

Abstract
The sialic acid-binding immunoglobulin type lectins (Siglecs) are expressed predominantly on white blood cells and participate in immune cell recognition of self. Most Siglecs contain cytoplasmic inhibitory immunoreceptor tyrosine-based inhibitory motifs (ITIMs) characteristic of inhibitory checkpoint co-receptors that suppress cell signaling when they are recruited to the immunological synapse of an activating receptor. Antibodies to activatory receptors typically activate immune cells by ligating the receptors on the cell surface. Here we report that the conjugationof high affinity ligands of Siglecs to anti-bodies targeting activatory immune receptors can suppress receptor mediated activation of immune cells. Indeed, B cell activation by antibodies to the B cell receptor (BCR) IgD is dramatically suppressed by conjugation of anti-IgD with high affinity ligands of a B cell Siglec CD22/Siglec-2. Similarly, degranulation of mast cells induced by antibodies to IgE, which ligate the IgE/FcεR1 receptor complex, is suppressed by conjugation of anti-IgE to high affinity ligands of a mast cell Siglec, CD33/Siglec-3 (CD33L). Moreover, the anti-IgE-CD33L suppresses anti-IgE mediated systemic anaphylaxis of sensitized humanized mice, and prevents anaphylaxis upon subsequent challenge with anti-IgE. The results demonstrate that attachment of ligands of inhibitory Siglecs to anti-receptor antibodies can suppress activation of immune cells and modulate unwanted immune responses.
Keywords: Sialic acid, Siglec, IgE, CD22, CD33, Mast cell, B cell
Graphical Abstract
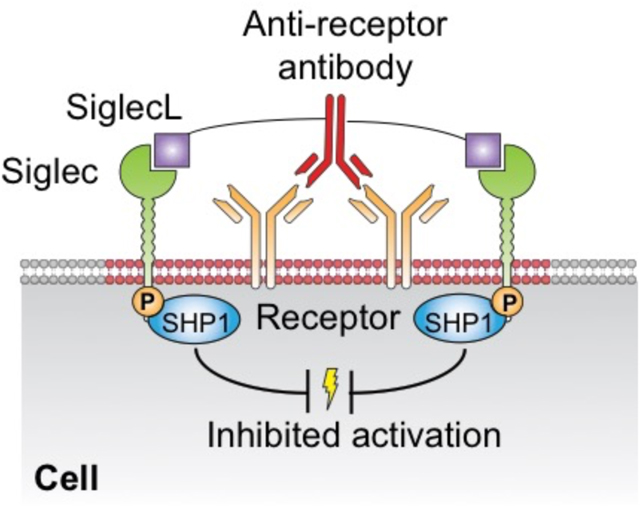
INTRODUCTION
Immune cells that mediate innate and adaptive immune responses are activated by cell surface receptors that recognize foreign antigens and chemical signatures presented by pathogens, allergens and inflammatory disease.1–5 These receptors are further regulated by co-receptors that can enhance activation or suppress activation to prevent unwanted immune responses.6–13 The Siglecs are a family of co-receptors expressed on immune cells that have in common recognition of sialic acid containing glycans as ligands. Since sialic acids are found on all mammalian cells, Siglecs can modulate signaling of immune cells that contact other self-cells that express their ligands as a result of their recruitment to the site of cell contact.8 Although some Siglecs enhance cell signaling by associating with activatory adaptor proteins such as DAP12 (e.g. Siglec-14, -15, 16), most Siglecs suppress cell signaling through immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in their cytoplasmic domains. When inhibitory Siglecs are present in an immunological synapse, their ITIMs become phosphorylated and recruit tyrosine phosphatases such as SHP1 and SHP2 that downregulate signaling cascades.8, 14
Because Siglecs are primarily expressed on immune cells they are recognized as attractive targets for cell specific therapies that can enhance desired immune responses or suppress unwanted immune responses.15–27 As one strategy to suppress unwanted immune responses by B cells and mast cells we developed Siglec tolerizing antigenic liposomes (STALs) that display both an antigen (or allergen) and glycan ligands of inhibitory Siglecs. For B cells the antigen is recognized by a B cell receptor (BCR) comprising membrane bound IgM and/or IgD, and a high affinity ligand of a B cell Siglec (CD22 or Siglec-G/10).23, 28–32 When administered in vivo, STALs suppress activation of B cells that recognize the antigen and cause apoptosis of the B cells resulting in tolerance to subsequent antigen challenge. For mast cells the STALs display an allergen, recognized by an IgE bound to the FcεRI receptor, and a high-affinity ligand of a mast cell inhibitory Siglec such as CD33/Siglec-3 or Siglec-8.33–34 When injected into a mouse sensitized to an anti-allergen IgE, mast cell degranulation mediated by the allergen is suppressed by recruitment of the Siglec, preventing severe systemic anaphylaxis, and desensitizing the animal to subsequent allergen challenge.33–34
While the STAL platform demonstrates the potential for modulating immune responses by recruitment of Siglecs, they are by design antigen specific, and the complexity of the nanoparticle platform limits their general utility. We reasoned that direct attachment of Siglec ligands to antigens or antibodies that bind directly to the activating receptor could potentially recruit Siglecs and suppress immune cell responses. Here, using anti-IgD that activates B cells by binding to the IgD BCR, or anti-IgE that activates mast cells by binding to the IgE/FcεRI receptor complex, we show that direct conjugation of Siglec ligands results in suppression of antibody mediated activation of the cells. Indeed, conjugation of anti-IgD with the ligand of the murine B cell Siglec CD22 (anti-IgD-CD22L) suppresses B cell activation in wild type B cells but has no effect on activation of B cells from a CD22 knockout mouse. Conjugation of an anti-human IgE (anti-IgE) with a high affinity ligand of human CD33 (anti-IgE-CD33L) suppresses anti-IgE mediated mast cell degranulation and systemic anaphylaxis in humanized mice. Moreover, mice treated with anti-IgE-CD33L are tolerized to subsequent challenge with anti-IgE. The results suggest the potential to regulate immune cell responses using Siglec-ligand conjugated antibodies that target activating receptors or receptor complexes on immune cells.35–36
RESULTS AND DISCUSSION
Conjugation of CD22L to anti-IgD causes CD22 mediated suppression of B cell activation.
We reasoned that conjugation of CD22L to anti-IgM or anti-IgD could suppress their activation of B cells by recruitment of CD22 to the BCR immunological synapse (Fig 1A). We focused our efforts on conjugation of ligands to surface lysines, an approach widely used for development of antibody drug conjugates (ADC).37–38 Our strategy was to employ azide/alkyne click chemistry to couple CD22L with a functionalized linker to an antibody conjugated with linkers containing the corresponding azide/alkyne as illustrated in Scheme 1. To this end we evaluated two different linkers, NHS-peg6-N3 and NHS-peg6-DBCO, both of which can participate in subsequent click chemistry, but differ in their hydrophilicity owing to polar -N3 and hydrophobic -DBCO functional groups. We selected anti-IgD instead of anti-IgM for experiments since the presence of IgM in the blood would abrogate future in vivo experiments. Anti-IgD conjugates were prepared using 20 molar equivalents (eq) of NHS-peg6-N3 and DBCO-peg6-NHS (Scheme 1, Scheme S1). The impact of linker conjugation on the ability of anti-IgD to activate murine B cells was then assessed using an in vitro calcium flux assay. While the anti-IgD modified with the azido-linker had minimal effect on B cell activation, the hydrophobic DBCO-linker strongly reduced activity (Fig. S1), a problem sometimes encountered with hydrophobic linkers.39 We therefore focused on the NHS-peg6-N3 linkers.
Figure 1.
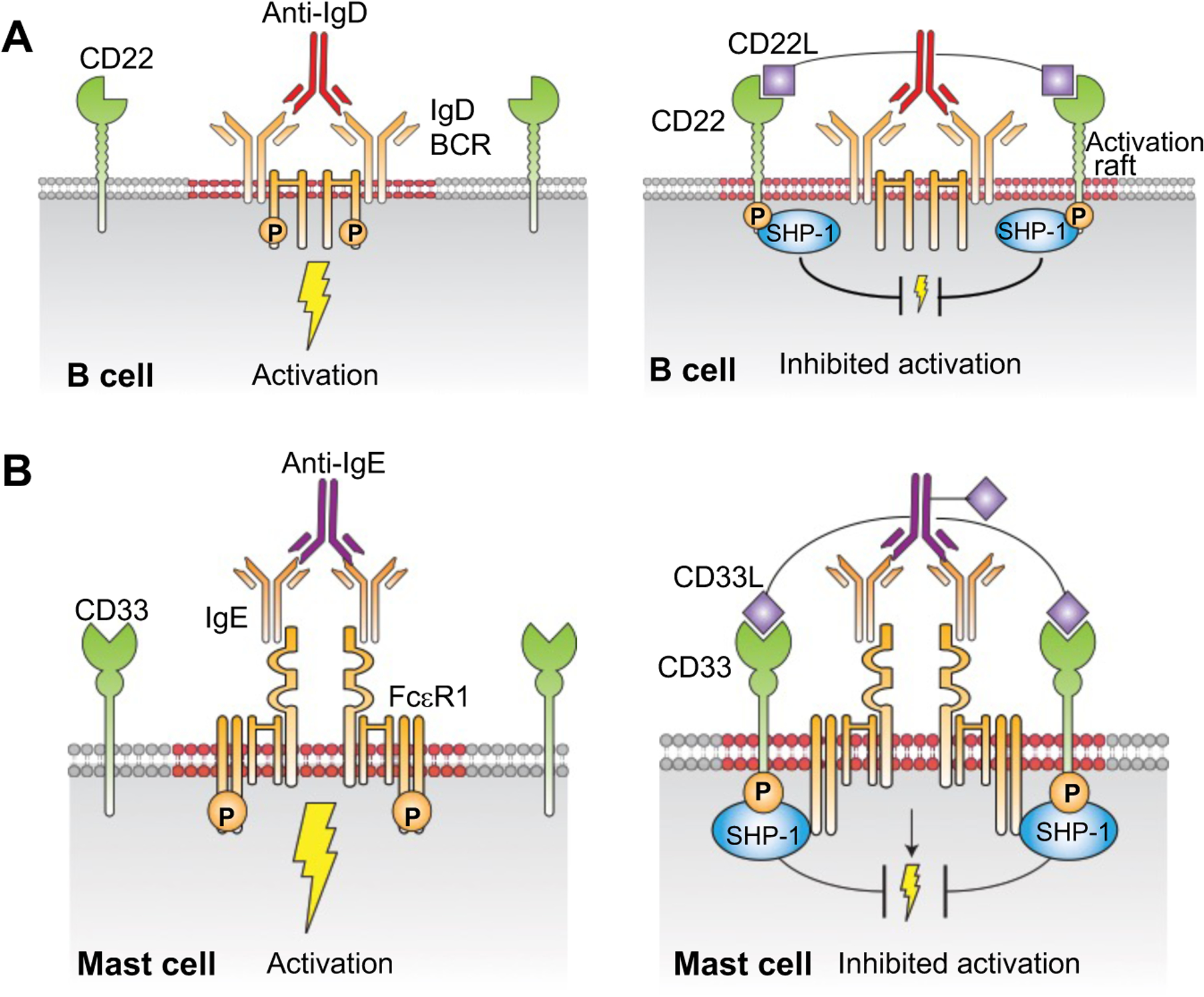
Proposed impact of inhibitory Siglec ligands conjugated to antibodies of immune cell activation receptors. (A) Anti-IgD serves as a surrogate antigen and cross-links the IgD-B cell receptor (IgD-BCR) on the surface of B cells to induce activation (left). When anti-IgD is conjugated to CD22L, ligand-mediated recruitment of CD22 to the IgD-BCR results in suppression of BCR signaling (right). (B) Anti-IgE ligates the mast cell IgE/FcεRI receptor complex causing mast cell activation (left). When anti-IgE is conjugated to CD33L, activation and degranulation of the mast cell is suppressed as a result of inhibitory CD33 being recruited to the IgE/FcεR1 receptor complex (right).
