

Abstract
Dyskeratosis congenita (DC) is a rare genetic disorder characterized by deficiencies in telomere maintenance leading to very short telomeres and the premature onset of certain age-related diseases, including pulmonary fibrosis (PF). PF is thought to derive from epithelial failure, particularly that of type II alveolar epithelial (AT2) cells, which are highly dependent on Wnt signaling during development and adult regeneration. We use human induced pluripotent stem cell-derived AT2 (iAT2) cells to model how short telomeres affect AT2 cells. Cultured DC mutant iAT2 cells accumulate shortened, uncapped telomeres and manifest defects in the growth of alveolospheres, hallmarks of senescence, and apparent defects in Wnt signaling. The GSK3 inhibitor, CHIR99021, which mimics the output of canonical Wnt signaling, enhances telomerase activity and rescues the defects. These findings support further investigation of Wnt agonists as potential therapies for DC-related pathologies.
Research organism: Human
Introduction
Dyskeratosis congenita (DC) is a rare genetic disorder characterized by bone marrow failure, skin abnormalities, elevated risk of certain cancers, and liver and pulmonary fibrosis (PF). These pathologies are caused by abnormally shortened and uncapped telomeres arising from deficiencies in telomere maintenance, typically due to defects in the action of telomerase. Significant progress has been made in treating the bone marrow failure of DC patients, particularly via transplantation, but PF remains a major life-limiting pathology (Agarwal, 2018; Dietz et al., 2011).
PF is a subtype of interstitial pneumonia that is chronic and progressive, replacing the normal lace-like alveolar architecture with patchy, hyperproliferative fibrous tissue (Lederer and Martinez, 2018). Current therapies are only modestly effective and do not reverse the underlying fibrosis, and lung transplantation is not always an option (King et al., 2014; Lederer and Martinez, 2018; Richeldi et al., 2014; Valapour et al., 2020). While much of the early work in PF pathogenesis focused on unraveling the contributions of fibroblasts, genetic studies of families with a predisposition to PF argue that defects in alveolar epithelial cells and telomeres are key drivers of disease (Alder et al., 2015; Armanios et al., 2007; Bullard et al., 2005; Cogan et al., 2015; Haschek and Witschi, 1979; Kropski et al., 2015; Maitra et al., 2010; Thomas et al., 2002; Wang et al., 2009). Recent work in mice shows that dysfunctional type II alveolar epithelial (AT2) cells, the putative stem cells of alveoli (Barkauskas et al., 2013), can lead to a progressive chronic fibrotic response similar to that seen in patients (Nureki et al., 2018). Furthermore, many of the same genes which when mutated cause DC have also been linked to familial PF (Alder et al., 2015; Armanios et al., 2007; Cogan et al., 2015; Kropski et al., 2017). In sporadic PF, both age and short telomeres are risk factors, and these risks are linked because age is associated with telomere shortening in the lung, particularly in AT2 cells (Alder et al., 2008; Everaerts et al., 2018). Consistent with a role for telomere dysfunction in driving PF, AT2 cells in sporadic PF express hallmarks of senescence and have shorter telomeres in fibrotic regions than those in non-fibrotic regions (Disayabutr et al., 2016; Kropski et al., 2015; Snetselaar et al., 2017). Additionally, two human Mendelian randomization studies argue that short telomeres are a cause of PF (Duckworth et al., 2020; Telomeres Mendelian Randomization Collaboration et al., 2017). Murine studies also argue that telomere dysfunction and senescence in AT2 cells can drive PF (Naikawadi et al., 2016; Povedano et al., 2015; Yao et al., 2019). Although causality is thus evident, exactly how AT2 cell telomere dysfunction leads to PF is poorly understood.
Previous work in our lab using mouse and human induced pluripotent stem (iPS) cell-derived organoid models of DC intestinal defects uncovered a positive feedback loop by which telomere capping and canonical Wnt signaling support one another under normal conditions to maintain the intestinal stem cell niche (Woo et al., 2016; Yang et al., 2017). In the setting of telomere dysfunction, this virtuous cycle becomes vicious: the resulting suppression of Wnt signaling interferes with stem cell function directly, and it also amplifies telomere dysfunction by diminishing Wnt-dependent expression of telomere maintenance factors, including the catalytic subunit of telomerase, TERT, and several of the telomere-protective shelterins. These studies demonstrated that Wnt pathway agonists (including GSK3 inhibitors, which mimic the inhibition of GSK3 by Wnts) can rescue these defects, raising the possibility that Wnt agonism could be of therapeutic benefit in DC. Given how telomere dysfunction in AT2 cells appears to drive PF, we wondered if Wnt agonism might be of benefit in PF. Wnt signaling is important for lung epithelial cell development (Frank et al., 2016; Goss et al., 2009; Li et al., 2002; Li et al., 2005; Maretto et al., 2003; Okubo and Hogan, 2004; Ostrin et al., 2018; Shu et al., 2005) and regeneration of the adult lung in response to injury (Nabhan et al., 2018; Zacharias et al., 2018). On the one hand, there is evidence that Wnts may drive PF; for example, they have been found to be upregulated in patients with PF (Chilosi et al., 2003; Königshoff et al., 2008; Königshoff et al., 2009). On the other hand, when the canonical Wnt transcriptional effector β-catenin is deleted in AT2 cells, mice are sensitized to bleomcyin induced PF (Tanjore et al., 2013). Wnt signaling is complex and context dependent (Wiese et al., 2018), and the exact spatial, temporal, and cell-type specificity of Wnt signaling in PF remains an area of intense investigation. It is difficult to extrapolate from observational pathologic studies of fully developed PF to the potential functional impact of Wnts at earlier stages of the disease, and the large number of interacting cell types in PF lungs also makes it challenging to identify primary drivers in such studies. Furthermore, differences between mouse and human telomere biology together with the generally lower susceptibility of mice to PF make mouse modeling difficult. We therefore generated AT2 cell organoids by directed differentiation of human iPSCs (iPSC-derived AT2 [iAT2] cells) to explore how telomere dysfunction might impact their function (Jacob et al., 2017; Jacob et al., 2019).
By comparing iAT2s that are isogenic except for an introduced mutation in the gene most often mutated in DC, X-linked DKC1, we show that mutant iAT2 cells become senescent in concert with telomere shortening and uncapping. iAT2 cells with short, uncapped telomeres exhibit gene expression changes consistent with decreased Wnt signaling, and treatment with GSK3 inhibitors, such as CHIR99021, rescues their growth and telomere dysfunction. These findings raise the possibility that Wnt agonists may be of benefit in rescuing the stem cell and telomere defects of AT2 cells associated with PF in DC patients.
Results
Engineering a DKC1 mutation into iPS cells
To model the AT2 cells from DC patients, we engineered a well-characterized, causal DC mutation in DKC1 (DKC1 A386T) (Agarwal et al., 2010; Batista et al., 2011; Woo et al., 2016) into the BU3 NKX2.1::GFP SFTPC::TdTomato (NGST) human iPS cell line (Jacob et al., 2017). We established an isogenic pair of cell lines: an introduced mutant line and a corresponding unedited wild-type (WT) line (Figure 1—figure supplement 1A-C). Both iPS lines maintained markers of pluripotency and normal karyotypes after the introduction of the DKC1 A386T mutation (Figure 1—figure supplement 1D).
Previous work (Agarwal et al., 2010; Batista et al., 2011; Woo et al., 2016) established that iPS cells with the DKC1 A386T mutation exhibit decreased telomerase activity resulting in telomere shortening with passage. We confirmed that telomerase activity was reduced, and telomeres shortened with successive passages, in the BU3 NGST DKC1 A386T iPS cell line when compared to its WT control (Figure 1—figure supplement 2A-C).
iAT2 cells with short telomeres fail to form alveolospheres and grow in size
We next differentiated these paired iPS cell lines into iAT2 cells using the protocol developed by Jacob et al. (see Figure 1A for differentiation strategy, Figure 1—figure supplement 3A, B for representative sorting strategies). Using iPSCs 35 passages after the introduction of the mutation yielded iAT2s that initially grew in a similar fashion to WT, but which over time developed a growth defect characterized by lower alveolosphere formation efficiency as well as smaller alveolospheres. The phenotype became apparent by 50 days of culture (D50) and was dramatic by D70 (Figure 1A–C). In contrast, using iPSCs only five passages after the introduction of the mutation yielded iAT2s with less dramatic defects in alveolosphere growth and size at D70 (Figure 1—figure supplement 4A-C). These data indicate that the defects observed were due to progressive telomere shortening after introducing the DKC1 mutation, and not the immediate effects of telomerase deficiency (or other potential deficiencies) caused by the DKC1 A386T mutation per se.
Figure 1. Dyskeratosis congenita induced pluripotent stem cell (iPSC)-derived type II alveolar epithelial cells (iAT2s) fail to form alveolospheres with successive passaging.
(A) Differentiation protocol used to probe the effects of the DKC1 A386T mutation on iAT2 cells. D.E., definitive endoderm specification; Ant., anteriorization. (B) Representative images of differentiating wild type and DKC1 A386T mutant bearing iAT2 alveolospheres (scale bars, 100 µm as indicated for iPS and NKX2.1+ progenitors; 1 mm for all alveolosphere images). (C) Quantifications of alveolosphere area and formation efficiency on D70 (n = 4; ** p<0.01, **** p<0.0001, and Student’s t-test). (D) Quantification of the percentage SFTPC+ cells and the number of SFTPC+ cells produced with passage of the iAT2 cells shows DKC1 A386T iAT2 alveolospheres accumulate fewer SFTPC+ cells (n = 4; ** p<0.01, and Student’s t-test). (E) RNA-seq of sorted SFTPC+ iAT2 cells at different passages shows AT2 cell genes are not grossly affected by the DKC1 A386T mutation (n = 4, W = wild type, M = mutant).

Figure 1—figure supplement 1. Introduction of the DKC1 A386T mutation into the BU3 NKX2.1::GFP SFTPC::TdTomato (NGST) induced pluripotent stem (iPS) cell line.

Figure 1—figure supplement 2. DKC1 A386T induced pluripotent stem (iPS) cells show decreased telomerase activity and telomeres shorten with passage.

Figure 1—figure supplement 3. Representative sorting strategies for NKX2.1+ progenitors and SFTPC+ induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cells.

Figure 1—figure supplement 4. Differentiation of induced pluripotent stem cell (iPSC)-derived type II alveolar epithelial cell (iAT2) alveolospheres from early passage iPS cells yields no growth defect.

Surfactant protein C (SFTPC) is a highly specific marker of AT2 cells (Kalina et al., 1992), and the yield of SFTPC::TdTomato+ (SFTPC+) cells was reduced significantly at D70 in DKC1 mutant cultures, while the percentage of SFTPC+ cells generated at each time point was not different, suggesting that there is a defect in AT2 cell proliferation or survival (Figure 1D). Sorted SFTPC+ cells from iAT2 cell alveolospheres maintained expression of multiple AT2 specific genes suggesting that the introduced mutation did not affect lineage specification (Figure 1E). Thus, DC iPS cells can generate iAT2 cell alveolospheres; however, these alveolospheres lose the capacity to self-renew with successive passaging.
