
Fig. 4 |. Overview of molecular pathways driving electropathology and AF.
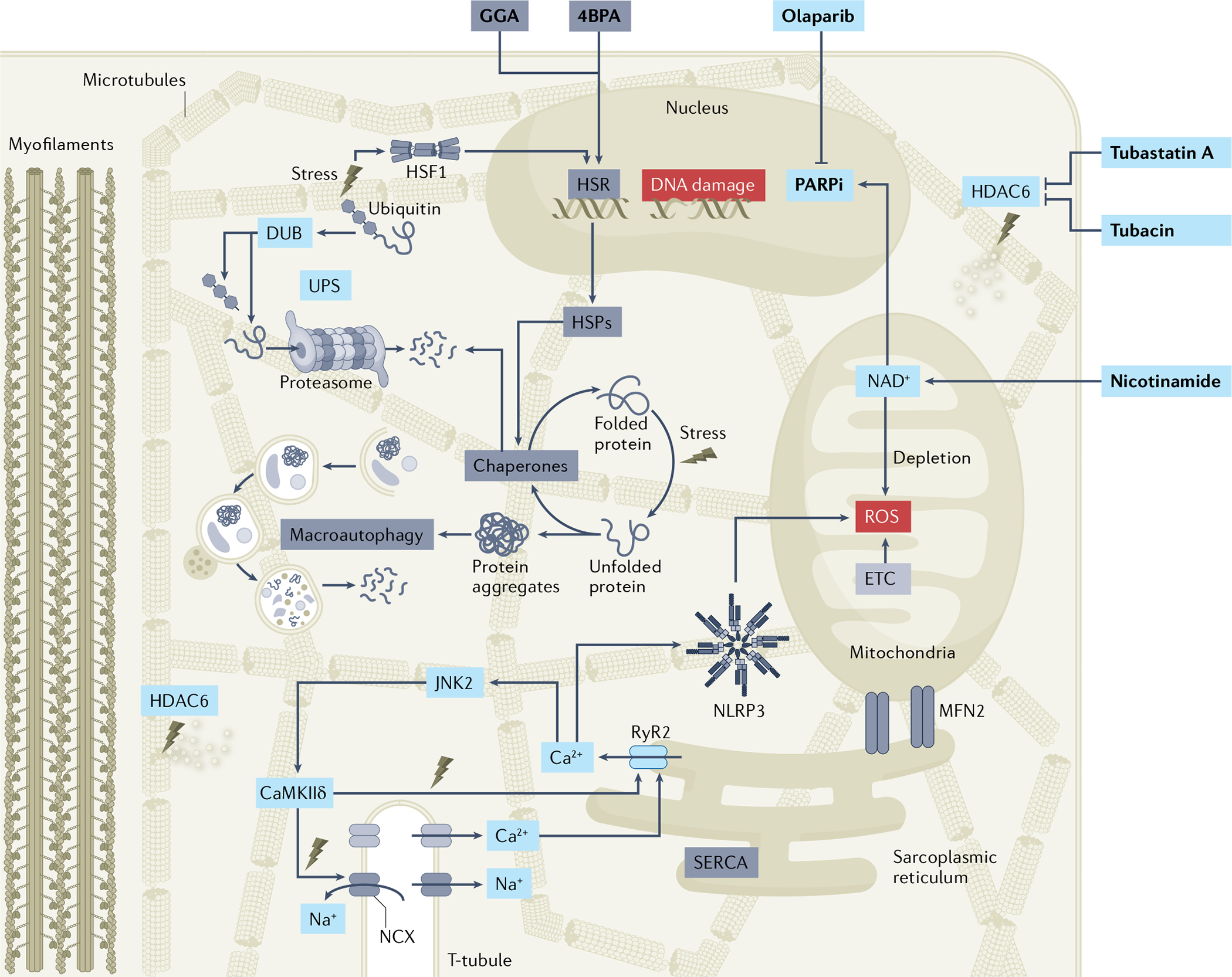
Atrial fibrillation (AF) causes loss of protein quality control via downregulation of the heat shock protein (HSP)-mediated heat shock response (HSR) and subsequent reduction in HSP expression levels and loss of chaperone activity. As HSPs represent the cell’s first line of defence against stress, the loss of stress response induces further endoplasmic reticulum stress and downstream excessive activation of the macroautophagy protein degradation pathway. AF also increases the activity of the histone deacetylase HDAC6, resulting in deacetylation of microtubules and destabilization of the microtubule network, Ca2+ handling alterations, and loss of cardiomyocyte contractile function (BOX 1). AF triggers DNA damage, poly(ADP-ribose) polymerase 1 (PARP1) activation and depletion of mitochondrial NAD+ levels, thereby causing electrophysiological and contractile impairment. Pharmacological treatments (in bold) that boost the HSR (for example, geranylgeranylacetone (GGA)), prevent endoplasmic reticulum stress (for example, 4-phenylbutyrate (4PBA)), inhibit HDAC6 (for example, tubastatin A and tubacin) or PARP1 (for example, olaparib) activity, or supplement NAD+ (for example, nicotinamide) protect against proteostasis dysfunction and AF progression in experimental model systems. As several of the proteoceutical compounds are marketed and off-patent, drug repurposing approaches might be within reach. The increased Ca2+ release from the sarcoplasmic reticulum (sarcoplasmic reticulum Ca2+ leak) in cardiomyocytes might directly activate the NLRP3 inflammasome by facilitating interactions between inflammasome components or might have indirect effects by promoting mitochondrial reactive oxygen species (ROS) production. In addition, increased levels of ROS can activate JUN N-terminal kinase 2 (JNK2) and Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ), resulting in excessive abnormal Ca2+ signalling. DUB, deubiquitylase; ETC, electron transport chain; MFN2, mitofusin 2; PARPi, PARP inhibitor; RyR2, ryanodine receptor 2; UPS, ubiquitin–proteasome system.