Scheme 1.
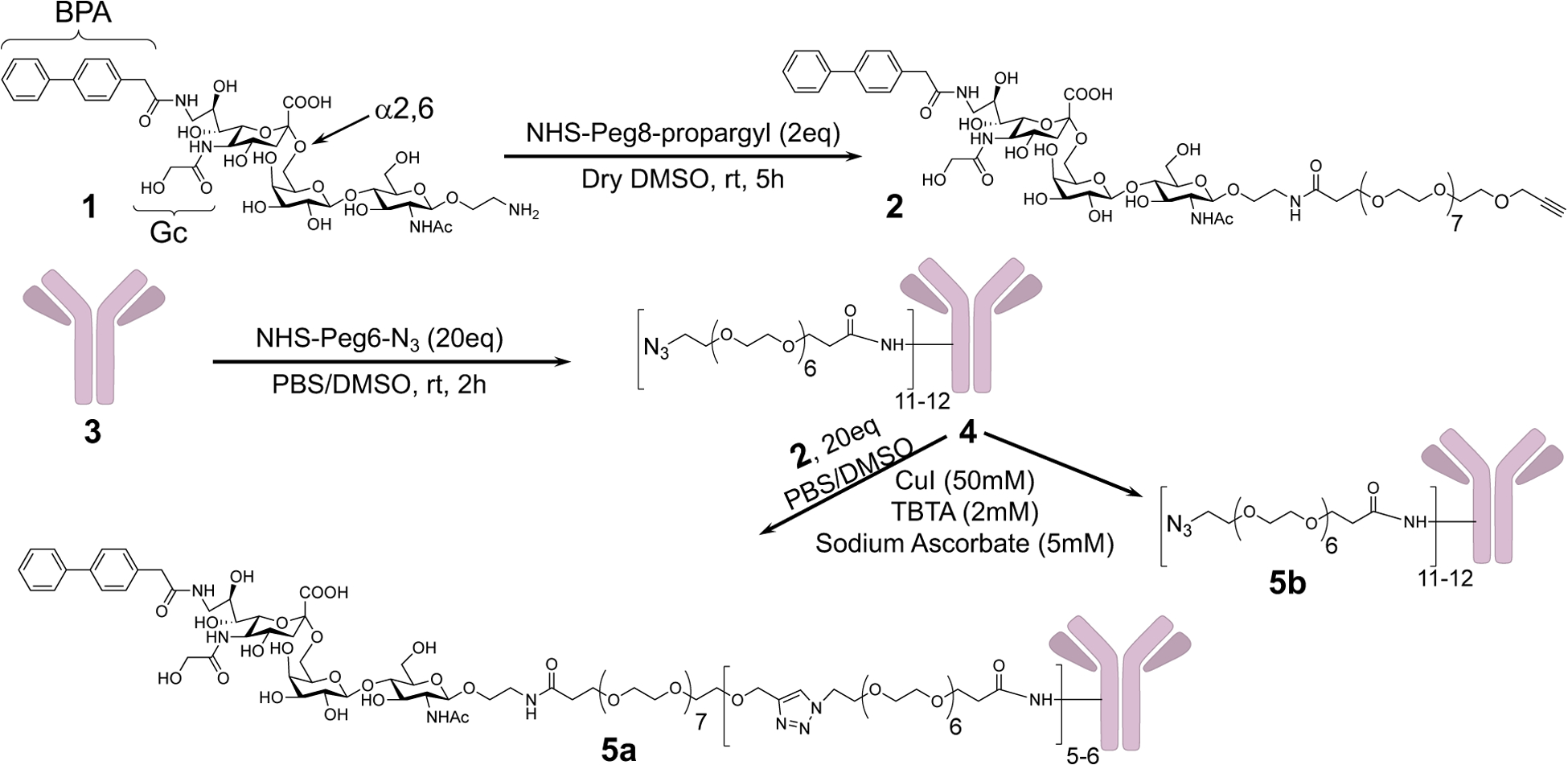
Synthesis of Propargyl-Peg8-mCD22L (2) and conjugation to anti-IgD (3)
To optimize linker conjugation, anti-IgD was reacted with several different linker stoichiometries, varying the amount of NHS-peg6-N3 used in the reaction (Scheme 1, Scheme S2). SDS gel electrophoresis and MALDI-TOF mass spectroscopy (Fig S2A–B) revealed that treatment of 1 eq of anti-IgD in phosphate buffered saline, pH7.4 (PBS) with either 5, 10, and 20 eq of NHS-peg6-N3 in DMSO afforded an average of 2–3, 5–8 and 11–12 linker/anti-IgD (Scheme S2). A stoichiometry of 20 was chosen as the favored stoichiometry since the goal was to avoid having anti-IgD with no CD22L.
For coupling to the azide functionalized antibody, the CD22L linker was modified to contain a propargyl group (Scheme 1). Propargyl-peg8-CD22L (2) was synthesized using 2 eq of NHS-peg8-propargyl with 1 eq of CD22 ligand (CD22L) in dry DMSO at room temperature for 5h. Next propargyl-peg8-CD22L were conjugated to anti-IgD-N3 (4) under Cu(I) catalyzed click chemistry to afford anti-IgD with 5–6 CD22L per antibody (anti-IgD-CD22L, 5a) as assessed by MALDI-TOF mass spectroscopy. Anti-IgD-N3 was similarly treated without propargyl-peg8-CD22L to afford 5b to serve as a control that was subjected to the same chemical treatments but without CD22L. Conjugation of the peg6-N3 linker and CD22L to anti-IgD was confirmed using SDS gel chromatography using a reducing agent, tris(2-carboxyethyl) phosphine (TCEP), to separate the antibody heavy (50 kDa) and light (25 kDa) chains (Fig S2B). This showed that both the heavy and light chains were modified.
To investigate if the presence of the CD22L on anti-IgD-CD22L was able to suppress the activation of B cells, we performed an in vitro assay using Ca2+ flux as a measure of activation. Treatment of B cells with anti-IgD (3) (10 μg/ml) strongly induced activation as did anti-IgD modified with the linkers only (5b). In contrast there was negligible activation with anti-IgD-CD22L (5a) (Fig. 2A). To confirm that suppressed activation with anti-IgD-CD22L was CD22 dependent we performed the same assay using B cells from CD22 knockout (CD22KO) mice that are deficient in CD22. As shown in Fig. 2B, anti-IgD-CD22L (5a) induced activation in CD22KO B cells equivalent to the anti-IgD (3) and anti-IgD-N3 (5b), strongly supporting the conclusion that suppression of B cell activation with anti-IgD-CD22L in wild type B cells is mediated by CD22 (Fig. 1A).
Figure 2.
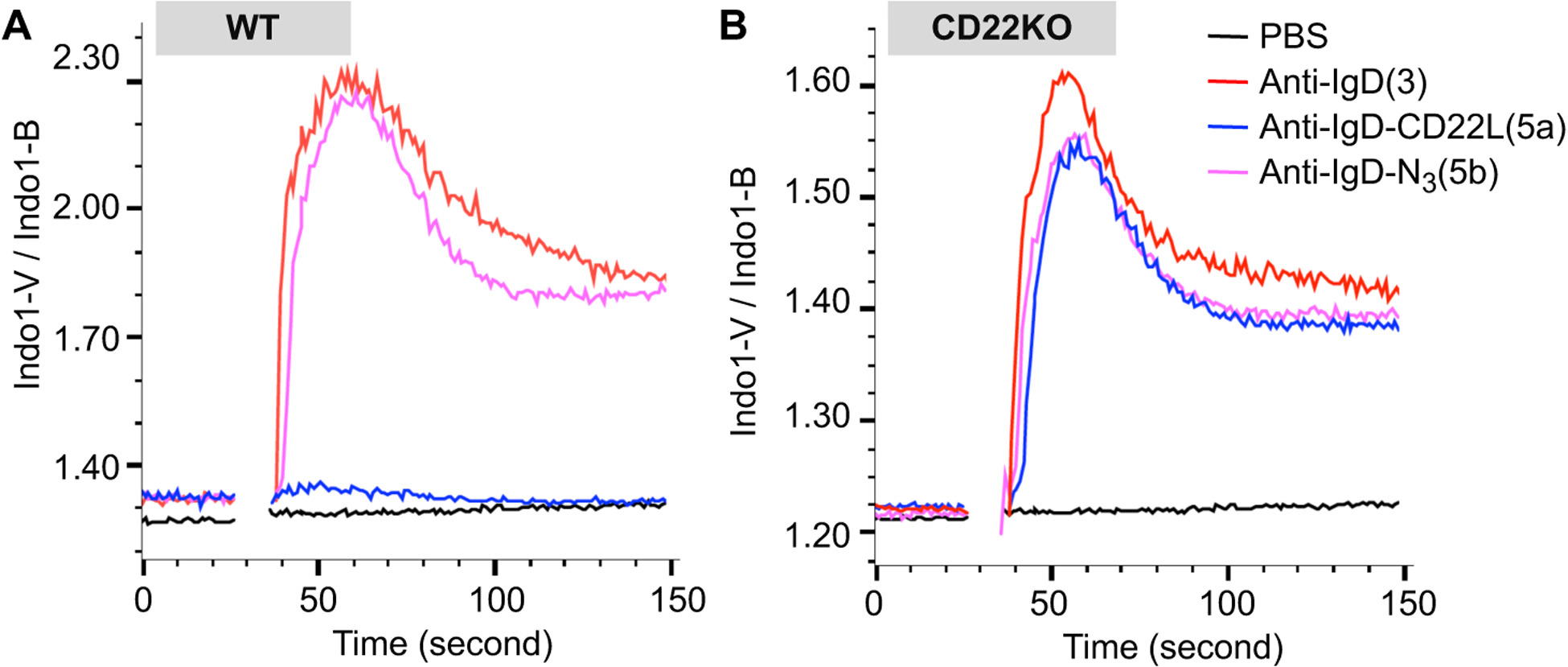
Impact of CD22L conjugated to anti-IgD on activation of murine B cells. Splenocytes from wildtype (WT; A) or CD22 knockout (CD22KO; B) mice on the C57BL/6J background were loaded with the fluorescent intracellular calcium indicator Indo-I AM, prior to staining with antibodies to analyze calcium flux in the B cells (B220+CD5−). Washed cells were resuspended in media containing CaCl2 and warmed to 37 °C prior to being stimulated with PBS or 10μg/ml of anti-IgD, (3) anti-IgD-N3, (5b) or anti-IgD-CD22L and analyzed by flow cytometry.