DC iAT2 cells derived from iPS cells 35 passages after introduction of the mutation develop hallmarks of senescence at late passage (D70)
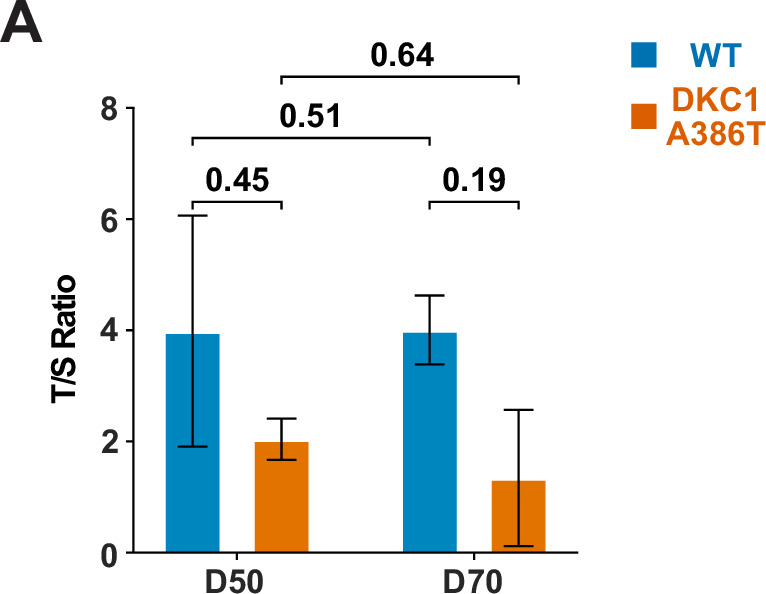
To better understand the AT2 cell defects, we further compared the WT and mutant iAT2 alveolospheres at different passages. RNA-seq-based gene expression analyses over successive passages of sorted SFTPC+ cells from iAT2 alveolospheres showed decreases in proliferation markers (MKI67 and MCM2) as well as an increase in expression of the cell cycle inhibitor CDKN1A (p21), and these changes were most pronounced in mutant alveolospheres (Figure 2A). Immunofluorescence microscopy based analyses of D70 mutant iAT2 alveolospheres showed an increase in DNA damage marked by 53BP1 foci (Figure 2B), and an increased fraction of cells expressing p21 protein (Figure 2C), but no increase in apoptosis (Figure 2—figure supplement 1A-C). Measuring telomere length using qPCR, DC, and WT iAT2 cells showed no significant change in average telomere length with passage, although average telomere lengths in mutants trended shorter than in WT (Figure 2—figure supplement 2A). However, measuring telomere lengths using TeSLA (Lai et al., 2017), which is more sensitive for the detection of short telomeres than most other techniques, revealed that DC iAT2 cells had a preponderance of short telomeres at D70 (Figure 2E–F). Consistent with this, DC iAT2 alveolospheres showed an increased number of telomere dysfunction induced foci (TIFs), a hallmark of uncapped telomeres (i.e., telomeres that signal DNA damage responses and cell cycle checkpoint arrest Takai et al., 2003; Figure 2D). These findings indicate that the short and uncapped telomeres that accumulate with passage of DC iAT2 cells lead them to senesce.
Figure 2. Dyskeratosis congenita induced pluripotent stem cell-derived type II alveolar epithelial cells (iAT2s) 35 passages after introduction of the mutation at D70 show hallmarks of senescence.
(A) Gene expression profiling of iAT2 cells at D28 and D50 shows no difference between wild type and DKC1 A386T in expression of markers of proliferation and a cell cycle inhibitor, while at D70 cells, there is a significant decrease in MCM2 and MKI67 as well as a significant increase in CDKN1A (p21) (n = 4, **** p<0.0001, DEseq2 pairwise contrast statistics). (B) At D70, DKC1 A386T mutant iAT2 cells have a higher fraction of cells with 53BP1 foci (n = 4, * p<0.05, and Student’s t-test; scale bars, 10 µm; insets highlight cells with 53BP1 foci as noted by the white arrowheads). (C) At D70, DKC1 A386T mutant iAT2s have a higher fraction of cells positive for p21 (n = 4, * p<0.05, and student’s t-test; scale bars, 10 µm). (D) At D70, DKC1 A386T mutant iAT2s have a higher fraction of cells with telomere dysfunction induced foci (TIFs) (n = 4, * p<0.05, and Student’s t-test; scale bars, 10 µm; insets highlight cells with TIFs, each one noted by a white arrowhead). (E) Representative TeSLA of DKC1 A386T iAT2 alveolospheres shows telomeres shorten with passage. (F) Quantification of DKC1 A386T iAT2 cell telomere lengths shows a preponderance of short telomeres appears as the iAT2 cells approach D70, red colored data points highlight telomeres under the 1.6 kb threshold (n = 2, ‘shortest 20%’ reports the 20th percentile of telomere length, in Kb).

Figure 2—figure supplement 1. Dyskeratosis congenita induced pluripotent stem cell-derived type II alveolar epithelial cells (iAT2s) 35 passages after introduction of the mutation at D70 do not show an increase in apoptosis.

Figure 2—figure supplement 2. Telomere qPCR reveals a trend toward shorter average telomere lengths in dyskeratosis congenita (DC) induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cells.
RNA-seq reveals pathways differentially expressed in DC iAT2 cells, including those related to Wnt signaling
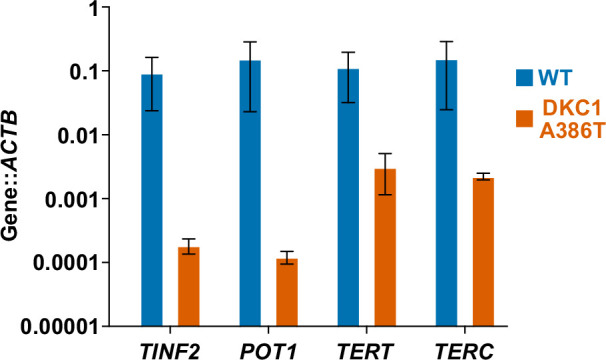
To further understand changes in the DC iAT2 cells, we more broadly evaluated RNA-seq-based gene expression changes of sorted SFTPC+ iAT2 cells. (Figure 3A). We found very few significantly differentially expressed genes when comparing WT and mutant cells at D28 and D50, but a large number of differentially expressed genes at D70 (Figure 3B), arguing that the gene expression changes seen at D70 are likely driven by uncapped telomeres. Gene set enrichment analyses (GSEA) revealed an upregulation of the DNA damage response, the unfolded protein response (UPR), mitochondrial related functions (oxidative phosphorylation, the respiratory electron chain), a downregulation of hypoxia related signaling, and hedgehog signaling along with other changes (see Supplementary file 1 for a full list). Ingenuity pathway analysis (IPA) revealed similar changes as well as defects in multiple pathways controlled by inflammatory cytokines like IL1β, IL6, IL17, and others (see Supplementary file 2 for the full lists). We found a marked upregulation in DC iAT2 cells of many pathways associated with PF (see Supplementary file 3 for curated list of PF related pathways, see Supplementary files 1 and 2 for the unedited analyses). These included the UPR (Lawson et al., 2008; Mulugeta et al., 2005), thyroid hormone metabolism (Yu et al., 2018), p53 signaling (Shetty et al., 2017), mitochondrial dysfunction and mitophagy (Chung et al., 2019; Yu et al., 2018), and caveolin function (Wang et al., 2006). This analysis also showed an upregulation of non-canonical Wnt signaling (Figure 3C), which correlated with a significant upregulation in WNT5A and WNT11, known non-canonical Wnt ligands (Figure 3D). Furthermore, almost every FZD gene, encoding co-receptors for canonical Wnt signaling, was downregulated in DC iAT2 cells (Figure 3E). Also, GSEA found genes with TCF7 targets in their promoters are downregulated in DC iAT2 cells at D70 (Figure 3F). IPA of master regulators at D70 revealed a decrease in genes controlled by lithium chloride (which can potentiate Wnt signaling by inhibiting GSK3) and TCF7 along with an upregulation of genes usually stimulated by Wnt pathway inhibitors (Figure 3G). GSEA also revealed a significant downregulation of targets of miR34a, a miRNA that we previously demonstrated negatively regulates many components of the Wnt pathway in response to uncapped telomeres (Figure 3H; Yang et al., 2017). The genes encoding four of the six shelterins, proteins that bind and help maintain normal telomere function, are direct targets of the canonical Wnt transcriptional effector β-catenin, and two of these, TINF2 and POT1, were downregulated in D70 DC iAT2 cells, which may contribute to telomere uncapping beyond simple telomere shortening (Figure 3I; Yang et al., 2017). These genes were confirmed to be downregulated by ddPCR, and furthermore, DC iAT2 cells expressed lower levels of both TERT and TERC (Figure 3—figure supplement 1). These data indicate that Wnt signaling in AT2 cells is greatly affected by shortened telomeres and that non-canonical Wnt signaling may be elevated while canonical (β-catenin-dependent) signaling may be diminished.
Figure 3. RNA-seq of passaged induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cells reveals a time dependent change in Wnt signaling.
(A) A schematic to show how cells were prepared for RNA-seq. (B) Volcano plots at D28, D50, and D70 show how the number of differentially expressed genes increases at D70. (C) Gene set enrichment analyses (GSEA) at D70 comparing DKC1 A386T iAT2 cells reveal an upregulation of non-canonical Wnt signaling and the planar cell polarity (PCP) pathway. (D) RNA-seq analysis shows upregulation of WNT5A and WNT11, non-canonical WNTs associated with pulmonary fibrosis (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, DEseq2 pairwise contrast statistics). (E) RNA-seq analysis shows broad downregulation of many FZD receptors (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, DEseq2 pairwise contrast statistics). (F) GSEA at D70 comparing DKC1 A386T iAT2 cells reveals a downregulation of genes with TCF7 bindings sites in their promoters. (G) Ingenuity pathway analysis reveals master regulators at D70 including downregulation of regulation associated with lithium chloride and TCF7, and upregulation of regulation associated with multiple Wnt inhibitors. (H) GSEA at D70 comparing DKC1 A386T iAT2 cells reveals a downregulation of genes with miR34A binding sites. TINF2 and POT1 are downregulated in DKC1 A386T iAT2 cells at D70 (* p<0.05, ** p<0.01).

Figure 3—figure supplement 1. ddPCR demonstrates key shelterin and telomerase related genes are downregulated in dyskeratosis congenita (DC) induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cells.
GSK3 inhibitors rescue the growth of DC iAT2 alveolospheres
Given our previous work showing that GSK3 inhibition reverses telomere dysfunction and associated defects in intestinal models of DC (Woo et al., 2016; Yang et al., 2017), we attempted to rescue the DC iAT2 alveolosphere formation defect by treatment with CHIR99021, a well-characterized GSK3 inhibitor, which thus stabilizes β-catenin and upregulates canonical Wnt target genes (Figure 4A). CHIR99021 partially rescued the alveolosphere growth defect of iAT2 cells in a dose dependent fashion (Figure 4B–C). Furthermore, if alveolospheres were cultured continuously prior to D70 with CHIR99021, this prevented the growth defect from emerging (Figure 4—figure supplement 1A-B). We also tested another GSK3 inhibitor, CHIR98014, which similarly rescued growth of the mutant iAT2 cells (Figure 4—figure supplement 2A-B). These findings suggest that GSK3 inhibition rescued the growth of DC iAT2 alveolospheres.
