARD1 functions as a ubiquitin ligase to promote site-specific ubiquitylation of residues in the unstructured C-terminal tail of H2A (K125/127/129), a unique and novel histone modification.
The BARD1 C-terminal Ank-BRCT domains recognize nucleosomes carrying H2A K13/15-Ub and H4K20me0, hallmarks of damaged chromatin in S/G2 phases when a newly replicated sister chromatid can be used as a template for homologous recombination.
Redundant BRCA1/BARD1 nucleosome-based recruitment pathways reveal specific requirements for BRCA1/BARD1 RING function and Ub ligase activity in DNA double-stranded break repair.
ACKNOWLEDGMENTS:
We thank J. Pruneda for his insightful feedback on this manuscript. W.Z. was supported by V Scholar V2019.Q13 from V Foundation for Cancer Research, Young Investigator Award from Max and Minnie Tomerlin Voelcker Fund, and NIH R01GM141091. S.R.W., P.S.B., and R.E.K. were supported by NIH R01 CA260834 and R.E.K. is the Edmond H. Fischer/WRF Endowed Chair in Biochemistry.
GLOSSARY
- BRCA1-A complex
A complex composed of ABRAXAS1, RAP80, BRCC36, BRCC45, and MERIT40 that recruits BRCA1/BARD1 to DNA DSBs. This is mediated by direct binding of phosphorylated ABRAXAS1 to the BRCTs of BRCA1, and an interaction between the RAP80 Ub interaction motif (UIM) and K63-linked poly-Ub chains deposited on linker histone H1 by the Ub ligase RNF8
- DNA double-stranded break (DSB) repair
a collection of biological pathways including (but not limited to) HR and NHEJ that recognize and repair double-stranded DNA breaks. DNA DSBs are caused by a variety of chemical and environmental mutagens in addition to normal cellular processes, resulting in fragmented chromosomes. Disruptions to normal DNA DSB repair processes can introduce mutations, sometimes leading to cancer phenotypes
- Haploinsufficiency
a genetic model of dominant gene action in diploid organisms where the presence of one wild-type allele and one mutant allele does not produce a wild-type phenotype
- Homologous recombination (HR)
a DNA repair pathway that utilizes the genetic information on a sister chromatid to perform template-based repair of the damaged chromosome. DNA repair by HR generally does not produce mutations, thereby promoting genome integrity and stability. HR is facilitated by BRCA1/BARD1 and associated repair factors in S/G2 phases of the cell-cycle when a sister chromatid is present. HR and non-homologous end-joining are competing DNA repair pathways
- Non-homologous end-joining (NHEJ)
a non-templated DNA repair pathway promoted by 53BP1. In NHEJ, broken DNA ends are rapidly ligated back together to avoid catastrophic chromosomal breakage. However, this process is error-prone and can lead to mutations
- Nucleosome
The fundamental organizing unit of chromatin. A mono-nucleosome is composed of a histone octamer formed by two copies each of histones H2A, H2B, H3, and H4 wrapped by about 147 base-pairs of double-stranded DNA. The histone octamer contains an ordered core region and dynamic histone tails that are subject to a wide variety of regulatory post-translational modifications (PTMs). Mono-nucleosomes are connected in chromatin via linker DNA and may contain linker histone H1. Many chromatin-regulating factors bind to nucleosomes directly, including BRCA1/BARD1 and 53BP1
- Poly (ADP-ribose) polymerase inhibitors (PARPi)
a class of small-molecule drugs that target PARP enzymes. These drugs exacerbate DNA damage by preventing early DNA damage signaling by PARP enzymes. Cells that are deficient in HR, such as those harboring certain BRCA1 and BARD1 mutations, are especially sensitive to PARPi drug treatment. Therefore, PARPi treatment can be used to assess the effects of mutations on BRCA1/BARD1 function in HR
- TP53 binding-protein 1 (53BP1)
a protein that promotes DNA DSB repair through NHEJ by directly competing with BRCA1/BARD1 to occupy damaged chromatin sites. 53BP1 uses its tandem Tudor domain and ubiquitin-directed recognition motif to bind to nucleosomes containing both H4K20me2 and H2A K15-Ub
- Transcriptional regulation
a collection of biological pathways that determine which and when genes are expressed in cells. In general, transcriptional regulation is influenced by the collection of histone post-translational modifications and regulatory enzymes/factors found at a particular genomic region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST STATEMENT: The authors declare no competing interests.