Conjugation of CD33L to anti-IgE suppresses IgE/FcεRI mediated activation of mast cells.
To establish the concept of conjugating ligands of inhibitory Siglecs to anti-receptor antibodies as a general platform for suppression of immune cell activation, we investigated conjugation of Siglec ligands to anti-IgE as a means to inhibit mast cell activation induced by ligation of the IgE/FcεRI complex (Fig 1B). Exposure of allergic individuals to allergen results in its binding to and ligation of the IgE/FcεRI receptor complex on mast cells resulting in degranulation and production of cytokines that lead to allergic symptoms and potentially severe anaphylaxis. Since anti-IgE also binds to and ligates the IgE/FcεRI complex, it can behave as a surrogate allergen inducing degranulation and IgE mediated systemic anaphylaxis when administered to animals.40–41 Mast cells are also known to express a variety of inhibitory receptors including Siglecs that bear the characteristic ITIMs in their cytoplasmic domains.8, 34, 42–44 Using various approaches, several groups have demonstrated that enforced recruitment of inhibitory receptors to the IgE/FcεRI complex can suppress FcεRI mediated mast cell activation.8, 14, 33, 45–51 In particular, we found that co-presentation of an allergen and a high affinity ligand of CD33 on liposomal nanoparticles (STALs) resulted in suppressed allergen mediated activation of human mast cells and mast cells from transgenic mice expressing human CD33.34 Based on these observations, we set out to conjugate CD33L to anti-IgE to assess its ability to suppress IgE/FcεRI complex mediated mast cell activation (Fig. 1B). We elected to use anti-human IgE since in vivo studies could be performed using transgenic mice with mast cells expressing both human FcεRI (hFcεRI) and human CD33 (hCD33 or CD33), or ‘humanized’ mice produced by injecting human CD34+ stem cells into immunodeficient NSG-SGM3 to generate a human immune system with human mast cells.
Accordingly, as illustrated in (Scheme 2) we prepared anti-IgE-CD33L (10a) using a strategy similar to that described for anti-IgD-CD22L (5a). CD33L with an ethyl-amine linker was coupled to NHS-peg8-propargyl to afford propargyl-peg8-CD33L (7). Human anti-IgE was reacted with 20 eq of NHS-peg6-N3 in DMSO to prepare azido-peg6-anti-IgE (anti-IgE-N3). These were then coupled by Cu(I) catalyzed click chemistry. To characterize conjugation efficiency, anti-IgE with or without CD33L were subjected to SDS gel electrophoresis after reduction with TCEP to separate the heavy and light chains (Fig. 3A). Clear increases in the apparent molecular weights suggest modification of the heavy and light chains of both the azide-linker and CD33L modified anti-IgEs. Additionally, MALDI-TOF mass spectroscopy was performed to determine the drug to antibody ratio (DAR) and was found to be 4–5 units of CD33L per anti-IgE.
Scheme 2.
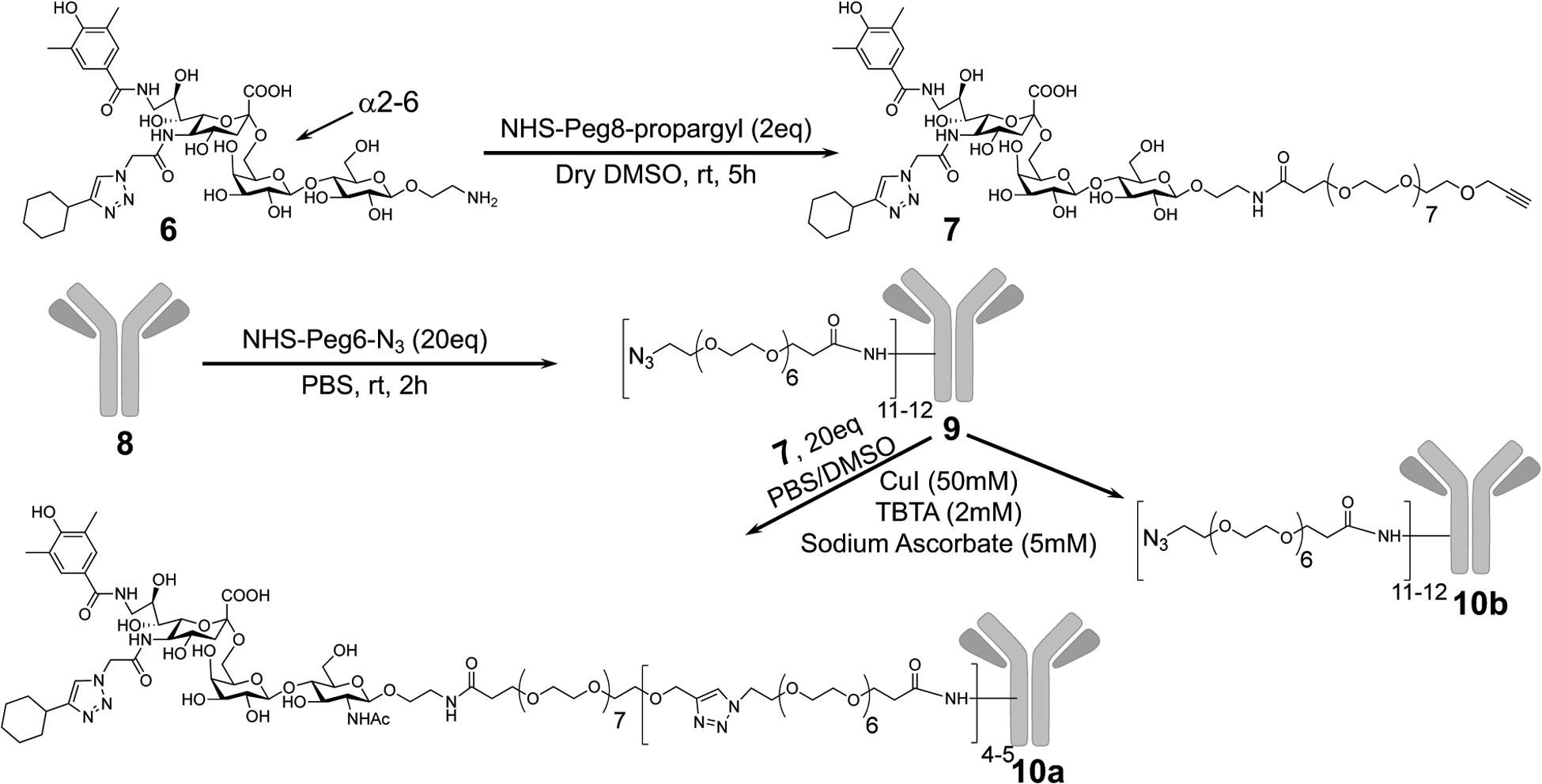
Synthesis of Propargyl-Peg8-CD33L (7) and conjugation to anti-IgE (8)
Figure 3.