Figure 4. CHIR99021 rescues growth and telomere defects in dyskeratosis congentia (DC) induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cell alveolospheres.
(A) Differentiation protocol used to test how CHIR99021 affects growth of DC iAT2s. (B) Representative images of differentiating wild type (WT) and DKC1 A386T mutant bearing cells with increasing amounts of CHIR99021 (scale bars, 1 mm for all alveolosphere images). (C) Quantifications of alveolosphere formation efficiency after treatment with differing concentrations of CHIR99021 (n = 4, * p<0.05, **** p<0.0001, Student’s t-test). (D) When D70 alveolospheres are cultured with 3 µM CHIR99021, DKC1 A386T mutant iAT2 cells have a lower fraction of cells with 53 BP1 foci. Note, data for no CHIR99021 bars are from Figure 2 (n = 3, * p<0.05, Student’s t-test; scale bars, 10 µm). (E) When D70 alveolospheres are cultured with 3 µM CHIR99021, DKC1 A386T mutant iAT2 cells have a lower fraction of p21 positive cells. Note, data for no CHIR99021 bars are from Figure 2 (n = 3, * p<0.05, Student’s t-test; scale bars, 10 µm). (F) When D70 alveolospheres are cultured with 3 µM CHIR99021, DKC1 A386T mutant iAT2 cells have a lower fraction of telomere dysfunction induced foci (TIF) positive cells. Note, data for no CHIR99021 bars are from Figure 2 (n = 3, * p<0.05, Student’s t-test; scale bars, 10 µm; insets highlight cells with TIFs, each one noted by the white arrowheads). (G) Telomeric repeat amplification protocol assay for telomerase activity in iAT2 cells using fivefold extract dilutions (n = 3).

Figure 4—figure supplement 1. The growth defect in dyskeratosis congenita (DC) induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cells is rescued by continuous treatment of CHIR99021 from D28 to D70 but not if withdrawn at D56.

Figure 4—figure supplement 2. CHIR98014 can rescue dyskeratosis congenita (DC) induced pluripotent stem cell-derived type II alveolar epithelial (iAT2) cell growth.

Figure 4—figure supplement 3. TeSLA revealed a modest increase in average telomere length but no apparent decrease in the frequency of shortest detectable telomeres.

CHIR99021 downregulates senescence markers and resolves TIFs in DC iAT2 alveolospheres
We ultimately investigated how CHIR99021 affects the telomere status of DC iAT2s. CHIR treated iAT2 cells showed fewer cells with 53BP1 foci, fewer p21+ cells, and fewer TIF+ cells (Figure 4D–F). TeSLA revealed a modest increase in average telomere length but no apparent decrease in the frequency of shortest detectable telomeres (Figure 4—figure supplement 3A-C). iAT2 cell telomerase activity was restored in mutant cells treated with CHIR99021 to levels similar to those seen in WT cells (Figure 4G). Together, these data argue that GSK3 inhibition not only rescues the growth of DC iAT2 cells, but it also rescues telomere defects, most likely through upregulation of telomerase activity that could extend telomeres that are shorter than those that can be detected by TeSLA; it is also possible that telomerase is contributing to telomere capping via lengthening-independent mechanisms (Perera et al., 2019). Of note, withdrawal of CHIR99021 during only 14 days prior to D70 was sufficient to elicit the growth defect (Figure 4—figure supplement 1A-B), consistent with an only modest increase in telomere length associated with the increased telomerase activity.
Discussion
We used isogenic human iPS cell lines to generate DC mutant iAT2 cells with shortened telomeres to interrogate how telomere dysfunction can affect AT2 cell function. We found that shortened and uncapped telomeres are associated with a defect in alveolosphere formation by iAT2 cells. This defect is characterized by senescent iAT2 cells that upregulate many pathways associated with PF including the UPR, mitochondrial biogenesis and function, thyroid hormone signaling, and p53 signaling. DC mutant iAT2 cells also suppress canonical Wnt signaling, and consistent with this, GSK3 inhibition rescues telomerase activity, telomere capping, and alveolosphere formation. This system provides a new preclinical model to better understand PF pathogenesis and how potential new PF therapeutics affect AT2 cell function in the context of telomere dysfunction.
Wnt signaling is complex and can be broken down into two major categories: β-catenin dependent (canonical) signaling and β-catenin independent (non-canonical) signaling. These distinctions can also be blurred as evidenced by studies that show how non-canonical ligands, such as WNT5A, can activate both arms of Wnt signaling (van Amerongen et al., 2012; Mikels and Nusse, 2006). These complexities therefore make the conflicting reports about whether β-catenin dependent Wnt signaling is of benefit (Tanjore et al., 2013) or of harm in PF (Douglas et al., 2006; Henderson et al., 2010; Kim et al., 2011; Königshoff et al., 2008; McDonough et al., 2019) unsurprising. Given the heterogeneity of the disease both in space and time and the context dependence of Wnt signaling, these studies can often only capture a snapshot of the fibrotic response. Furthermore, there are clear differences between mice and humans with regard to telomere and lung biology (Basil and Morrisey, 2020; Gomes et al., 2011). These limitations highlight the need for human models capable of assessing the spatial, temporal, and cell type specific properties of Wnt signaling in PF pathogenesis.
Our study also provides evidence of upregulation of β-catenin-independent signaling in DC iAT2 cells and that re-activating β-catenin dependent Wnt signaling using GSK3 inhibitors might provide support for AT2 cell proliferation in the context of telomere dysfunction. It is tempting to extrapolate from other models of lung disease to understand how the activity of the β-catenin dependent Wnt pathway might be of benefit in PF. β-catenin dependent Wnt signaling improved regeneration and survival in a model of emphysema (Kneidinger et al., 2011), and inhibition of WNT5A, and thus presumably some component of β-catenin independent Wnt signaling, improved repair in a model of COPD (Baarsma et al., 2017). Our work, consistent with previous studies, argues that β-catenin dependent Wnt signaling supports AT2 cell telomere capping and proliferation, which may be of benefit during regeneration and repair (Nabhan et al., 2018; Uhl et al., 2015; Zacharias et al., 2018).
Our previous work uncovered a positive feedback loop between Wnt signaling and telomeres in the intestine (Woo et al., 2016; Yang et al., 2017). Here, we show that aspects of the Wnt-telomere feedback loop appear to be at play in AT2 cells (Fernandez and Johnson, 2018), arguing that this connection between Wnt and telomeres is present not just in proliferative tissues such as the intestine, but also in lung cells, which are normally quiescent but proliferate in response to injury. Previous work highlighted the importance of telomerase during alveolar regeneration (Driscoll et al., 2000; Lee et al., 2009). Furthermore, given previous demonstrations that Wnt can stimulate, directly or indirectly, TERT and TERC expression as well as telomerase activity in several cell types (Baena-Del Valle et al., 2018; Hoffmeyer et al., 2012; Jaitner et al., 2012; Zhang et al., 2012), we anticipated and were able to demonstrate (i) that TERT and TERC transcripts are reduced in senescent iAT2 cells and (ii) that GSK3 inhibition can restore telomerase activity in this context. Of note, CHIR99021 is known to induce proliferation in a wide variety of cell types (Hesselbarth et al., 2021; Jacob et al., 2017), which likely contributes to the growth stimulation observed in both WT and DKC1 mutant iAT2s shown in Figure 4—figure supplement 1. This raises the possibility that GSK3 inhibition has additional effects independent of telomeres. We also note that the effects of CHIR99021 could differ in some ways from canonical Wnt signaling, by impacting targets other than GSK3 or by differentially affecting the numerous pathways that are downstream of GSK3. However, the Wnt-related transcriptional changes in the mutant iAT2s, the high selectivity of CHIR99021 for GSK3 and its common use as a replacement for Wnt ligands in cell culture differentiation protocols, along with the fact that two different GSK3 inhibitors rescued growth of the mutant iAT2s suggest that the rescue provided by CHIR99021 is indeed related to GSK3 and Wnt (An et al., 2012; Loh et al., 2016; Pagliuca et al., 2014; Rezania et al., 2014; Ring et al., 2003; Teo et al., 2014).
DKC1 mutant iAT2 cells displayed a senescent phenotype that included upregulation of the p21 cyclin-dependent kinase inhibitor. Recent observations have revealed similar p21 upregulation in the AT2 cells of human PF lungs and have also demonstrated a causal role for p21 in driving experimental PF in mice (Lee et al., 2021; Lv et al., 2022). Senescence of the mutant iAT2 cells manifested only after serial passaging and telomere shortening, demonstrating that the senescence is not a direct result of DKC1 mutation per se. The senescence-associated growth defect emerged gradually, becoming more pronounced at later passages. Similarly gradual onset of telomere-driven senescence has been observed in other settings and has been attributed to (i) the stochastic nature of telomere shortening (causing some cells in a population to senesce prior to others) and (ii) graded increases in DNA damage responses in proportion to the extent of telomere shortening (Cesare et al., 2009; Cesare et al., 2013; Kaul et al., 2011; Martin-Ruiz et al., 2004; Ruis and Boulton, 2021; Suram and Herbig, 2014; Van Ly et al., 2018).
This Wnt-telomere feedback loop might not be the only regulatory loop at play in AT2 cells. Indeed, we hypothesize that telomeres might be an integrator and amplifier of multiple cellular stress responses. For example, our evidence of upregulation of mitochondrial processes in DC iAT2 cells is consistent with established feedback loops between telomeres and mitochondria: telomere dysfunction can drive mitochondrial dysfunction (Sahin et al., 2011) and mitochondrial dysfunction can drive telomere dysfunction (Guha et al., 2018; Passos et al., 2007; Qian et al., 2019). Furthermore, our finding of the upregulation of genes associated with the UPR is consistent with reports linking cellular senescence and the UPR (Pluquet et al., 2015). We hypothesize that the UPR might in turn drive telomere dysfunction, and it would thus be of interest to investigate telomere status in familial PF driven by misfolded proteins (Katzen and Beers, 2020). These multiple integrated loops might help explain how these various vital cellular processes combine to cause dysfunction in AT2 cells in PF.
Recent work, using mouse AT2 cell organoids, has elucidated a developmental trajectory by which AT2 cells can differentiate via an intermediate state into type I alveolar epithelial (AT1) cells in response to bleomycin injury (Choi et al., 2020; Kobayashi et al., 2020; Strunz et al., 2020). Of note, the intermediate cells are characterized by high levels of p53 signaling and DNA damage which resolve with their final transition to an AT1 cell fate (Kobayashi et al., 2020), arguing that repairing DNA damage, potentially at telomeres, is an important step in the transition to an AT1 cell. Furthermore, many of the pathways that promote the differentiation of AT2 cells into AT1 cells are downregulated in DC iAT2 cells, including those involving IL1β, glycolysis, and HIF1α (Choi et al., 2020). We speculate that DC AT2 cells with short uncapped telomeres may have trouble suppressing DNA damage at telomeres as they differentiate into AT1 cells. This may lock these cells into the transition state and may help explain their persistence in PF lungs. These persistent transitioning AT2 cells may thereby contribute to fibrosis. Testing this idea in the human iPSC-derived alveolosphere model will require technical advances to enable the generation of AT1 cells. Regardless, our DC iAT2 cell model recapitulates many hallmarks of PF AT2 cells and offers a new system to probe the underlying biology of PF.