REFERENCES
- 1.Friedman LS et al. (1994) Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet 8, 399–404 [DOI] [PubMed] [Google Scholar]
- 2.Hall J et al. (1990) Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250, 1684–1689 [DOI] [PubMed] [Google Scholar]
- 3.King M-C et al. (2003) Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302, 643–646 [DOI] [PubMed] [Google Scholar]
- 4.Miki Y et al. (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71 [DOI] [PubMed] [Google Scholar]
- 5.Stewart MD et al. (2018) BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proc. Natl. Acad. Sci. U. S. A 115, 1316–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W et al. (2021) A synergetic effect of BARD1 mutations on tumorigenesis. Nat. Commun 12, 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber-Lassalle N et al. (2019) Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res 21, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullan PB et al. (2006) The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 25, 5854–5863 [DOI] [PubMed] [Google Scholar]
- 9.Zhao W et al. (2019) The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem 88, 221–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarsounas M and Sung P (2020) The antitumorigenic roles of BRCA1–BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol 21, 284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brzovic PS et al. (2001) Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol 8, 833–837 [DOI] [PubMed] [Google Scholar]
- 12.Hashizume R et al. (2001) The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem 276, 14537–14540 [DOI] [PubMed] [Google Scholar]
- 13.Lorick KL et al. (1999) RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci 96, 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savage KI and Harkin DP (2015) BRCA1, a “complex” protein involved in the maintenance of genomic stability. FEBS J 282, 630–646 [DOI] [PubMed] [Google Scholar]
- 15.Zhao W et al. (2017) BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550, 360–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scully R et al. (1997) BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. U. S. A 94, 5605–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scully R et al. (1997) Dynamic Changes of BRCA1 Subnuclear Location and Phosphorylation State Are Initiated by DNA Damage. Cell 90, 425–435 [DOI] [PubMed] [Google Scholar]
- 18.Kolas Nadine K et al. (2007) Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 318, 1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bijan Sobhian et al. (2007) RAP80 Targets BRCA1 to Specific Ubiquitin Structures at DNA Damage Sites. Science 316, 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mattiroli F et al. (2012) RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195 [DOI] [PubMed] [Google Scholar]
- 21.Pruneda JN et al. (2012) Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell 47, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzger MB et al. (2014) RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Ubiquitin-Proteasome Syst 1843, 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witus SR et al. (2021) The BRCA1/BARD1 ubiquitin ligase and its substrates. Biochem. J DOI: 10.1042/BCJ20200864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reid LJ et al. (2008) E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl. Acad. Sci. U. S. A 105, 20876–20881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shakya R et al. (2011) BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drost R et al. (2011) BRCA1 RING Function Is Essential for Tumor Suppression but Dispensable for Therapy Resistance. Cancer Cell 20, 797–809 [DOI] [PubMed] [Google Scholar]
- 27.Shakya R et al. (2008) The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci 105, 7040–7045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Q et al. (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Q et al. (2018) Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Mol. Cell 70, 842–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalb R et al. (2014) BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep 8, 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vignesh Kasinath et al. (2021) JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 371, eabc3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fradet-Turcotte A et al. (2013) 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu PL et al. (2019) Structural Basis of H2B Ubiquitination-Dependent H3K4 Methylation by COMPASS. Mol. Cell 76, 712–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Worden EJ et al. (2019) Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell 176, 1490–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fierz B et al. (2011) Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol 7, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Debelouchina GT et al. (2017) Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat. Chem. Biol 13, 105–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shah S et al. (2020) Histone H2A isoforms: Potential implications in epigenome plasticity and diseases in eukaryotes. J. Biosci 45, 4. [PubMed] [Google Scholar]
- 38.Paull TT et al. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. CB 10, 886–895 [DOI] [PubMed] [Google Scholar]
- 39.Hu Q et al. (2021) Mechanisms of BRCA1–BARD1 nucleosome recognition and ubiquitylation. Nature 596, 438–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim B-J et al. (2017) The Histone Variant MacroH2A1 Is a BRCA1 Ubiquitin Ligase Substrate. Cell Rep 19, 1758–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebastian R et al. (2020) Epigenetic Regulation of DNA Repair Pathway Choice by MacroH2A1 Splice Variants Ensures Genome Stability. Mol. Cell 79, 836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Douet J et al. (2017) MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci 130, 1570–1582 [DOI] [PubMed] [Google Scholar]