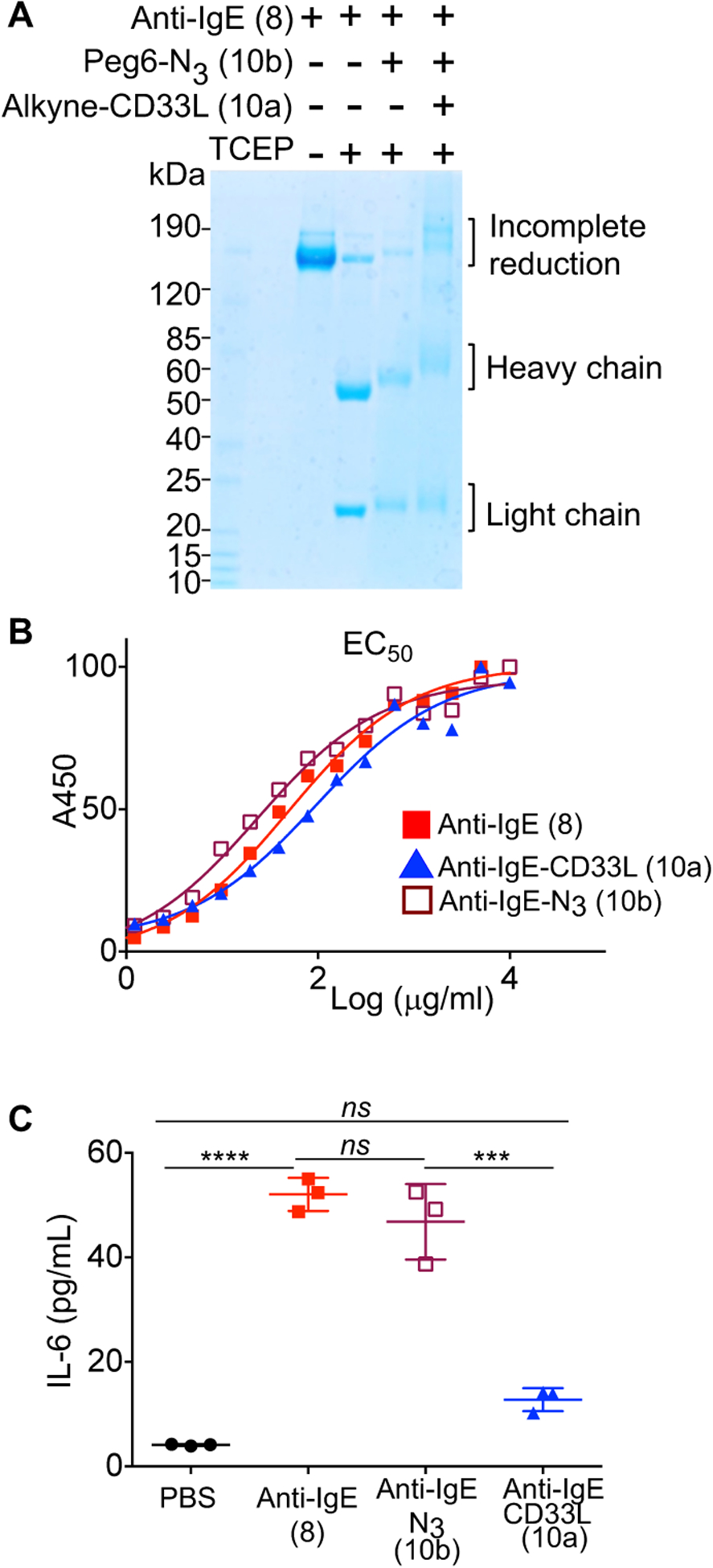
Impact of CD33L conjugation to anti-IgE on the binding to IgE and induction of mast cell activation. (A) Analysis of anti-IgE conjugates by SDS gels. Samples were reduced with tris(2-carboxyethyl) phosphine (TCEP) at 37 °C for 30 minutes prior to electrophoresis to separate the IgE heavy (~50KDa) and light (~25KDa) chains. (B) ELISA assays to assess EC50 for binding of anti-IgE (8), anti-IgE-N3 (10b), and anti-IgE-CD33L (10a) to human IgE. Anti-IgE (8), anti-IgE-N3 (10b), and anti-IgE-CD33L (10a) were serially diluted in 96 well plates coated with human IgE and incubated for 1.5h at 37 °C. Plates were washed and incubated with secondary antibody conjugated to horse radish peroxidase (HRP) (anti-hIgG2a-HRP), washed again and incubated with HRP substrate (TMB) to detect bound anti-IgE by absorbance at 450 nm. (C) Impact of anti-IgE induced production of IL-6 by bone marrow derived mast cells (BMMCs). BMMCs from hFcεRI+ × hCD33+ mice were sensitized with human IgE (1μg/106cells) overnight at 37 °C. Cells were washed, aliquoted to 105 cells/100μl in a 96 well plate and then mixed at 37° C with PBS as a buffer control or with anti-IgE (8), anti-IgE-N3(10b), or CD33L-anti-IgE (10a) in PBS to give a final concentration of 10 μg/ml. IL-6 cytokine production was assessed in an ELISA by measuring absorbance at 450 nm. Results in (C) are representative of 3 independent experiments and data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison test (***P< 0.001 and ****P< 0.0001); BMMCs, bone marrow derived mast cells.
Since binding of the antibody to IgE is critical for its ability to induce ligation of the IgE/FcεRI complex and activate mast cells, we next tested the impact of the modifications on binding activity by ELISA. Accordingly, anti-IgE (8), anti-IgE-N3 (10b), and anti-IgE-CD33L (10a) were titrated in 96 well plates coated with human IgE (hIgE) to assess the amount needed to achieve half maximal binding (EC50). As shown in Fig. 3B, the addition of N3-linkers and CD33L had minimal impact on binding to IgE, with the EC50 values within a factor of two of unmodified anti-IgE.
To evaluate the effect of human anti-IgE-CD33L on IgE/FcεRI-mediated mast cell activation, bone marrow derived mast cells (BMMCs) were prepared from transgenic mice that express both hFceR1 and hCD33. BMMCs were sensitized with hIgE overnight, and then used to test the ability of anti-IgE (8), anti-IgE-N3 (10b), or anti-IgE-CD33L (10a) to activate the cells by measuring the production of the cytokine interleukin-6 (IL-6) by ELISA. As shown in Fig. 3C, relative to the buffer (PBS) control, anti-IgE (8) and anti-IgE-N3 (10b) induced robust activation and production of IL-6, while anti-IgE-CD33L (10a) strongly suppressed activation.
Anti-IgE-CD33L suppresses anaphylaxis in IgE sensitized humanized mice.
To assess the ability of anti-IgE-CD33L to inhibit anaphylaxis mediated by the IgE/FcεRI receptor complex in vivo, we used the passive cutaneous anaphylaxis (PC) and passive systemic anaphylaxis (PS) models with transgenic and ‘humanized’ mice, respectively. In the PC assay two closely related strains of transgenic mice were used. The control strain of mice had mast cells expressing hFcεRI on a murine FcεRI knockout background but no hCD33 (referred to as hFcεRI+ × hCD33−). The related transgenic strain had mast cells expressing both hFcεRI and hCD33 (referred to as hFcεRI+ × hCD33+).
For the PC assay mice were injected intradermally in the ear with PBS as a control, or with hIgE (1μg) to sensitize mast cells. The next day mice were injected i.v. with either anti-IgE (8), anti-IgE-CD33L (10b) or no antibody (PBS) in a PBS buffer containing Evans blue dye to detect vascular leakage (Fig 4A). After 1h ears were harvested and dye extracted to assess the degree of mast cell mediated vascular leakage. In hFcεRI+ × hCD33− control mice (Fig. 4B) strong anaphylaxis was induced in ears sensitized with IgE and treated with either anti-IgE (8) or anti-IgE-CD33L (10a), but exhibited minimal anaphylaxis in ears injected with PBS. The results show that in mast cells that do not express hCD33, anti-IgE with or without CD33L were equally able to induce IgE mediated anaphylaxis in these mice. However, in the hFcεRI+ × hCD33+ mice with mast cells expressing both hFcεRI and hCD33, while the same degree of anaphylaxis was induced by anti-IgE (8), anaphylaxis was strongly suppressed in mice treated with anti-IgE-CD33L (10a) (Fig. 4C). Taken together, the results suggest that the CD33L conjugated to the anti-IgE suppresses mast cell activation and vascular leakage in this PC assay when the mast cells express hCD33.
Figure 4.
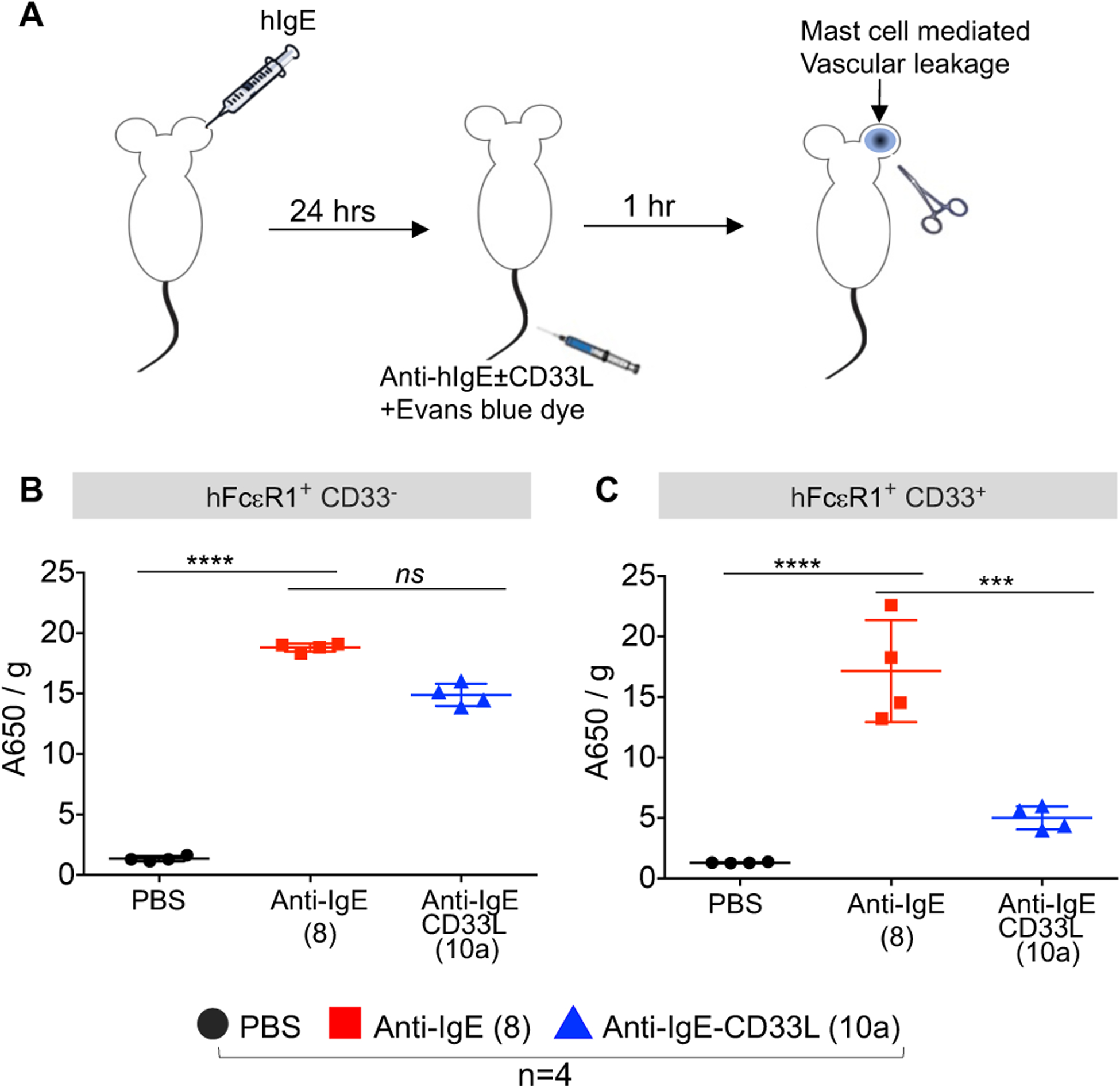
Impact of CD33L conjugation to anti-IgE on the induction of passive cutaneous anaphylaxis (PC) in the mouse ear. (A) Schematic representation of PC assay. One ear of each mouse is sensitized with hIgE (1μg/mice) followed by intravenous injection (i.v.) one day later with 0.2 ml of PBS containing Evans blue dye to detect vascular leakage, and either no antibody (PBS) or 10 μg of either anti-IgE (8) or anti-IgE-CD33L (10a). After 1h treated ears were removed and assessed for anaphylaxis by extracting the blue dye (OD A460/g tissue). (B) Assessment of anaphylaxis induced in hFcεRI+ × hCD33− mice. (C) Assessment of anaphylaxis induced in hFcεRI+ × hCD33+ mice. Results in B-C were analyzed by one-way ANOVA followed by Tukey’s multiple comparison test (***P< 0.001 and ****P< 0.0001).