Materials and methods
See Appendix 1 for Key Resources Table.
iPSC line generation and maintenance
The BU3 NGST line was a generous gift from Dr. Darrell Kotton at Boston University. iPS cells used for differentiation were maintained on growth factor reduced Matrigel (Corning) coated plates in StemMACS iPS-Brew XF medium (Miltenyi Biotec). Cells were cultured in clusters and passaged every 4–5 days using StemMACS Dissociation reagent (Miltenyi Biotec). All iPS lines were genotyped using an RFLP at the relevant important loci, and the sequence was confirmed by Sanger sequencing. All cells were routinely screened for mycoplasma contamination using a PCR-based assay (Uphoff and Drexler, 2014).
CRISPR editing for generating paired DKC1 mutant cell lines in BU3 NGST
To generate the introduced BU3 NGST line harboring the DKC1 mutation, we used the CRISPR track on the UCSC genome browser to select candidate guideRNAs (gRNAs) that targeted as close to the individual mutation as possible, had easily mutable PAM sites and would introduce a new restriction site to make screening easier (see the Key Resources Table for exact sequences and Figure S1 for details). gRNAs were ordered as oligos from IDT and cloned into pX458, a gift from Dr. Feng Zhang’s lab (Addgene # 48138). The candidate guides were tested for cutting efficiency by transfecting them into HEK293T cells and assaying cutting efficiency using T7E1 digestion of the PCR amplified locus. The most efficient guides were chosen and ssODN HDR templates were designed to eliminate the PAM. iPS cells were then nucleofected with the Amaxa Nucleofection system using the following program (P3, CA-137) (Lonza). The cells were allowed to recover for 36–48 hr at high density in the presence of ROCK inhibitor and then isolated by FACS for GFPhi cells. They were plated at low density (2500 cells/10 cm plate) and allowed to form single colonies. After 7–10 days, individual clones were selected and transferred to 96 well plates and screened for introduction of the restriction site for each mutation. Restriction enzyme positive clones were expanded and then subjected to sanger sequencing for identification of correctly edited clones. Successfully edited clones were checked for normal karyotype by G-banding (Cell Line Genetics), mycoplasma contamination, and pluripotency marker expression by immunofluorescence and were subsequently passaged for at least five passages before being re-genotyped to ensure that the clones were not mixed. During differentiations, all iAT2 cells were genotyped by restriction digest to ensure that the mutation was not lost with passage.
Directed differentiation into NKX2.1+ lung progenitors and SFTPC+ iPS-derived AT2 cells
A modified version of the protocol described in Jacob et al., 2017 was used to generate SFTPC expressing iAT2 cells. In brief, iPS cells were seeded at 500,000 cells per well on a six-well plate with ROCK inhibitor for 24 hr and incubated at 5% O2 | 5% CO2 | 90% N2. Definitive endoderm was induced using the StemDiff Definitive Endoderm kit for 3 days. Next, the cells were split at a ratio of 1:3 onto fresh Matrigel plates and anteriorized using dorsomorphin (2 µM) and SB431542 (10 µM) in complete serum-free differentiation media (cSFDM) for 3 days. Cells were then differentiated into NKX2.1+ progenitors by incubating in CBRa media (cSFDM containing CHIR99021 [3 µM], BMP4 [10 ng/mL], and retinoic acid [100 nM]) for 7 days changing media every 2 days at first and then increasing to every day media changes when the media became more acidic. On day 15 or 16, NKX2.1+ progenitors were isolated using a FACSJazz sorter using the endogenous NKX2.1::GFP reporter.
NKX2.1+ sorted cells were re-plated at a density of 400,000 cells/mL in 90% Matrigel supplemented with 10% of CK + DCI + TZV media (cSFDM containing 3 µM CHIR99021, 10 ng/mL KGF, 100 nM dexamethasone, 100 µM 8Br-cAMP and 100 µM IBMX, and 2 µM TZV) (from now on referred to as 90/10 Matrigel). The Matrigel droplets were allowed to cure at 37°C for 20–30 min and then overlaid with an appropriate amount of CK + DCI + TZV media. These alveolosphere containing Matrigel droplets were incubated at 37°C at 20% O2 | 5% CO2 | 75% N2 (room air) for 14 days changing with fresh media every other day. Per Hurely et al., CHIR99021 was withdrawn from the media on day 17 for 4 days and re-added back on day 21 to optimize generation and specification of SFTPC+ cells. On day 28, the iAT2 containing alveolospheres were sorted on a FACSJazz sorter for SFTPC+ cells using the endogenous SFTPC::TdTomato reporter. These sorted SFTPC+ cells were replated at a concentration of 65,000 cells/mL in 90/10 Matrigel drops and grown in K + DCI + TZV at 37°C in an ambient air incubator supplemented to 5% CO2 for 3 weeks changing media every other day.
Alveolosphere counting and formation efficiency calculations
Alveolosphere images were taken on a Leica Thunder widefield microscope using a 1.25× objective. Z-stacks were maximum projected and then thresholded using ImageJ to create a binary file. Binary files were eroded and dilated to ensure maximum determination of the alveolosphere size. Finally, the binary images were separated by watershedding, and alveolospheres were counted using Analyze Particles in ImageJ.
Immunofluorescence microscopy of iAT2 alveolospheres
Alveolospheres were washed with PBS and then fixed in place using 2% paraformaldehyde (PFA) at room temperature for 30 min and then dehydrated and paraffin embedded and sectioned. Once cut, slides were de-paraffinized, rehydrated, permeabilized, and antigens were retrieved by steaming for 15 min in a citrate buffer (Vector Labs). After blocking, each slide was incubated with a primary antibody using the concentrations listed in the Key Resource Table. Slides were incubated with primary antibody at 37°C for 2 hr. After washing, slides were incubated with appropriate fluorochrome conjugated secondary antibodies (see Key Resources Table for antibody details). Slides were then washed, counterstained with DAPI, and mounted. Images were acquired using a Leica Thunder widefield microscope.
TIFs were stained as described in Suram et al., 2012. In brief, cut slides were de-paraffinized, rehydrated, permeabilized, and antigens were retrieved as for other immunofluoresence stains. Slides were blocked and stained for 53BP1 and then stained with an appropriate fluorochrome conjugated secondary antibody. Slides were then re-fixed with PFA, quenched with glycine, re-dehydrated in an ethanol series, and air dried. The slides were then stained with the PNA probe. The slides were washed, rehydrated in an ethanol series, and stained with a tertiary fluorochrome conjugated antibody. Slides were then washed, counterstained with DAPI, and mounted. TIF images were acquired using a Leica SP8 confocal microscope. Quantification of nuclei was carried out in ImageJ in a blinded fashion.
Measurement of telomerase activity with TRAP
iPS cells or iAT2 cells were cultured as indicated in each figure legend. 100,000 cells were harvested using methods described and lysed using NP-40 lysis buffer and processed as described in Herbert et al., 2006. In brief, lysates were incubated with a telomerase substrate and incubated at 30°C for telomerase to add telomere repeats. The reactions were then PCR amplified. Telomere repeats were resolved on a 4–20% TBE polyacrylamide gel and visualized by staining with SYBR Green nucleic acid gel stain. Relative telomerase activity was quantified using ImageJ focusing on the first six amplicons averaged across the dilutions.
Measurement of telomere lengths by TRF and TeSLA
Telomere lengths were measured as described in Lai et al., 2016; Lai et al., 2017. DNA was isolated from cells using a Gentra Puregene kit (Qiagen). DNA was quantified by fluorometry using Qubit 2.0 (Invitrogen). For terminal restriction fragment (TRF) analysis in brief, 500 ng of DNA was digested with CviAII overnight followed by digestion with a mixture of BfaI, MseI, and NdeI overnight. For TeSLA in brief, 50 ng of DNA was ligated to telorette adapters, then digested with CviAII, then digested with a combination of BfaI, MseI, and NdeI, dephosphorylated, and TeSLA adapters (AT/TA Adapters) were ligated on. These TeSLA libraries were PCR amplified using Lucigen’s FailSafe polymerase kit with Pre-Mix H.
Southern blotting was carried out using previously established protocols with some modification (Kimura et al., 2010; Lai et al., 2017). TRFs and TeSLA PCR reactions were separated on a 0.7% agarose gel at 0.833 V/cm for 24 hr. The gel was depurinated and denatured and then transferred to a Hybond XL membrane (Cytiva) by capillary transfer using denaturation buffer. The Hybond membrane was hybridized using a DIG-labeled telomere probe overnight. The blot was then washed and exposed using CDP-Star on an LAS-4000 Image Quant imager (Cytiva). TRFs were analyzed using ImageQuant while TeSLAs were analyzed using the MatLab software developed previously (Lai et al., 2017).
Measurement of telomere lengths by qPCR
Average telomere length was measured by qPCR as described in Cawthon, 2002; Joglekar et al., 2020 with some modifications. In brief, isolated genomic DNA was quantified by Qubit fluorometry (Invitrogen) and diluted to within the range of a standard curve constructed from a mixture of all samples analyzed. Triplicate qPCR reactions of the telomeric (T) product and the single copy gene (S) (HBB) were amplified using a Roche LightCycler 480 II (Roche) using the following programs: T PCR program 95°C for 10 min, 40 cycles of 95°C for 15 s, 56°C for 1 min; S PCR program 95°C for 10 min, 40 cycles of 95°C for 15 s, 58°C for 1 min. Cq values were computed using the second derivative method, and T/S ratios were calculated using the 2-∆∆Ct method.
RNA-sequencing and data analysis
SFTPC + sorted cells from the indicated times during, counted and harvested in TRIzol and stored at –80°C until further processing. The RNA was isolated using a Direct-Zol kit (Zymo Research). RNA concentration was obtained by Qubit fluorometry (Invitrogen), and the integrity was checked by tape station analysis (Agilent Technologies). All samples had RINs >8, and the libraries were prepared by poly-A selection and sequenced by GeneWiz, LLC.
RNA sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina following manufacturer’s instructions (NEB). Briefly, mRNAs were first enriched with oligo (dT) beads. Enriched mRNAs were fragmented for 15 min at 94°C. First strand and second strand cDNAs were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies) and quantified using a Qubit 2.0 Fluorometer (Invitrogen) as well as by quantitative PCR (KAPA Biosystems).
The sequencing libraries were pooled and clustered on one lane of a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq instrument (4000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2 × 150 bp paired end configuration. Image analysis and base calling were conducted by the HiSeq control software. Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Fastq files were checked for quality using FastQC. Raw sequence files (fastq) for 22 samples were mapped using salmon (https://combine-lab.github.io/salmon/; Patro et al., 2017) against the human transcripts described in Gencode (version v33, built on the human genome GRCm38, https://www.gencodegenes.org), with a 70.5% average mapping rate yielding 30.4 M average total input reads per sample. Transcript counts were summarized to the gene level using tximport (available here) and normalized and tested for differential expression using DESeq2 (available here). Subsets of time-matched samples were used to compute pair-wise contrast statistics for mutant vs WT at each time. Raw FASTQ files and analyzed counts are available on GEO under the accession number GSE160871.