- 43.Gamble MJ et al. (2010) The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev 24, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witus SR et al. (2021) BRCA1/BARD1 site-specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nat. Struct. Mol. Biol 28, 268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elizabeth Sarcinella et al. (2007) Monoubiquitylation of H2A.Z Distinguishes Its Association with Euchromatin or Facultative Heterochromatin. Mol. Cell. Biol 27, 6457–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christensen DE et al. (2007) E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol 14, 941–948 [DOI] [PubMed] [Google Scholar]
- 47.Harper JW and Schulman BA (2021) Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis. Annu. Rev. Biochem 90, 403–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng N and Shabek N (2017) Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem 86, 129–157 [DOI] [PubMed] [Google Scholar]
- 49.Horn V et al. (2019) Structural basis of specific H2A K13/K15 ubiquitination by RNF168. Nat. Commun 10, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGinty RK et al. (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallego LD et al. (2016) Structural mechanism for the recognition and ubiquitination of a single nucleosome residue by Rad6-Bre1. Proc. Natl. Acad. Sci. U. S. A 113, 10553–10558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H et al. (2004) Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878 [DOI] [PubMed] [Google Scholar]
- 53.Cao R et al. (2005) Role of Bmi-1 and Ring1A in H2A Ubiquitylation and Hox Gene Silencing. Mol. Cell 20, 845–854 [DOI] [PubMed] [Google Scholar]
- 54.Mattiroli F et al. (2014) The nucleosome acidic patch plays a critical role in RNF168-dependent ubiquitination of histone H2A. Nat. Commun 5, 3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGinty RK and Tan S (2021) Principles of nucleosome recognition by chromatin factors and enzymes. Current Opinion in Structural Biology 71, 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monteiro ANA et al. (1996) Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc. Natl. Acad. Sci 93, 13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chapman MS and Verma IM (1996) Transcriptional activation by BRCA1. Nature 382, 678–679 [DOI] [PubMed] [Google Scholar]
- 58.Samavat H and Kurzer MS (2015) Estrogen metabolism and breast cancer. Cancer Lett 356, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mungenast F and Thalhammer T (2014) Estrogen Biosynthesis and Action in Ovarian Cancer. Front. Endocrinol 5, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fuhrman BJ et al. (2012) Estrogen Metabolism and Risk of Breast Cancer in Postmenopausal Women. JNCI J. Natl. Cancer Inst 104, 326–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Savage KI et al. (2014) BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res 74, 2773–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J et al. (2020) 53BP1 loss rescues embryonic lethality but not genomic instability of BRCA1 total knockout mice. Cell Death Differ 27, 2552–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bouwman P et al. (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol 17, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaspers JE et al. (2013) Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov 3, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Escribano-Díaz C et al. (2013) A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 49, 872–883 [DOI] [PubMed] [Google Scholar]
- 66.Thorslund T et al. (2015) Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527, 389–393 [DOI] [PubMed] [Google Scholar]
- 67.Li M and Yu X (2013) Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakamura K et al. (2019) H4K20me0 recognition by BRCA1–BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol 21, 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Becker JR et al. (2021) BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination. Nature 596, 433–437 [DOI] [PubMed] [Google Scholar]
- 70.Krais JJ et al. (2021) RNF168-mediated localization of BARD1 recruits the BRCA1-PALB2 complex to DNA damage. Nat. Commun 12, 5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson MD et al. (2016) The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103 [DOI] [PubMed] [Google Scholar]
- 72.Saredi G et al. (2016) H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 534, 714–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dai L et al. (2021) Structural insight into BRCA1-BARD1 complex recruitment to damaged chromatin. Mol. Cell 81, 2765–2777.e6 [DOI] [PubMed] [Google Scholar]
- 74.Fox D 3rd et al. (2008) Crystal structure of the BARD1 ankyrin repeat domain and its functional consequences. J. Biol. Chem 283, 21179–21186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winget JM and Mayor T (2010) The Diversity of Ubiquitin Recognition: Hot Spots and Varied Specificity. Mol. Cell 38, 627–635 [DOI] [PubMed] [Google Scholar]
- 76.Walser F et al. (2020) Ubiquitin Phosphorylation at Thr12 Modulates the DNA Damage Response. Mol. Cell 80, 423–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sherker A et al. (2021) Two redundant ubiquitin-dependent pathways of BRCA1 localization to DNA damage sites. EMBO Rep 22, e53679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Densham RM et al. (2016) Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol 23, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jonathan Markert et al. SMARCAD1 is an ATP-dependent histone octamer exchange factor with de novo nucleosome assembly activity. Sci. Adv 7, eabk2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uckelmann M et al. (2018) USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun 9, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Poepsel S et al. (2018) Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol 25, 154–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wojcik F et al. (2018) Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat. Commun 9, 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.