To assess the impact of the anti-IgE-CD33L in the PS model, we employed NSG-SGM3-hCD34+ humanized mice. These mice are created by injecting (i.v.) 3–4 week old NSG-SGM3 mice with human cord blood CD34+ stem/progenitor cells and waiting approximately 12 weeks until they acquire a differentiated human immune system including mast cells.52 To conduct the PS model, NSG-SGM3-hCD34+ mice were first sensitized with hIgE (1 μg/mouse i.v.) (Fig. 5A). The next day groups of 5 mice were treated with anti-IgE (8), anti-IgE-N3 (10b), or anti-IgE-CD33L (10a), and anaphylaxis was assessed by measuring rectal temperature every 10 minutes for 1h. As shown in Fig. 5B mice treated with both anti-IgE (8) and anti-IgE-N3 (10b) developed strong anaphylaxis as evidenced by a rapid decrease in rectal temperature, and in each case 3 out of 5 mice died after 30 minutes. In contrast little or no change in rectal temperature was found in mice when treated with anti-IgE-CD33L (10a) demonstrating that the presence of the CD33L prevents mast cell induced anaphylaxis.
Figure 5.
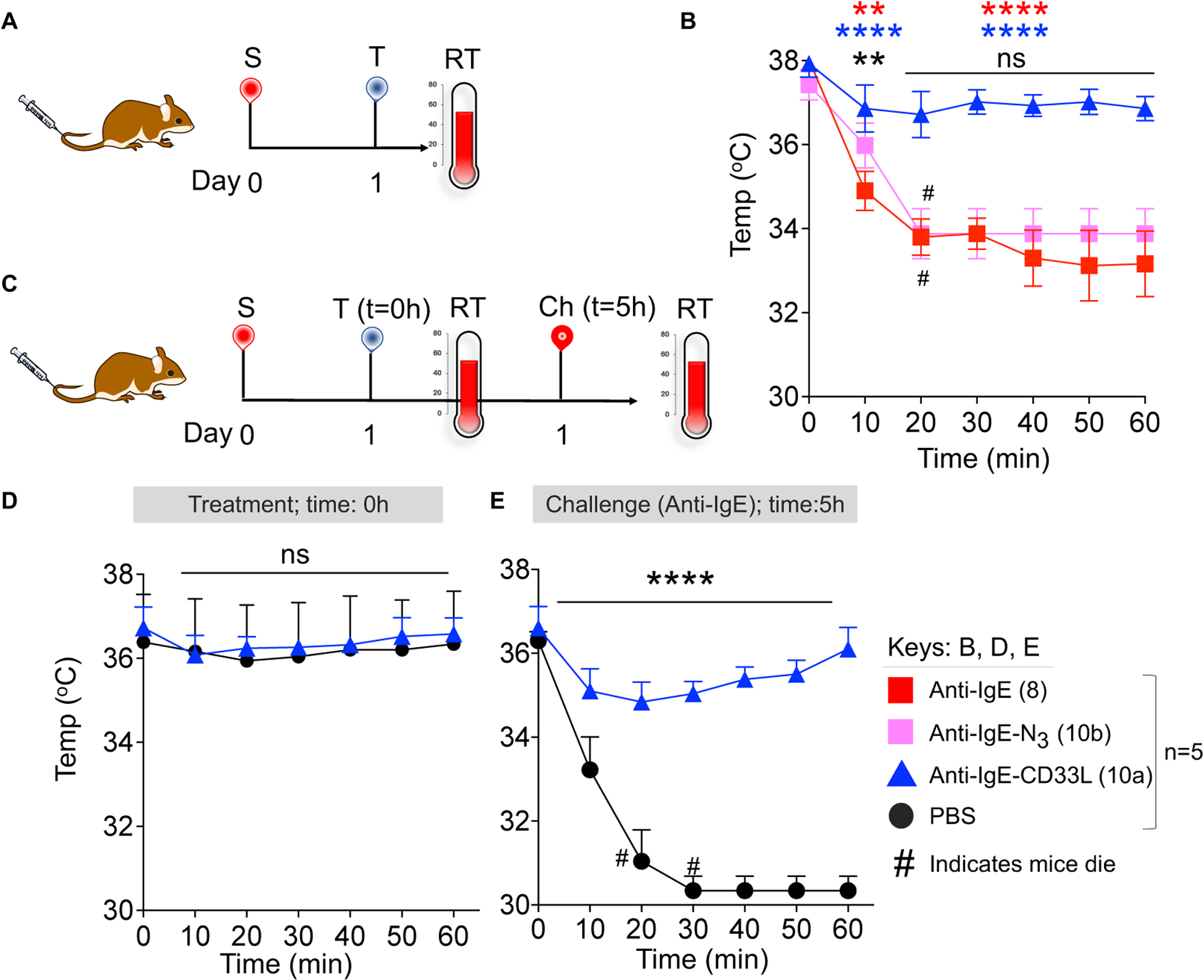
Impact of CD33L conjugation to anti-IgE on induction of passive systemic anaphylaxis (PS) and desensitization to subsequent challenge. (A) Schematic representation of the PS model in NSG-SGM3-hCD34+ humanized mice. On Day 0, mice were sensitized (S) with hIgE (1μg/mouse i.v.). The next day (Day 1) mice were treated (T) with anti-IgE (8), N3-anti-IgE (10b), or CD33L-anti-IgE (10a) (1μg/mouse i.v.) and rectal temperature (RT) was measured at 10 minutes intervals for 1h. (B) CD33L-anti-IgE (10a) suppresses passive systematic anaphylaxis in NSG-SGM3-hCD34+ mice. (C) Schematic presentation of the PS model to test for mast cell desensitization. Mice were sensitized with hIgE as described in panel A. On day 1 mice were treated (T) with either anti-IgE-CD33L (10a) (1μg/mouse i.v.) or PBS and rectal temperature (RT) was measured at 10 minute intervals for 1h. After 5h, mice were then challenged (Ch) with anti-IgE (2μg/mouse i.v.). (D) Anti-IgE-CD33L treatment protected mice from passive systematic anaphylaxis with subsequent challenge of anti-IgE. Results in (B) were analyzed by Two-way ANOVA followed by Tukey’s test (*P<0.05, **P<0.01, ***P<0.001 & ****P<0.0001). At 10 minutes **(black) is for anti-IgE vs anti-IgE-N3, ****(blue) is for anti-IgE vs anti-IgE-CD33L and **(red) is for anti-IgE-N3 vs anti-IgE-CD33L. Results in (D) were analyzed by Two-way ANOVA followed by Sidak’s multiple test (***P=0.0002 and ****P<0.0001), # indicates that mice died.
Finally, we investigated whether treatment with anti-IgE-CD33L (10a) desensitized mast cells to subsequent challenge with anti-IgE (Fig. 5C). In this case, NSG-SGM3-hCD34+ mice were first sensitized with hIgE as described above. The next day mice were treated with either PBS or anti-IgE-CD33L (1μg/mouse) and then after 5h all mice were challenged with anti-IgE (2μg/mice). No anaphylaxis was observed in the initial treatment (T=0h) with either PBS or anti-IgE-CD33L (10a) (Fig. 5d). However, upon challenge with anti-IgE 5 hours later there was profound anaphylaxis in the PBS treated mice, but no significant decrease of rectal temperature in the mice treated with anti-IgE-CD33L (10a) (Fig. 5e). The results demonstrate that treatment with anti-IgE-CD33L not only suppressed mast cell activation, but also desensitized the mast cells to subsequent challenge with anti-IgE.
CONCLUSIONS
Antibodies to activatory receptors on immune cells typically activate the cell as a result of ligating the receptor, resulting in the activation of protein kinases that initiate cell signaling pathways.53–54 Here we have demonstrated that coupling high affinity glycan ligands of inhibitory Siglecs to the antibodies of two exemplary activation receptors, IgD (BCR) on B cells and the IgE/FcεRI complex on mast cells, results in suppression of antibody mediated activation of the cells. We hypothesize that suppression of cell signaling results in recruitment of inhibitory Siglecs to the targeted receptor, implying that they are not already co-localized as has been already documented for CD22 and the BCR on B cells,10, 55 and for CD33 and the FcεRI on mast cells.8, 34, 51 We suggest that immune cell activation by antibodies to other activatory receptors may be similarly suppressed by conjugation of ligands to inhibitory Siglecs on the same cell. However, for some activatory receptors such as for toll like receptors on B cells, Siglecs are already strongly colocalized.56–57 Thus, the degree of suppression of cell signaling achieved by coupling Siglec ligands to anti-receptor antibodies may depend on the microdomain localization of the receptor and Siglec being targeted.
An FDA approved anti-IgE, omalizumab, is used therapeutically for treatment of allergic asthma and urticaria and other IgE mediated diseases.58–60 It works by removing IgE from the blood, which over time lessens the sensitivity to allergens. It does not bind to the IgE/FcεRI complex because the epitope recognized by omalizumab is blocked when IgE is bound to the hFceR1 receptor. Thus, omalizumab does not ligate the IgE/FcεRI complex, and leaves mast cells sensitized to allergens until removal of IgE from the blood over time reduces the degree of mast cell sensitization.58–60 Our demonstration that anti-IgE-CD33L suppresses activation of mast cells and desensitizes them to subsequent challenge with anti-IgE suggests the possibility for development of an anti-IgE that would both remove IgE from the blood and desensitize mast cells from subsequent allergen challenge. However, the desensitization would need to be extremely robust to ensure there would be no inadvertent anaphylaxis.