GSEA (Subramanian et al., 2005) was carried out in R (v4.0.2) (R Development Core Team, 2020) using RStudio (v1.3.1056) (RStudio Team, 2020), the tidyverse (v1.3.0) (Wickham et al., 2019), and the readxl package (v1.3.1) (Hadley Wickham, 2019). GSEA was run for contrasts of interest in preranked mode using the DESeq2 statistic as the ranking metric (Love et al., 2014). Annotated molecular signatures from the Hallmark Collection (H), Curated Collection (C2), and Regulatory Target Molecular Collection (C3) maintained by the Molecular Signatures Database were accessed in RStudio using the msigdbr package (v7.1.1) (Dolgalev, 2020; Liberzon et al., 2011; Liberzon et al., 2015). The clusterProfiler package (v3.17.1) was used to perform GSEA on the unfiltered, sorted gene lists (Yu et al., 2012). GSEA results were viewed using the DT package (v0.15) (Yihui Xie, 2020). GSEA plots were generated using the enrichplot package (v1.9.1) (Yu, 2020).
Genes that differed in expression >twofold and were associated with an adjusted p-value <0.05 from the D70 time point were also analyzed through the use of IPA (Ingenuity Systems, https://www.ingenuity.com) (Krämer et al., 2014) through the University of Pennsylvania Molecular Profiling Facility.
ddPCR
RNA was isolated as listed above for the RNA-sequencing. 140 ng of RNA was first treated with ezDNAse (Invitrogen) and converted to cDNA using the SuperScript IV First-Strand Synthesis System (Invitrogen). The equivalent of 12.5 ng of original RNA was used as template in the reactions, and the ddPCR assays as well as the ddPCR supermix used are listed in the Key Resources Table (Bio-Rad). ddPCR was performed according to manufacturer’s instructions using QX200 AutoDG Droplet Digital PCR System (Bio-Rad). Results were analyzed using Quanta-Soft Version 1.7.4.0917 (Bio-Rad). Copies/µL were used and normalized to ACTB.
Statistical methods
Statistical methods are outlined in each of the figure legends. Each replicate ‘n’ represents an entirely separate differentiation from the pluripotent stem cell stage. For experiments with iPS cells, ‘n’ represents separate wells of the same iPS line (specifically for Figure S1 and S2). Quantitative data is represented as the mean with error bars representing the standard error of the mean. Student’s t-tests (unpaired and two-tailed) were used for determining statistical significance for all comparisons unless otherwise noted.
Acknowledgements
We would like to thank Drs. Eric Witze, Peter Klein, Timothy Olson, Andrew Vaughan, Leo Cardenas, and Aravind Sivakumar for fruitful discussions. We would like to thank Dr. Dong Hun Woo, Carla Hoge, John McCormick, and Dr. Rachel Truitt for help culturing iPS cells, and Dr. Darrell Kotton for providing the BU3 NGST line. We also thank Drs. Erica Carpenter and Jacob Till for help with the ddPCR. This work was also supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Center for Molecular Studies in Digestive and Liver Diseases (P30DK050306) and its Molecular Profiling and Imaging core facilities, by the University of Pennsylvania induced Pluripotent Stem Cell Core and the Penn Genomic Analysis Core, by the Flow Cytometry Core Laboratory, the Human Pluripotent Stem Cell Core at the Children’s Hospital of Philadelphia Research Institute, and the Abramson Cancer Center BioRepository Shared Resource and the Cancer Center Support Grant from the NCI, P30 CA016520.
Appendix 1
Key resources table
Appendix 1—key resources table.
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (Homo sapiens) | BU3 NGST | Jacob et al., 2017 | iPS cell line | |
| Cell line (Homo sapiens) | BU3 NGST – WT | This Paper | iPS cell line, WT control passaged with the DKC1 A386T cell line. See Figure 1—figure supplement 1 & 2 for more details. Contact the corresponding authors for the cell line. | |
| Cell line (Homo sapiens) | BU3 NGST - DKC1 A386T | This Paper | iPS cell line, DKC1 A386T passaged with the WT cell line. See Figure 1—figure supplement 1 & 2 for more details. Contact the corresponding authors for the cell line. | |
| Cell line (Homo sapiens) | AG04646 | Woo et al., 2016 | iPS cell line | |
| Recombinant DNA reagent | pX458 | Feng Zhang | Addgene # 48,138 | |
| Chemical compound, drug | Thiazovivin (TZV) | Cayman Chemical compound, drug | CAT# 14,245 | |
| Chemical compound, drug | SB431542 | Cayman Chemical compound, drug | CAT# 13,031 | |
| Chemical compound, drug | Dorsomorphin | Cayman Chemical compound, drug | CAT# 21,207 | |
| Chemical compound, drug | CHIR99021 | Cayman Chemical compound, drug | CAT# 13,122 | |
| Chemical compound, drug | CHIR98014 | Cayman Chemical compound, drug | CAT# 15,578 | |
| Chemical compound, drug | Retinoic Acid | Sigma | CAT# R2625-50MG | |
| Peptide, recombinant protein | rhBMP4 | R&D Systems | CAT# 314 BP-050 | |
| Peptide, recombinant protein | rhKGF | R&D Systems | CAT# 251 KG-050 | |
| Chemical compound, drug | Dexamethasone | Sigma | CAT# D4902-100MG | |
| Chemical compound, drug | 8Br-cAMP | Sigma | CAT# 7880–250 MG | |
| Chemical compound, drug | IBMX | Sigma | CAT# I5879-250MG | |
| Peptide, recombinant protein | DNaseI | Roche | CAT# 4716728001 | |
| Chemical compound, drug | 7-AAD | BD Biosciences | CAT# 51-68981E | |
| Chemical compound, drug | Propidium Iodide | BD Biosciences | CAT# 556,463 | |
| Peptide, recombinant protein | Dispase II | Gibco | CAT# 17105041 | |
| Chemical compound, drug | SYBR Green I Nucleic Acid Gel Stain | Invitrogen | CAT# S7563 | |
| Peptide, recombinant protein | Matrigel, Growth Factor Reduced | Corning | CAT# 354,230 | |
| Chemical compound, drug | Stem MACS iPS-Brew XF | Miltenyi Biotec | CAT# 130-104-368 | |
| Peptide, recombinant protein | Accutase | Innovative Cell Technologies | CAT# AT104-500 | |
| Commercial assay, kit | P3 Primary Cell 4D-NucleofectorTM X Kit L | Lonza | CAT# V4XP-3012 | |
| Chemical compound, drug | IMDM | Corning/Mediatech | CAT# MT10-016-CV | |
| Chemical compound, drug | Ham’s F12 Nutrient Mix | Corning/Mediatech | CAT# MT10-080-CV | |
| Chemical compound, drug | B27 | Gibco | CAT# 17504044 | |
| Chemical compound, drug | N2 | Gibco | CAT# 17502048 | |
| Chemical compound, drug | BSA Fraction V | Gibco | CAT# 15260037 | |
| Chemical compound, drug | Monothioglycerol | Sigma | CAT# M6145-25ML | |
| Chemical compound, drug | GlutaMAX | Gibco | CAT# 35050061 | |
| Chemical compound, drug | Ascorbic Acid | Sigma | CAT# A4544-25G | |
| Chemical compound, drug | Primocin | Invivogen | CAT# ANT-PM-2 | |
| Chemical compound, drug | Hank’s Balanced Salt Solution | Gibco | CAT# 14175079 | |
| Chemical compound, drug | HEPES | Gibco | CAT# 15630080 | |
| Chemical compound, drug | EDTA | Gibco | CAT# 15575039 | |
| Peptide, recombinant protein | 0.05% trypsin-EDTA | Gibco | CAT# 25300054 | |
| Commercial assay, kit | Direct-zol RNA Micro Kit | Zymo | CAT# R2061 | |
| Commercial assay, kit | Gentra Puregene Cell Kit | Qiagen | CAT# 158,745 | |
| Antibody | Anti-NANOG (Rabbit polyclonal) | Reprocell/Stemgent | CAT# 09–0020 | IF (1:100 37°C for 2 hr) |
| Antibody | Anti-53BP1 (Rabbit monoclonal) | Novus | CAT# NB100-304 | IF (1:100 37°C for 2 hr) |
| Antibody | Anti-P21 (Mouse monoclonal) | Santa Cruz | CAT# sc-6246 | IF (1:50 37°C for 2 hr) |
| Antibody | Anti-Cleaved Caspase 3 (CC3) (Rabbit monoclonal) | Cell Signaling Technologies | CAT# 9,664 | IF (1:2000 37°C for 2 hr) |
| Sequence-based reagent | Anti-Cy3-Telo-C Probe | PNA Bio | CAT# F1002 | Anti-telomeric |
| Antibody | Anti-Digoxigenin-AP, Fab fragments | Roche | CAT# 11093274910 | |
| Antibody | Goat anti-Rabbit IgG Secondary Antibody, Alexa Fluor 647 (Donkey polyclonal) | Invitrogen | CAT# A-21244 | IF (1:250 RT for 1 hr) |
| Antibody | Donkey anti-Mouse IgG Secondary Antibody, Alexa Fluor 647 (Donkey polyclonal) | Invitrogen | CAT# A-31571 | IF (1:250 RT for 1 hr) |
| Antibody | Donkey anti-Goat IgG Secondary Antibody, Alexa Fluor 647 (Donkey polyclonal) | Invitrogen | CAT# A-21447 | IF(1:250 RT for 1 hr) |
| Chemical compound, drug | TriZOL | Invitrogen | CAT# 15596018 | |
| Peptide, recombinant protein | FailSafe Polymerase Enzyme Mix | Lucigen | CAT# FS9901K | |
| Chemical compound, drug | FailSafe Buffer H | Lucigen | CAT# FSP995H-INCL | |
| Peptide, recombinant protein | CviAII | NEB | CAT# R0640L | |
| Peptide, recombinant protein | BfaI | NEB | CAT# R0568L | |
| Peptide, recombinant protein | MseI | NEB | CAT# R0525L | |
| Peptide, recombinant protein | NdeI | NEB | CAT# R0111L | |
| Peptide, recombinant protein | rSAP | NEB | CAT# M0371L | |
| Chemical compound, drug | CDP-Star | Roche | CAT# 11759051001 | |
| Chemical compound, drug | DIG-11-dUTP | Roche | CAT# 11558706910 | |
| Chemical compound, drug | DIG Easy Hybridization Granules | Roche | CAT# 11796895001 | |
| Chemical compound, drug | Blocking Reagent | Roche | CAT# 11096176001 | |
| Sequence-based reagent | DIG Labeld DNA Molecular Weight Marker II | Roche | CAT# 11218590910 | |
| Chemical compound, drug | Hybond-XL | Cytiva/Amersham | CAT# RPN303S | |
| Peptide, recombinant protein | Klenow Fragment | NEB | CAT# M0212S | |
| Peptide, recombinant protein | Lambda Exonuclease | NEB | CAT# M0262S | |
| Peptide, recombinant protein | Go-TAQ Flexi | Promega | CAT# M8298 | |
| Peptide, recombinant protein | ddPCR Multiplex Supermix | BioRad | CAT# 12005909 | |
| Software | FIJI | NIH | RRID:SCR_002285 | https://imagej.net/Fiji |
| Software | Salmon | Patro et al., 2017 | ||
| Software | Ingenuity pathway analysis | Krämer et al., 2014 | ||
| Software | FlowJo | BD Biosciences | RRID:SCR_008520 | |
| Software | ImageQuant TL 8.2 | Cytiva | RRID:SCR_018374 | |
| Software | TeSLAQuant | Lai et al., 2017 |
Appendix 2
Primer Sequences
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Sequence-based reagent | gRNA-DKC1A386-5-F | This Paper | gRNA Cloning Primers designed to target DKC1 A386 – See Figure 1—figure supplement 1 for details | CAC CGC TTC TTC TGA CTT GCC TGG |
| Sequence-based reagent | gRNA-DKC1A386-5-R | This Paper | gRNA Cloning Primers designed to target DKC1 A386 – See Figure 1—figure supplement 1 for details | AAA CCC AGG CAA GTC AGA AGA AGC |
| Sequence-based reagent | DKC1-Geno-F | Woo et al., 2016 | Genotyping primers for DKC1 Gene | AAA AGT AGA GTG GCT GCG GT |
| Sequence-based reagent | DKC1-Geno-R | Woo et al., 2016 | Genotyping primers for DKC1 Gene | ACA ACC TCC ATG CTC ACC TG |
| Sequence-based reagent | TeSLA-T1 | Lai et al., 2017 | Telorette | ACT GGC CAC GTG TTT TGA TCG ACC CTA AC |
| Sequence-based reagent | TeSLA-T2 | Lai et al., 2017 | Telorette | ACT GGC CAC GTG TTT TGA TCG ATA ACC CT |
| Sequence-based reagent | TeSLA-T3 | Lai et al., 2017 | Telorette | ACT GGC CAC GTG TTT TGA TCG ACC TAA CC |
| Sequence-based reagent | TeSLA-T4 | Lai et al., 2017 | Telorette | ACT GGC CAC GTG TTT TGA TCG ACT AAC CC |
| Sequence-based reagent | TeSLA-T5 | Lai et al., 2017 | Telorette | ACT GGC CAC GTG TTT TGA TCG AAA CCC TA |
| Sequence-based reagent | TeSLA-T6 | Lai et al., 2017 | Telorette | ACT GGC CAC GTG TTT TGA TCG AAC CCT AA |
| Sequence-based reagent | TeSLA Adapter Short | Lai et al., 2017 | GGT TAC TTT GTA AGC CTG TC[SpcC3] | |
| Sequence-based reagent | TeSLA Adapter TA | Lai et al., 2017 | [Phos] TAG ACA GGC TTA CAA AGT AAC CAT GGT GGA GAA TTC TGT CGT CTT CAC GCT ACA TT [SpcC3] | |
| Sequence-based reagent | TeSLA Adapter AT | Lai et al., 2017 | [Phos] ATG ACA GGC TTA CAA AGT AAC CAT GGT GGA GAA TTC TGT CGT CTT CAC GCT ACA TT [SpcC3] | |
| Sequence-based reagent | AP | Lai et al., 2017 | TGT AGC GTG AAG ACG ACA GAA | |
| Sequence-based reagent | TeSLA-TP | Lai et al., 2017 | TGG CCA CGT GTT TTG ATC GA | |
| Sequence-based reagent | G-Rich ONT | Lai et al., 2016 | [Phos] CCC TAA CCC TAA CCC TAA CCC TAA CCC TAA CCC TAA CCC TAG ATA GTT GAG AGT C | |
| Sequence-based reagent | Branched Universal Sequence-based reagent | Lai et al., 2016 | [Phos] GAC TCT CAA CTA TC +T + A | |
| Sequence-based reagent | TS | Herbert et al., 2006 | AAT CCG TCG AGC AGA GTT | |
| Sequence-based reagent | ACX | Herbert et al., 2006 | GCG CGG CTT ACC CTT ACC CTT ACC CTA ACC | |
| Sequence-based reagent | Tel1B | Luo et al. 2020 | CGG TTT GTT TGG GTT TGG GTT TGG GTT TGG GTT TGG GTT | |
| Sequence-based reagent | Tel2B | Luo et al. 2020 | GGC TTG CCT TAC CCT TAC CCT TAC CCT TAC CCT TAC CCT | |
| Sequence-based reagent | HBG1 | Cawthon, 2002 | GCT TCT GAC ACA ACT GTG TTC ACT AGC | |
| Sequence-based reagent | HBG2 | Cawthon, 2002 | CAC CAA CTT CAT CCA CGT TCA CC | |
| Sequence-based reagent | TINF2 | BioRad | dHsaCPE5027564 | |
| Sequence-based reagent | POT1 | BioRad | dHsaCPE50447370 | |
| Sequence-based reagent | TERT | BioRad | dHsaCPE5048434 | |
| Sequence-based reagent | TERC | BioRad | dHsaCNS923142164 | |
| Sequence-based reagent | ACTB | BioRad | dHsaCPE5190200 |
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Christopher J Lengner, Email: lengner@vet.upenn.edu.
F Brad Johnson, Email: johnsonb@pennmedicine.upenn.edu.
Weiwei Dang, Baylor College of Medicine, United States.
Jessica K Tyler, Weill Cornell Medicine, United States.
Funding Information
This paper was supported by the following grants:
National Institute on Aging R21AG054209 to Christopher J Lengner, F Brad Johnson.
National Institute on Aging 5T32AG000255 to Rafael Jesus Fernandez.
Team Telomere/Penn Orphan Disease Center to Christopher J Lengner, F Brad Johnson.
National Heart, Lung, and Blood Institute R01HL148821 to Christopher J Lengner, F Brad Johnson.
Additional information
Competing interests
No competing interests declared.
No competing interests declared.
Reviewing editor, eLife.
Author contributions
Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review and editing.
Data curation, Formal analysis, Investigation, Methodology, Visualization.
Methodology.
Methodology, Writing – review and editing.
Investigation.
Methodology, Resources.
Investigation.
Investigation.
Investigation.
Investigation.
Investigation.
Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Visualization.
Data curation, Formal analysis, Resources, Software, Visualization.
Data curation, Formal analysis, Resources, Software, Visualization.
Resources.
Resources.
Conceptualization, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review and editing.
Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review and editing.
Additional files
These tables provide the unedited output of the GSEA analysis using the C2 curated gene sets, H hallmark gene sets, and C3 regulatory target gene sets when comparing D70 mutant to WT iAT2 cells. The table reports the name of the gene set (ID), the size of the gene set (setSize), the raw enrichment score (enrichmentScore), the normalized enrichment score, along with the p-value, the adjusted p-value (p.adjust) and false discovery rate q-value (q-values). The ‘core enrichment’ column displays the genes in the gene set.
These tables provide the unedited output of the IPA. The summary tab lists metadata associated with the analysis. The ‘analysis ready molecules’ lists the differentially expressed genes that differed in expression greater than twofold and were associated with an adjusted p-value < 0.05 from D70 mutant iAT2 cells (DEG list). The ‘canonical pathways tab’ lists pathways curated from the literature and the p-value for enrichment using the DEG list used in the IPA. The ‘upstream regulators’ tab lists the transcription factors, cytokines, and other genetic regulators whose target genes are in the DEG list. ‘Causal networks’ seeks to build a regulatory network based off of the ‘upstream regulators’ to identify master regulators of the DEG list. For more information on interpreting ingenuity analysis results see Krämer et al., 2014.
These tables display selected results from gene set enrichment analysis (GSEA) and ingenuity pathway analyses (IPAs) that highlight pathways found to be differentially regulated in mutant iAT2 cells at D70 when compared with wild type cells. The first table displays GSEA results along with the pathway name, normalized enrichment score, and adjusted p-value (D70 p-adj). The second table displays IPA results from the canonical pathways analysis. These are gene sets that are differentially regulated in mutant iAT2 cells at D70 when compared with wild type cells. The p-value reports the significance of enrichment of the molecules in that gene set, and the activation score reports how concordant the gene expression changes are with what is predicted from the literature embedded in IPA (a negative z-score argues that the gene set is down regulated in the mutant iAT2 cells, whereas a positive z-score argues that the gene set is upregulated in mutant iAT2 cells; the lack of a z-score is indicative there was insufficient evidence to provide a z-score.) The ‘molecules’ column lists the genes that were in that gene set that were also found in our differentially expressed gene list when comparing mutant iAT2 cells to wildtype cells.
Data availability
Sequencing data was deposited in GEO: GSE160871.