There is still much to be learned about the natural roles of Siglecs in regulation of immune cell signaling. With increasing interest in the potential to modulate the activities of Siglecs to enhance immune responses (e.g. for treatment of cancer)61–63 or suppress unwanted immune cell responses (e.g. allergies, autoimmune diseases)8, 10, 64, a concerted effort to better understand their natural functions will also facilitate efforts to modulate their functions to therapeutic benefit.
MATERIALS AND METHODS
Materials.
Unless otherwise specified, chemical and biological reagents were purchased from Fisher Scientific or Sigma-Aldrich and used directly without further purification. NHS-peg6-N3, NHS-peg8-propargyl, and NHS-peg6-DBCO were obtained from Broadpharm (San Diego, CA). Anti-IgD and anti-IgE were produced from hybridoma cell lines HB-161™ and HB-121™ (ATCC). Human anti-OVA IgE (clone 11B6) was provided by Prof. Scott A. Smith (Vanderbilt University Medical Center, Nashville, Tennessee, USA). Murine CD22L and human CD33L were prepared as previously described.65–66 1HNMR spectra were recorded on Bruker DRX-600 (600 M Hz) instruments at 25 °C and are reported in parts per million (d) relative to HOD (4.79 ppm, D2O). Coupling constants (J) are reported in Hertz. 13C NMR spectra were recorded on Bruker DRX-600 (150 MHz) instruments at 25 °C. NMR data were processed with Mnova software. ESI-TOF high resolution data were collected either on negative ion mode or positive ion mode (performed by TSRI MS center). Flow cytometry data were acquired on a LSR II flow cytometer (BD Biosciences) and analyzed using the FlowJo software. SDS gel imaging was performed on a ChemiDoc™ MP (Bio-Rad). Data from biological assays were processed and graphed with Prism software (GraphPad) for the curve-fitting and calculations. Data are presented as the average ± Standard Error of the Mean (SEM) of triplicate determinations.
Mice:
Mice were maintained in a specific pathogen free environment, and experimental procedures were approved by the Institutional Animal Care and Use Committee at The Scripps Research Institute. Wild type (WT) cells used for the B cell activation assays with anti-IgDa (HB-161) were from Hy10 mice on the C57BL/6J background that have the immunoglobulin heavy chain (IgH) locus of the ‘a’ allotype and can be activated by anti-IgDa.67 WT Hy10 mice were then crossed with CD22 knockout (CD22 KO) mice on the C57BL/6J background for a source of CD22 KO B cells compatible with anti-IgDa.68 To study the impact of anti-IgE-CD33L on hIgE and hFcεRIα signaling, hFcεRIα transgenic mice were mated with mFcεRIα knockout mice to produce hCD33− controls with the hFcεRIα+ mFcεRIα−/− genotype.69–70 These mice were then crossed with previously described transgenic mice that express hCD33 in connective tissue mast cells (Mcpt5-Cre+Rosa26-Stopfl/fl-hCD33+/+) to generate mice bearing the genotype of hCD33+/+ Mcpt5-Cre+ hFcεRIα+ mFcεRIα−/−.34, 71 Passive cutaneous anaphylaxis (PC) experiments were performed in hCD33+/+ Mcpt5-Cre+ hFcεRIα+ mFcεRIα−/− with mast cells that express hCD33 and in hFcεRIα+ mFcεRIα−/− control mice that do not express hCD33 due to the lack of Mcpt5-Cre, all on the C57BL/6J background. CD34+ humanized mice used for the PS model were generated by intravenously injecting 3–4 week-old NSG-SGM3 mice (The Jackson Laboratory, stock 013062) with 0.5–1 × 105 human CD34+ cells (Lonza, #2C-101) in 200 ul PBS. Mice used for experiments had >5% hCD45+ cells in the blood after 12–16 weeks as determined by flow cytometry.
Conjugation of SIGLEC ligand to anti-IgD/anti-IgE antibody:
NHS-peg6-N3 in dry DMSO (20eq in 2 μl) was added to the anti-IgD or anti-IgE antibody in PBS (1eq, 5mg/500μl, pH=7.4) and shaken (500rpm) at room temperature for 2h. After the reaction, excess of NHS-peg6-N3 was removed by exclusion column chromatography (SEC) using a column (1.0 × 20 cm) of Sephadex G-100. In the next step, propargyl-peg8-CD22L or proparygy-peg8-CD33L (20eq) was mixed with respective anti-IgD-N3 or anti-IgE-N3 antibody (1eq, 2mg) in a total volume of 500 μl PBS. The click reaction was performed by adding 2 μl DMSO containing CuI (50mM), TBTA [Tris(benzyltriazolylmethyl)amine, 2mM], sodium ascorbate (5mM). After allowing the reaction to proceed for 2h at room temperature, all the organic and inorganic reagents were removed by Sephadex G-100 SEC. Typical yields of antibody siglec ligand conjugate was 1.6 mg.
MALDI-TOF of antibodies:
Prior to MALDI-TOF analysis, all antibodies were desalted using G-100 size exclusion column and PBS as an eluent. MALDI-TOF was performed using sinapinic acid as the matrix and antibody in PBS (Conc. 1mg/ml).
Culture of BMMCs:
Cells were collected in RPMI-1640 from femurs of hCD33+/+ Mcpt5-Cre+ hFcεRIα+ mFcεRIα−/− mice and cultured as previously described.34 Briefly, bone marrow cells were cultured for 4 weeks in 50% RPMI 1640 media supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 50 μM β-mercaptoethanol and 50% of the same media previously cultured with IL-3 producing WEHI-3B hybridoma cells. Maturation of bone marrow cells into BMMCs was determined by flow cytometry using c-Kit and FcεRI double-positive staining. In vitro assays were performed after sorting hCD33+ (GFP+) BMMCs using a FACSAria (BD Biosciences).
Calcium flux assay:
Splenocytes containing B cells were isolated from mouse spleen in HBSS and wash with PBS. Prior to assay, splenocytes (15×106 cells/10ml) were incubated with the Indo-1 intracellular dye (1 μM) in RPMI media supplemented with 1%FBS, 10mM HEPES, 1mM MgCl2 1mM EGTA (loading buffer) at 37 °C for 45 minutes. After incubation cells were washed with RPMI loading buffer without Indo-1. stained with B220 PECy7(1:300, v/v) and CD5-PE (1:300, v/v) and kept at 0 °C for 20 minutes. After washing, cells were resuspended with HBSS media supplemented with 1%FBS, 1mM MgCl2 and 1mM CaCl2 (running buffer, 2×106 cells/ml). The Ca++ flux assay was performed 200 μl of the cell mixture (105 cells). The cells were warmed to 37 °C for 5 minutes. Induction of Ca++ flux was then assessed one sample at a time by adding 10 μl of either PBS as a background control or with anti-IgD (3) anti-IgD-N3 (5b) or anti-IgE-CD22L (5a) in PBS to give a final concentration of 10 μg/ml. Each sample was immediately mixed and Indo-I fluorescence (violet vs blue) was monitored by flow cytometry for 2.5 minutes at 37 °C. The data was analyzed using Flowjo software (version 10.8.0) with kinetic parameter.
SDS gel electrophoresis:
Antibody samples (10 μl of 0.5mg/ml) were reduced by mixing with 5 μl tris(carboxyethyl)phosphine, TCEP (50mg/ml) for 30 minutes at 37 °C. Samples were then mixed with SDS sample buffer (5μl, Bolt™) and gel electrophoresis was performed at 150 volts for 1.2 h using precast SDS gesl (Invitrogen, BoltTM, 4–12%, bis-tris plus). Proteins were detected using Coomassie blue staining for 90 minutes followed by washing with water: methanol: acetic acid = 5:4:1 for 5h.
Anti-IgE binding ELISA assay:
To assess binding activity of anti-IgE samples wells of a 96 well microplate (high binding, Greiner Bio-one) were coated with human IgE (60 μl of 5 μg/ml anti-OVA clone 11B6) overnight at 4 °C. The next the day plate was washed with PBS-Tween (PBS with 0.5ml tween-20/L) and blocked with 1% BSA (100μl) at room temperature for 1h. Serial dilutions of anti-IgE (8), anti-IgE-N3 (10b), CD33L-anti-IgE (10a) (50 μl) were added and incubated at 37 °C for 1.5h. The plate was then washed again with PBS-Tween and secondary antibody (50 μl) anti-IgG2a-HRP (1:1000, v/v, goat anti-mouse IgG2a-HRP, Southern Biotech, Cat. No. 1081–05) was added. After washing the plate, the ELISA was developed using TMB peroxidase substrate (75μl/well, Rockland) for 4 min and quenched with 2M H2SO4 (75μl/well) and absorbance was measured at 450nm using plate reader (Synergy H1, BioTek). EC50 curve was generated using GraphPad Prism (8.4.3).
BMMC production of IL-6:
BMMCs from hCD33+/+ Mcpt5-Cre+ hFcεRIα+ mFcεRIα−/− mice were sensitized with human IgE (anti-OVA-hIgE clone 11B6, 1μg/106 cells/ml) in 50% IL-3 conditioned media at 37 °C /overnight. The next day, the cells were washed and suspended in RPMI without IL-3 (106 cells/ml) and aliquoted (100 μl) to wells of a 96 well plate. Cells were then mixed with 1 μl of PBS as a control or 1μl anti-IgE (8), anti-IgE-N3 (10b), CD33L-anti-IgE (10a) in PBS to make final conc. 10μg/ml at 37° C. After centrifugation (300g, 5 min), the supernatant was collected, and IL-6 cytokine production was measured by sandwich ELISA. For ELISA, 96 well microplate (high binding, Greiner Bio-one) was incubated with 1μg/ml(50μl/well) of anti-IL-6 capturing antibody in PBS (biolegend, clone MP5–32C11) for overnight at 4 °C. The next the day plate was washed with PBS-Tween (PBS with 0.5ml tween-20/L) and blocked with 1% BSA (100μl) at room temperature for 1h. Supernatant(50μl) from BMMCs treated with PBS, anti-IgE (8), anti-IgE-N3 (10b) and CD33L-anti-IgE (10a) were incubated at 4°C for overnight. Next, plate was washed again with PBS-Tween (PBS with 0.5ml tween-20/L) and anti-IL-6-biotin detection antibody in 1% BSA (biolegend, clone MP5–32C11) was incubated at room temperature for 1h. Next, plate was washed with PBS-Tween (PBS with 0.5ml tween-20/L) followed by PBS and streptavidin-HRP in 1% BSA (biolegend, Cat. No. 405210) was incubated at room temperature for 30 minutes. ELISA was developed using TMB peroxidase substrate (75μl/well, Rockland) for 4 min, quenched with 2M H2SO4 (75μl/well) and absorbance was measured at 450nm using plate reader (Synergy H1, BioTek).