The following dataset was generated:
Fernandez RJ, Johnson FB, Tobias JW. 2021. Transcriptional profiling of short telomere iPS-derived sorted SFTPC+ iAT2 cells. NCBI Gene Expression Omnibus. GSE160871
References
- Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD, Huo H, Okuka M, Dos Reis RM, Loewer S, Ng HH, Keefe DL, Goldman FD, Klingelhutz AJ, Liu L, Daley GQ. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis Congenita. Hematology/Oncology Clinics of North America. 2018;32:669–685. doi: 10.1016/j.hoc.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder JK, Chen JJL, Lancaster L, Danoff S, Su S, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA, Lansdorp PM, Loyd JE, Armanios MY. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. PNAS. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015;147:1361–1368. doi: 10.1378/chest.14-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WF, Germain AR, Bishop JA, Nag PP, Metkar S, Ketterman J, Walk M, Weiwer M, Liu X, Patnaik D. Discovery of Potent and Highly Selective Inhibitors of GSK3b, Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information (US; 2012. [PubMed] [Google Scholar]
- Armanios MY, Chen JJL, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. The New England Journal of Medicine. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- Baarsma HA, Skronska-Wasek W, Mutze K, Ciolek F, Wagner DE, John-Schuster G, Heinzelmann K, Günther A, Bracke KR, Dagouassat M, Boczkowski J, Brusselle GG, Smits R, Eickelberg O, Yildirim AÖ, Königshoff M. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. The Journal of Experimental Medicine. 2017;214:143–163. doi: 10.1084/jem.20160675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena-Del Valle JA, Zheng Q, Esopi DM, Rubenstein M, Hubbard GK, Moncaliano MC, Hruszkewycz A, Vaghasia A, Yegnasubramanian S, Wheelan SJ, Meeker AK, Heaphy CM, Graham MK, De Marzo AM. MYC drives overexpression of telomerase RNA (hTR/TERC) in prostate cancer. The Journal of Pathology. 2018;244:11–24. doi: 10.1002/path.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM. Type 2 alveolar cells are stem cells in adult lung. The Journal of Clinical Investigation. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basil MC, Morrisey EE. Lung regeneration: a tale of mice and men. Seminars in Cell & Developmental Biology. 2020;100:88–100. doi: 10.1016/j.semcdb.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista LFZ, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, Giri N, Wernig M, Alter BP, Cech TR, Savage SA, Reijo Pera RA, Artandi SE. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–402. doi: 10.1038/nature10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. American Journal of Respiratory and Critical Care Medicine. 2005;172:1026–1031. doi: 10.1164/rccm.200503-504OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Research. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nature Structural & Molecular Biology. 2009;16:1244–1251. doi: 10.1038/nsmb.1725. [DOI] [PubMed] [Google Scholar]
- Cesare AJ, Hayashi MT, Crabbe L, Karlseder J. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Molecular Cell. 2013;51:141–155. doi: 10.1016/j.molcel.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilosi M, Poletti V, Zamò A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A, Maestro R, Semenzato G, Doglioni C. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. The American Journal of Pathology. 2003;162:1495–1502. doi: 10.1016/s0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, Lee JH. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell. 2020;27:366–382. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K-P, Hsu C-L, Fan L-C, Huang Z, Bhatia D, Chen Y-J, Hisata S, Cho SJ, Nakahira K, Imamura M, Choi ME, Yu C-J, Cloonan SM, Choi AMK. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nature Communications. 2019;10:3390. doi: 10.1038/s41467-019-11327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C, Garnett ET, Montgomery KH, Mason WR, McKean DF, Powers J, Murphy E, Olson LM, Choi L, Cheng DS, Blue EM, Young LR, Lancaster LH, Steele MP, Brown KK, Schwarz MI, Fingerlin TE, Schwartz DA, Lawson WE, Loyd JE, Zhao Z, Phillips JA, Blackwell TS. Rare variants in RTEL1 are associated with familial interstitial pneumonia. American Journal of Respiratory and Critical Care Medicine. 2015;191:646–655. doi: 10.1164/rccm.201408-1510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz AC, Orchard PJ, Baker KS, Giller RH, Savage SA, Alter BP, Tolar J. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplantation. 2011;46:98–104. doi: 10.1038/bmt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disayabutr S, Kim EK, Cha S-I, Green G, Naikawadi RP, Jones KD, Golden JA, Schroeder A, Matthay MA, Kukreja J, Erle DJ, Collard HR, Wolters PJ. miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLOS ONE. 2016;11:e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgalev I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. Msigdbr 2020
- Douglas IS, Diaz del Valle F, Winn RA, Voelkel NF. Beta-catenin in the fibroproliferative response to acute lung injury. American Journal of Respiratory Cell and Molecular Biology. 2006;34:274–285. doi: 10.1165/rcmb.2005-0277OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll B, Buckley S, Bui KC, Anderson KD, Warburton D. Telomerase in alveolar epithelial development and repair. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2000;279:L1191–L1198. doi: 10.1152/ajplung.2000.279.6.L1191. [DOI] [PubMed] [Google Scholar]
- Duckworth A, Gibbons MA, Allen RJ, Almond H, Beaumont RN, Wood AR, Lunnon K, Lindsay MA, Wain LV, Tyrrell J, Scotton CJ. Evidence That Telomere Length Is Causal for Idiopathic Pulmonary Fibrosis but Not Chronic Obstructive Pulmonary Disease: A Mendelian Randomisation Study. bioRxiv. 2020 doi: 10.1101/2020.02.05.20019653. [DOI] [PubMed]
- Everaerts S, Lammertyn EJ, Martens DS, De Sadeleer LJ, Maes K, van Batenburg AA, Goldschmeding R, van Moorsel CHM, Dupont LJ, Wuyts WA, Vos R, Gayan-Ramirez G, Kaminski N, Hogg JC, Janssens W, Verleden GM, Nawrot TS, Verleden SE, McDonough JE, Vanaudenaerde BM. The aging lung: tissue telomere shortening in health and disease. Respiratory Research. 2018;19:95. doi: 10.1186/s12931-018-0794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez RJ, Johnson FB. A regulatory loop connecting WNT signaling and telomere capping: possible therapeutic implications for dyskeratosis congenita. Annals of the New York Academy of Sciences. 2018;1418:56–68. doi: 10.1111/nyas.13692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DB, Peng T, Zepp JA, Snitow M, Vincent TL, Penkala IJ, Cui Z, Herriges MJ, Morley MP, Zhou S, Lu MM, Morrisey EE. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Reports. 2016;17:2312–2325. doi: 10.1016/j.celrep.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes NMV, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR, Austad SN, Venditti C, Pagel M, Shay JW, Wright WE. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell. 2011;10:761–768. doi: 10.1111/j.1474-9726.2011.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, Yamaguchi TP, Morrisey EE. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Developmental Cell. 2009;17:290–298. doi: 10.1016/j.devcel.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M, Srinivasan S, Johnson FB, Ruthel G, Guja K, Garcia-Diaz M, Kaufman BA, Glineburg MR, Fang J, Nakagawa H, Basha J, Kundu T, Avadhani NG. hnRNPA2 mediated acetylation reduces telomere length in response to mitochondrial dysfunction. PLOS ONE. 2018;13:e0206897. doi: 10.1371/journal.pone.0206897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley Wickham JB. readxl: Read Excel Files. Readxl 2019
- Haschek WM, Witschi H. Pulmonary fibrosis--a possible mechanism. Toxicology and Applied Pharmacology. 1979;51:475–487. doi: 10.1016/0041-008x(79)90372-7. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Chi EY, Ye X, Nguyen C, Tien Y, Zhou B, Borok Z, Knight DA, Kahn M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. PNAS. 2010;107:14309–14314. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert BS, Hochreiter AE, Wright WE, Shay JW. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nature Protocols. 2006;1:1583–1590. doi: 10.1038/nprot.2006.239. [DOI] [PubMed] [Google Scholar]
- Hesselbarth R, Esser TU, Roshanbinfar K, Schrüfer S, Schubert DW, Engel FB. CHIR99021 Promotes hiPSC-Derived Cardiomyocyte Proliferation in Engineered 3D Microtissues. Advanced Healthcare Materials. 2021;10:e2100926. doi: 10.1002/adhm.202100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science (New York, N.Y.) 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, Na CL, Weaver TE, Vedaie M, Hurley K, Hinds A, Russo SJ, Kook S, Zacharias W, Ochs M, Traber K, Quinton LJ, Crane A, Davis BR, White FV, Wambach J, Whitsett JA, Cole FS, Morrisey EE, Guttentag SH, Beers MF, Kotton DN. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell. 2017;21:472–488. doi: 10.1016/j.stem.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A., Vedaie M, Roberts DA, Thomas DC, Villacorta-Martin C, Alysandratos KD, Hawkins F, Kotton DN. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nature Protocols. 2019;14:3303–3332. doi: 10.1038/s41596-019-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitner S, Reiche JA, Schäffauer AJ, Hiendlmeyer E, Herbst H, Brabletz T, Kirchner T, Jung A. Human telomerase reverse transcriptase (hTERT) is a target gene of β-catenin in human colorectal tumors. Cell Cycle (Georgetown, Tex.) 2012;11:3331–3338. doi: 10.4161/cc.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar MV, Satoor SN, Wong WKM, Cheng F, Ma RCW, Hardikar AA. An Optimised Step-by-Step Protocol for Measuring Relative Telomere Length. Methods and Protocols. 2020;3:E27. doi: 10.3390/mps3020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalina M, Mason RJ, Shannon JM. Surfactant protein C is expressed in alveolar type II cells but not in Clara cells of rat lung. American Journal of Respiratory Cell and Molecular Biology. 1992;6:594–600. doi: 10.1165/ajrcmb/6.6.594. [DOI] [PubMed] [Google Scholar]
- Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. The Journal of Clinical Investigation. 2020;130:5088–5099. doi: 10.1172/JCI139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Reports. 2011;13:52–59. doi: 10.1038/embor.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Kim SH, Seo JY, Chung H, Kwak HJ, Lee SK, Yoon HJ, Shin DH, Park SS, Sohn JW. Blockade of the Wnt/β-catenin pathway attenuates bleomycin-induced pulmonary fibrosis. The Tohoku Journal of Experimental Medicine. 2011;223:45–54. doi: 10.1620/tjem.223.45. [DOI] [PubMed] [Google Scholar]
- Kimura M, Stone RC, Hunt SC, Skurnick J, Lu X, Cao X, Harley CB, Aviv A. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nature Protocols. 2010;5:1596–1607. doi: 10.1038/nprot.2010.124. [DOI] [PubMed] [Google Scholar]
- King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW, ASCEND Study Group A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. The New England Journal of Medicine. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- Kneidinger N, Yildirim AÖ, Callegari J, Takenaka S, Stein MM, Dumitrascu R, Bohla A, Bracke KR, Morty RE, Brusselle GG, Schermuly RT, Eickelberg O, Königshoff M. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. American Journal of Respiratory and Critical Care Medicine. 2011;183:723–733. doi: 10.1164/rccm.200910-1560OC. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, Banovich NE, Kropski JA, Tata PR. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nature Cell Biology. 2020;22:934–946. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Königshoff M., Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, Eickelberg O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLOS ONE. 2008;3:e2142. doi: 10.1371/journal.pone.0002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Königshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, Rose F, Fink L, Seeger W, Schaefer L, Günther A, Eickelberg O. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. The Journal of Clinical Investigation. 2009;119:772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (Oxford, England) 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET, Degryse AL, Mitchell DB, Polosukhin VV, Rickman OB, Choi L, Cheng D-S, McConaha ME, Jones BR, Gleaves LA, McMahon FB, Worrell JA, Solus JF, Ware LB, Lee JW, Massion PP, Zaynagetdinov R, White ES, Kurtis JD, Johnson JE, Groshong SD, Lancaster LH, Young LR, Steele MP, Phillips Iii JA, Cogan JD, Loyd JE, Lawson WE, Blackwell TS. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. American Journal of Respiratory and Critical Care Medicine. 2015;191:417–426. doi: 10.1164/rccm.201406-1162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropski JA, Reiss S, Markin C, Brown KK, Schwartz DA, Schwarz MI, Loyd JE, Phillips JA, Blackwell TS, Cogan JD. Rare Genetic Variants in PARN Are Associated with Pulmonary Fibrosis in Families. American Journal of Respiratory and Critical Care Medicine. 2017;196:1481–1484. doi: 10.1164/rccm.201703-0635LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TP, Wright WE, Shay JW. Generation of digoxigenin-incorporated probes to enhance DNA detection sensitivity. BioTechniques. 2016;60:306–309. doi: 10.2144/000114427. [DOI] [PubMed] [Google Scholar]