Passive cutaneous anaphylaxis (PC) assay:
One ear of each mouse (male) was sensitized intradermally with human IgE (1μg/50 μl). The next day mice were given intravenous injection (i.v.) of 0.2 ml PBS containing 10 μg of either anti-IgE (8) or anti-IgE-CD33L (10a) and 1% Evan blue dye to detect vascular leakage. After 1h mice were euthanized, ears were excised, cut into small pieces, and shaken (500rpm) at 37 °C for overnight in dimethyl formamide (500μl). Aliquots (100 μl) were added to wells of 96 well plates and absorbance of the Evans blue dye was measured at 650nm using Synergy H1 plate reader (BioTeK).
Passive systemic anaphylaxis (PS) assay:
Humanized mice (male, NSG-SGM3-hCD34+) were sensitized intravenously (i.v.) with human IgE (1μg in 200 μl PBS). The next day, after measuring baseline rectal temperature, mice were given intravenous injections (i.v.) of PBS (200 μl) containing 1μg anti-IgE (8), anti-IgE-N3 (10b), anti-IgE-CD33L (10a) or no antibody (PBS) and rectal temperature was measured at 10 minutes interval for 60 minutes using a PhysiTemp Instrument.
Supplementary Material
ACKNOWLEDGMENT
We thank Jonathan Cartmell (Glyconet, Univ. of Alberta) for providing CD33L, Jason Cyster (UCSF) for Hy10 mice, Lars Nitschke (University of Erlangen-Nuernberg) for CD22 knockout mice, Bruce Bochner (Northwestern University) for Mcpt5-Cre mice, and Robert Anthony (Harvard Medical School) for hFcεRIα transgenic and mFcεRIα knockout mice.
Funding Sources
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID), NIH grants U19AI136443, R01AI132790 and R21AI123307) and Department of Defense (DOD) grants W81XWH-16-1-0303 and W81XWH-21-1-0315.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https:
Information on the construction and characterization of anti-IgD constructs, a listing of antibodies used in experiments and their sources, experimental methods for synthesis of Siglec ligands with propargyl functionalized linkers, NMR and mass spectrometry spectra (PDF).
While the authors declare no financial interest, a patent has been filed on this subject with several authors listed as inventors (M.I., F.P., B.A., S.D. and J.C.P.), and there is potential for future financial benefits to the inventors.
REFERENCES
- (1).Biram A; Davidzohn N; Shulman Z, T cell interactions with B cells during germinal center formation, a three-step model. Immunol Rev 2019, 288 (1), 37–48. [DOI] [PubMed] [Google Scholar]
- (2).Bournazos S; Gupta A; Ravetch JV, The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol 2020, 20 (10), 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chauhan P; Nair A; Patidar A; Dandapat J; Sarkar A; Saha B, A primer on cytokines. Cytokine 2021, 145, 155458. [DOI] [PubMed] [Google Scholar]
- (4).Djaoud Z; Parham P, HLAs, TCRs, and KIRs, a Triumvirate of Human Cell-Mediated Immunity. Annu Rev Biochem 2020, 89, 717–739. [DOI] [PubMed] [Google Scholar]
- (5).Lind NA; Rael VE; Pestal K; Liu B; Barton GM, Regulation of the nucleic acid-sensing Toll-like receptors. Nat Rev Immunol 2021, 22, 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bochner BS; Zimmermann N, Role of siglecs and related glycan-binding proteins in immune responses and immunoregulation. J Allergy Clin Immunol 2015, 135 (3), 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen Z; Wang JH, How the Signaling Crosstalk of B Cell Receptor (BCR) and Co-Receptors Regulates Antibody Class Switch Recombination: A New Perspective of Checkpoints of BCR Signaling. Front Immunol 2021, 12, 663443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Duan S; Paulson JC, Siglecs as Immune Cell Checkpoints in Disease. Annu Rev Immunol 2020, 38, 365–395. [DOI] [PubMed] [Google Scholar]
- (9).Khan M; Arooj S; Wang H, NK Cell-Based Immune Checkpoint Inhibition. Front Immunol 2020, 11, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Macauley MS; Crocker PR; Paulson JC, Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol 2014, 14 (10), 653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Müller J; Nitschke L, The role of CD22 and Siglec-G in B-cell tolerance and autoimmune disease. Nat Rev Rheumatol 2014, 10 (7), 422–8. [DOI] [PubMed] [Google Scholar]
- (12).Pan C; Liu H; Robins E; Song W; Liu D; Li Z; Zheng L, Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol 2020, 13 (1), 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tsubata T, Ligand Recognition Determines the Role of Inhibitory B Cell Co-receptors in the Regulation of B Cell Homeostasis and Autoimmunity. Front Immunol 2018, 9, 2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Crocker PR; Paulson JC; Varki A, Siglecs and their roles in the immune system. Nat Rev Immunol 2007, 7 (4), 255–66. [DOI] [PubMed] [Google Scholar]
- (15).Adams OJ; Stanczak MA; von Gunten S; Läubli H, Targeting sialic acid-Siglec interactions to reverse immune suppression in cancer. Glycobiology 2018, 28 (9), 640–647. [DOI] [PubMed] [Google Scholar]
- (16).Bärenwaldt A; Läubli H, The sialoglycan-Siglec glyco-immune checkpoint - a target for improving innate and adaptive anticancer immunity. Expert Opin Ther Targets 2019, 23 (10), 839–853. [DOI] [PubMed] [Google Scholar]
- (17).Büll C; Collado-Camps E; Kers-Rebel ED; Heise T; Søndergaard JN; den Brok MH; Schulte BM; Boltje TJ; Adema GJ, Metabolic sialic acid blockade lowers the activation threshold of moDCs for TLR stimulation. Immunol Cell Biol 2017, 95 (4), 408–415. [DOI] [PubMed] [Google Scholar]
- (18).Edgar LJ; Kawasaki N; Nycholat CM; Paulson JC, Targeted Delivery of Antigen to Activated CD169(+) Macrophages Induces Bias for Expansion of CD8(+) T Cells. Cell Chem Biol 2019, 26 (1), 131–136.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Farid S; Mirshafiey A; Razavi A, Siglec-8 and Siglec-F, the new therapeutic targets in asthma. Immunopharmacol Immunotoxicol 2012, 34 (5), 721–6. [DOI] [PubMed] [Google Scholar]
- (20).Fry TJ; Shah NN; Orentas RJ; Stetler-Stevenson M; Yuan CM; Ramakrishna S; Wolters P; Martin S; Delbrook C; Yates B; Shalabi H; Fountaine TJ; Shern JF; Majzner RG; Stroncek DF; Sabatino M; Feng Y; Dimitrov DS; Zhang L; Nguyen S; Qin H; Dropulic B; Lee DW; Mackall CL, CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2018, 24 (1), 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Herrmann M; Krupka C; Deiser K; Brauchle B; Marcinek A; Ogrinc Wagner A; Rataj F; Mocikat R; Metzeler KH; Spiekermann K; Kobold S; Fenn NC; Hopfner KP; Subklewe M, Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018, 132 (23), 2484–2494. [DOI] [PubMed] [Google Scholar]
- (22).Kim MY; Yu KR; Kenderian SS; Ruella M; Chen S; Shin TH; Aljanahi AA; Schreeder D; Klichinsky M; Shestova O; Kozlowski MS; Cummins KD; Shan X; Shestov M; Bagg A; Morrissette JJD; Sekhri P; Lazzarotto CR; Calvo KR; Kuhns DB; Donahue RE; Behbehani GK; Tsai SQ; Dunbar CE; Gill S, Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173 (6), 1439–1453.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Srivastava A; Arlian BM; Pang L; Kishimoto TK; Paulson JC, Tolerogenic Nanoparticles Impacting B and T Lymphocyte Responses Delay Autoimmune Arthritis in K/BxN Mice. ACS Chem Biol 2021, 16 (10), 1985–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stanczak MA; Siddiqui SS; Trefny MP; Thommen DS; Boligan KF; von Gunten S; Tzankov A; Tietze L; Lardinois D; Heinzelmann-Schwarz V; von Bergwelt-Baildon M; Zhang W; Lenz HJ; Han Y; Amos CI; Syedbasha M; Egli A; Stenner F; Speiser DE; Varki A; Zippelius A; Läubli H, Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest 2018, 128 (11), 4912–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tang X; Sui D; Liu M; Zhang H; Liu M; Wang S; Zhao D; Sun W; Liu M; Luo X; Lai X; Liu X; Deng Y; Song Y, Targeted delivery of zoledronic acid through the sialic acid - Siglec axis for killing and reversal of M2 phenotypic tumor-associated macrophages - A promising cancer immunotherapy. Int J Pharm 2020, 590, 119929. [DOI] [PubMed] [Google Scholar]
- (26).Wang J; Sun J; Liu LN; Flies DB; Nie X; Toki M; Zhang J; Song C; Zarr M; Zhou X; Han X; Archer KA; O’Neill T; Herbst RS; Boto AN; Sanmamed MF; Langermann S; Rimm DL; Chen L, Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med 2019, 25 (4), 656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xiao H; Woods EC; Vukojicic P; Bertozzi CR, Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci U S A 2016, 113 (37), 10304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bednar KJ; Nycholat CM; Rao TS; Paulson JC; Fung-Leung WP; Macauley MS, Exploiting CD22 To Selectively Tolerize Autoantibody Producing B-Cells in Rheumatoid Arthritis. ACS Chem Biol 2019, 14 (4), 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bednar KJ; Shanina E; Ballet R; Connors EP; Duan S; Juan J; Arlian BM; Kulis MD; Butcher EC; Fung-Leung WP; Rao TS; Paulson JC; Macauley MS, Human CD22 Inhibits Murine B Cell Receptor Activation in a Human CD22 Transgenic Mouse Model. J Immunol 2017, 199 (9), 3116–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Macauley MS; Paulson JC, Siglecs induce tolerance to cell surface antigens by BIM-dependent deletion of the antigen-reactive B cells. J Immunol 2014, 193 (9), 4312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Macauley MS; Pfrengle F; Rademacher C; Nycholat CM; Gale AJ; von Drygalski A; Paulson JC, Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest 2013, 123 (7), 3074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Orgel KA; Duan S; Wright BL; Maleki SJ; Wolf JC; Vickery BP; Burks AW; Paulson JC; Kulis MD; Macauley MS, Exploiting CD22 on antigen-specific B cells to prevent allergy to the major peanut allergen Ara h 2. J Allergy Clin Immunol 2017, 139 (1), 366–369.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Duan S; Arlian BM; Nycholat CM; Wei Y; Tateno H; Smith SA; Macauley MS; Zhu Z; Bochner BS; Paulson JC, Nanoparticles Displaying Allergen and Siglec-8 Ligands Suppress IgE-FcεRI-Mediated Anaphylaxis and Desensitize Mast Cells to Subsequent Antigen Challenge. J Immunol 2021, 206 (10), 2290–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Duan S; Koziol-White CJ; Jester WF Jr.; Smith SA; Nycholat CM; Macauley MS; Panettieri RA Jr.; Paulson JC, CD33 recruitment inhibits IgE-mediated anaphylaxis and desensitizes mast cells to allergen. J Clin Invest 2019, 129 (3), 1387–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Al-Halifa S; Gauthier L; Arpin D; Bourgault S; Archambault D, Nanoparticle-Based Vaccines Against Respiratory Viruses. Front Immunol 2019, 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li X; Gao J; Yang Y; Fang H; Han Y; Wang X; Ge W, Nanomaterials in the application of tumor vaccines: advantages and disadvantages. Onco Targets Ther 2013, 6, 629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Joubert N; Beck A; Dumontet C; Denevault-Sabourin C, Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals (Basel) 2020, 13 (9), 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nejadmoghaddam MR; Minai-Tehrani A; Ghahremanzadeh R; Mahmoudi M; Dinarvand R; Zarnani AH, Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J Med Biotechnol 2019, 11 (1), 3–23. [PMC free article] [PubMed] [Google Scholar]
- (39).Tsuchikama K; An Z, Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 2018, 9 (1), 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang K; Liu J; Truong T; Zukin E; Chen W; Saxon A, Blocking Allergic Reaction through Targeting Surface-Bound IgE with Low-Affinity Anti-IgE Antibodies. J Immunol 2017, 198 (10), 3823–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang K; Elias M; Zhang H; Liu J; Kepley C; Bai Y; Metcalfe DD; Schiller Z; Wang Y; Saxon A, Inhibition of Allergic Reactivity through Targeting FcεRI-Bound IgE with Humanized Low-Affinity Antibodies. J Immunol 2019, 203 (11), 2777–2790. [DOI] [PubMed] [Google Scholar]
- (42).Li L; Yao Z, Mast cell and immune inhibitory receptors. Cell Mol Immunol 2004, 1 (6), 408–15. [PubMed] [Google Scholar]
- (43).Migalovich-Sheikhet H; Friedman S; Mankuta D; Levi-Schaffer F, Novel identified receptors on mast cells. Front Immunol 2012, 3, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shibuya A; Nakahashi-Oda C; Tahara-Hanaoka S, Inhibitory Immunoreceptors on Mast Cells in Allergy and Inflammation. In Innovative Medicine: Basic Research and Development, Nakao K; Minato N; Uemoto S, Eds. Springer Copyright 2015, The Author(s). Tokyo, 2015; pp 95–107. [PubMed] [Google Scholar]
- (45).Bachelet I; Munitz A; Levi-Schaffer F, Abrogation of allergic reactions by a bispecific antibody fragment linking IgE to CD300a. J Allergy Clin Immunol 2006, 117 (6), 1314–20. [DOI] [PubMed] [Google Scholar]
- (46).Eggel A; Buschor P; Baumann MJ; Amstutz P; Stadler BM; Vogel M, Inhibition of ongoing allergic reactions using a novel anti-IgE DARPin-Fc fusion protein. Allergy 2011, 66 (7), 961–8. [DOI] [PubMed] [Google Scholar]
- (47).Liu Y; Sun Y; Chang LJ; Li N; Li H; Yu Y; Bryce PJ; Grammer LC; Schleimer RP; Zhu D, Blockade of peanut allergy with a novel Ara h 2-Fcγ fusion protein in mice. J Allergy Clin Immunol 2013, 131 (1), 213–21.e1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zellweger F; Gasser P; Brigger D; Buschor P; Vogel M; Eggel A, A novel bispecific DARPin targeting FcγRIIB and FcεRI-bound IgE inhibits allergic responses. Allergy 2017, 72 (8), 1174–1183. [DOI] [PubMed] [Google Scholar]
- (49).Zhu D; Kepley CL; Zhang K; Terada T; Yamada T; Saxon A, A chimeric human-cat fusion protein blocks cat-induced allergy. Nat Med 2005, 11 (4), 446–9. [DOI] [PubMed] [Google Scholar]
- (50).Krauth MT; Böhm A; Agis H; Sonneck K; Samorapoompichit P; Florian S; Sotlar K; Valent P, Effects of the CD33-targeted drug gemtuzumab ozogamicin (Mylotarg) on growth and mediator secretion in human mast cells and blood basophils. Exp Hematol 2007, 35 (1), 108–16. [DOI] [PubMed] [Google Scholar]
- (51).Yokoi H; Myers A; Matsumoto K; Crocker PR; Saito H; Bochner BS, Alteration and acquisition of Siglecs during in vitro maturation of CD34+ progenitors into human mast cells. Allergy 2006, 61 (6), 769–76. [DOI] [PubMed] [Google Scholar]
- (52).Dispenza MC; Krier-Burris RA; Chhiba KD; Undem BJ; Robida PA; Bochner BS, Bruton’s tyrosine kinase inhibition effectively protects against human IgE-mediated anaphylaxis. J Clin Invest 2020, 130 (9), 4759–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Alberola-Ila J; Takaki S; Kerner JD; Perlmutter RM, Differential signaling by lymphocyte antigen receptors. Annu Rev Immunol 1997, 15, 125–54. [DOI] [PubMed] [Google Scholar]
- (54).Saunders KO, Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front Immunol 2019, 10, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Collins BE; Smith BA; Bengtson P; Paulson JC, Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat Immunol 2006, 7 (2), 199–206. [DOI] [PubMed] [Google Scholar]
- (56).Kawasaki N; Rademacher C; Paulson JC, CD22 regulates adaptive and innate immune responses of B cells. J Innate Immun 2011, 3 (4), 411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Jellusova J; Nitschke L, Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front Immunol 2011, 2, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).D’Amato G; Piccolo A; Salzillo A; Noschese P; D’Amato M; Liccardi G, A recombinant humanized anti-IgE monoclonal antibody (omalizumab) in the therapy of moderate-to-severe allergic asthma. Recent Pat Inflamm Allergy Drug Discov 2007, 1 (3), 225–31. [DOI] [PubMed] [Google Scholar]
- (59).Frix AN; Schleich F; Paulus V; Guissard F; Henket M; Louis R, Effectiveness of omalizumab on patient reported outcomes, lung function, and inflammatory markers in severe allergic asthma. Biochem Pharmacol 2020, 179, 113944. [DOI] [PubMed] [Google Scholar]
- (60).Guntern P; Eggel A, Past, present, and future of anti-IgE biologics. Allergy 2020, 75 (10), 2491–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Gray MA; Stanczak MA; Mantuano NR; Xiao H; Pijnenborg JFA; Malaker SA; Miller CL; Weidenbacher PA; Tanzo JT; Ahn G; Woods EC; Laubli H; Bertozzi CR, Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat Chem Biol 2020, 16 (12), 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Laubli H; Kawanishi K; George Vazhappilly C; Matar R; Merheb M; Sarwar Siddiqui S, Tools to study and target the Siglec-sialic acid axis in cancer. FEBS J 2021, 288 (21), 6206–6225. [DOI] [PubMed] [Google Scholar]
- (63).van de Wall S; Santegoets KCM; van Houtum EJH; Bull C; Adema GJ, Sialoglycans and Siglecs Can Shape the Tumor Immune Microenvironment. Trends Immunol 2020, 41 (4), 274–285. [DOI] [PubMed] [Google Scholar]
- (64).Kiwamoto T; Kawasaki N; Paulson JC; Bochner BS, Siglec-8 as a drugable target to treat eosinophil and mast cell-associated conditions. Pharmacol Ther 2012, 135 (3), 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Rillahan CD; Macauley MS; Schwartz E; He Y; McBride R; Arlian BM; Rangarajan J; Fokin VV; Paulson JC, Disubstituted Sialic Acid Ligands Targeting Siglecs CD33 and CD22 Associated with Myeloid Leukaemias and B Cell Lymphomas. Chem Sci 2014, 5 (6), 2398–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Peng W; Paulson JC, CD22 Ligands on a Natural N-Glycan Scaffold Efficiently Deliver Toxins to B-Lymphoma Cells. J Am Chem Soc 2017, 139 (36), 12450–12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Allen CD; Okada T; Tang HL; Cyster JG, Imaging of germinal center selection events during affinity maturation. Science 2007, 315 (5811), 528–31. [DOI] [PubMed] [Google Scholar]
- (68).Samardzic T; Marinkovic D; Danzer CP; Gerlach J; Nitschke L; Wirth T, Reduction of marginal zone B cells in CD22-deficient mice. Eur J Immunol 2002, 32 (2), 561–7. [DOI] [PubMed] [Google Scholar]
- (69).Dombrowicz D; Brini AT; Flamand V; Hicks E; Snouwaert JN; Kinet JP; Koller BH, Anaphylaxis mediated through a humanized high affinity IgE receptor. J Immunol 1996, 157 (4), 1645–51. [PubMed] [Google Scholar]
- (70).Angata K; Nakayama J; Fredette B; Chong K; Ranscht B; Fukuda M, Human STX polysialyltransferase forms the embryonic form of the neural cell adhesion molecule. Tissue-specific expression, neurite outgrowth, and chromosomal localization in comparison with another polysialyltransferase, PST. J Biol Chem 1997, 272 (11), 7182–90. [DOI] [PubMed] [Google Scholar]
- (71).Scholten J; Hartmann K; Gerbaulet A; Krieg T; Müller W; Testa G; Roers A, Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res 2008, 17 (2), 307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.