- Lai TP, Zhang N, Noh J, Mender I, Tedone E, Huang E, Wright WE, Danuser G, Shay JW. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nature Communications. 2017;8:1356. doi: 10.1038/s41467-017-01291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. The New England Journal of Medicine. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- Lee J, Reddy R, Barsky L, Scholes J, Chen H, Shi W, Driscoll B. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2009;296:L57–L70. doi: 10.1152/ajplung.90411.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, La J, Aziz S, Dobrinskikh E, Brownell R, Jones KD, Achtar-Zadeh N, Green G, Elicker BM, Golden JA, Matthay MA, Kukreja J, Schwartz DA, Wolters PJ. Molecular markers of telomere dysfunction and senescence are common findings in the usual interstitial pneumonia pattern of lung fibrosis. Histopathology. 2021;79:67–76. doi: 10.1111/his.14334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xiao J, Hormi K, Borok Z, Minoo P. Wnt5a participates in distal lung morphogenesis. Developmental Biology. 2002;248:68–81. doi: 10.1006/dbio.2002.0729. [DOI] [PubMed] [Google Scholar]
- Li C, Hu L, Xiao J, Chen H, Li JT, Bellusci S, Delanghe S, Minoo P. Wnt5a regulates Shh and Fgf10 signaling during lung development. Developmental Biology. 2005;287:86–97. doi: 10.1016/j.ydbio.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics (Oxford, England) 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Systems. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, Shyh-Chang N, Fernhoff NB, George BM, Wernig G, Salomon REA, Chen Z, Vogel H, Epstein JA, Kundaje A, Talbot WS, Beachy PA, Ang LT, Weissman IL. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell. 2016;166:451–467. doi: 10.1016/j.cell.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X, Liu C, Liu S, Li Y, Wang W, Li K, Hua F, Cui B, Zhang X, Yu J, Yu J, Hu Z. The cell cycle inhibitor P21 promotes the development of pulmonary fibrosis by suppressing lung alveolar regeneration. Acta Pharmaceutica Sinica B. 2022;12:735–746. doi: 10.1016/j.apsb.2021.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant Protein A2 Mutations Associated with Pulmonary Fibrosis Lead to Protein Instability and Endoplasmic Reticulum Stress. Journal of Biological Chemistry. 2010;285:22103–22113. doi: 10.1074/jbc.M110.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/β-catenin signaling during mouse development and in colorectal tumors. PNAS. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Ruiz C, Saretzki G, Petrie J, Ladhoff J, Jeyapalan J, Wei W, Sedivy J, von Zglinicki T. Stochastic variation in telomere shortening rate causes heterogeneity of human fibroblast replicative life span. The Journal of Biological Chemistry. 2004;279:17826–17833. doi: 10.1074/jbc.M311980200. [DOI] [PubMed] [Google Scholar]
- McDonough JE, Ahangari F, Li Q, Jain S, Verleden SE, Herazo-Maya J, Vukmirovic M, DeIuliis G, Tzouvelekis A, Tanabe N, Chu F, Yan X, Verschakelen J, Homer RJ, Manatakis DV, Zhang J, Ding J, Maes K, De Sadeleer L, Vos R, Neyrinck A, Benos PV, Bar-Joseph Z, Tantin D, Hogg JC, Vanaudenaerde BM, Wuyts WA, Kaminski N. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight. 2019;4:131597. doi: 10.1172/jci.insight.131597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLOS Biology. 2006;4:040115. doi: 10.1371/journal.pbio.0040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. American Journal of Respiratory Cell and Molecular Biology. 2005;32:521–530. doi: 10.1165/rcmb.2005-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science (New York, N.Y.) 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, Rock JR, Looney MR, Wolters PJ. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1:e86704. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, Beers MF. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. The Journal of Clinical Investigation. 2018;128:4008–4024. doi: 10.1172/JCI99287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo T, Hogan BLM. Hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm. Journal of Biology. 2004;3:11. doi: 10.1186/jbiol3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrin EJ, Little DR, Gerner-Mauro KN, Sumner EA, Ríos-Corzo R, Ambrosio E, Holt SE, Forcioli-Conti N, Akiyama H, Hanash SM, Kimura S, Huang SXL, Chen J. β-Catenin maintains lung epithelial progenitors after lung specification. Development (Cambridge, England) 2018;145:dev160788. doi: 10.1242/dev.160788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TBL, von Zglinicki T. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLOS Biology. 2007;5:e050110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera ON, Sobinoff AP, Teber ET, Harman A, Maritz MF, Yang SF, Pickett HA, Cesare AJ, Arthur JW, MacKenzie KL, Bryan TM. Telomerase promotes formation of a telomere protective complex in cancer cells. Science Advances. 2019;5:eaav4409. doi: 10.1126/sciadv.aav4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. American Journal of Physiology. Cell Physiology. 2015;308:C415–C425. doi: 10.1152/ajpcell.00334.2014. [DOI] [PubMed] [Google Scholar]
- Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Reports. 2015;12:286–299. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, Watkins SC, Kolodieznyi D, Bruchez MP, Van Houten B. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. PNAS. 2019;116:18435–18444. doi: 10.1073/pnas.1910574116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team . Vienna, Austria: R Foundation for Statistical Computing; 2020. http://www.r-project.org [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YHC, Johnson JD, Kieffer TJ. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nature Biotechnology. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR, INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. The New England Journal of Medicine. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, Ma ST, Reeder JW, Samuels I, Slabiak T, Wagman AS, Hammond M-EW, Harrison SD. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- RStudio Team RStudio: Integrated Development Environment for R. RStudio, PBC 2020
- Ruis P, Boulton SJ. The end protection problem-an unexpected twist in the tail. Genes & Development. 2021;35:1–21. doi: 10.1101/gad.344044.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty SK, Tiwari N, Marudamuthu AS, Puthusseri B, Bhandary YP, Fu J, Levin J, Idell S, Shetty S. p53 and miR-34a Feedback Promotes Lung Epithelial Injury and Pulmonary Fibrosis. The American Journal of Pathology. 2017;187:1016–1034. doi: 10.1016/j.ajpath.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM, Piccolo S, Birchmeier W, Whitsett JA, Millar SE, Morrisey EE. Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Developmental Biology. 2005;283:226–239. doi: 10.1016/j.ydbio.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T, van der Vis JJ, Goldschmeding R, Grutters JC, van Moorsel CHM. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLOS ONE. 2017;12:e0189467. doi: 10.1371/journal.pone.0189467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O’Reilly M, Lehmann M, Burgstaller G, Königshoff M, Chapman HA, Theis FJ, Schiller HB. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nature Communications. 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL, Hande MP, d’Adda di Fagagna F, Herbig U. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. The EMBO Journal. 2012;31:2839–2851. doi: 10.1038/emboj.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suram A., Herbig U. The replicometer is broken: telomeres activate cellular senescence in response to genotoxic stresses. Aging Cell. 2014;13:780–786. doi: 10.1111/acel.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Current Biology. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- Tanjore H, Degryse AL, Crossno PF, Xu XC, McConaha ME, Jones BR, Polosukhin VV, Bryant AJ, Cheng D-S, Newcomb DC, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. β-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. American Journal of Respiratory and Critical Care Medicine. 2013;187:630–639. doi: 10.1164/rccm.201205-0972OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telomeres Mendelian Randomization Collaboration. Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, Wade KH, Timpson NJ, Evans DM, Willeit P, Aviv A, Gaunt TR, Hemani G, Mangino M, Ellis HP, Kurian KM, Pooley KA, Eeles RA, Lee JE, Fang S, Chen WV, Law MH, Bowdler LM, Iles MM, Yang Q, Worrall BB, Markus HS, Hung RJ, Amos CI, Spurdle AB, Thompson DJ, O’Mara TA, Wolpin B, Amundadottir L, Stolzenberg-Solomon R, Trichopoulou A, Onland-Moret NC, Lund E, Duell EJ, Canzian F, Severi G, Overvad K, Gunter MJ, Tumino R, Svenson U, van Rij A, Baas AF, Bown MJ, Samani NJ, van t’Hof FNG, Tromp G, Jones GT, Kuivaniemi H, Elmore JR, Johansson M, Mckay J, Scelo G, Carreras-Torres R, Gaborieau V, Brennan P, Bracci PM, Neale RE, Olson SH, Gallinger S, Li D, Petersen GM, Risch HA, Klein AP, Han J, Abnet CC, Freedman ND, Taylor PR, Maris JM, Aben KK, Kiemeney LA, Vermeulen SH, Wiencke JK, Walsh KM, Wrensch M, Rice T, Turnbull C, Litchfield K, Paternoster L, Standl M, Abecasis GR, SanGiovanni JP, Li Y, Mijatovic V, Sapkota Y, Low S-K, Zondervan KT, Montgomery GW, Nyholt DR, van Heel DA, Hunt K, Arking DE, Ashar FN, Sotoodehnia N, Woo D, Rosand J, Comeau ME, Brown WM, Silverman EK, Hokanson JE, Cho MH, Hui J, Ferreira MA, Thompson PJ, Morrison AC, Felix JF, Smith NL, Christiano AM, Petukhova L, Betz RC, Fan X, Zhang X, Zhu C, Langefeld CD, Thompson SD, Wang F, Lin X, Schwartz DA, Fingerlin T, Rotter JI, Cotch MF, Jensen RA, Munz M, Dommisch H, Schaefer AS, Han F, Ollila HM, Hillary RP, Albagha O, Ralston SH, Zeng C, Zheng W, Shu X-O, Reis A, Uebe S, Hüffmeier U, Kawamura Y, Otowa T, Sasaki T, Hibberd ML, Davila S, Xie G, Siminovitch K, Bei J-X, Zeng Y-X, Försti A, Chen B, Landi S, Franke A, Fischer A, Ellinghaus D, Flores C, Noth I, Ma S-F, Foo JN, Liu J, Kim J-W, Cox DG, Delattre O, Mirabeau O, Skibola CF, Tang CS, Garcia-Barcelo M, Chang K-P, Su W-H, Chang Y-S, Martin NG, Gordon S, Wade TD, Lee C, Kubo M, Cha P-C, Nakamura Y, Levy D, Kimura M, Hwang S-J, Hunt S, Spector T, Soranzo N, Manichaikul AW, Barr RG, Kahali B, Speliotes E, Yerges-Armstrong LM, Cheng C-Y, Jonas JB, Wong TY, Fogh I, Lin K, Powell JF, Rice K, Relton CL, Martin RM, Davey Smith G. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncology. 2017;3:636–651. doi: 10.1001/jamaoncol.2016.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo AKK, Valdez IA, Dirice E, Kulkarni RN. Comparable generation of activin-induced definitive endoderm via additive Wnt or BMP signaling in absence of serum. Stem Cell Reports. 2014;3:5–14. doi: 10.1016/j.stemcr.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AQ, Lane K, Phillips J, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, Roberts R, Haines J, Stahlman M, Loyd JE. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. American Journal of Respiratory and Critical Care Medicine. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- Uhl FE, Vierkotten S, Wagner DE, Burgstaller G, Costa R, Koch I, Lindner M, Meiners S, Eickelberg O, Königshoff M. Preclinical validation and imaging of Wnt-induced repair in human 3D lung tissue cultures. The European Respiratory Journal. 2015;46:1150–1166. doi: 10.1183/09031936.00183214. [DOI] [PubMed] [Google Scholar]
- Uphoff CC, Drexler HG. Detection of Mycoplasma contamination in cell cultures. Current Protocols in Molecular Biology. 2014;106:28. doi: 10.1002/0471142727.mb2804s106. [DOI] [PubMed] [Google Scholar]
- Valapour M, Lehr CJ, Skeans MA, Smith JM, Uccellini K, Goff R, Foutz J, Israni AK, Snyder JJ, Kasiske BL. OPTN/SRTR 2018 Annual Data Report: Lung. American Journal of Transplantation. 2020;20 Suppl s1:427–508. doi: 10.1111/ajt.15677. [DOI] [PubMed] [Google Scholar]
- van Amerongen R, Fuerer C, Mizutani M, Nusse R. Wnt5a can both activate and repress Wnt/β-catenin signaling during mouse embryonic development. Developmental Biology. 2012;369:101–114. doi: 10.1016/j.ydbio.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ly D, Low RRJ, Frölich S, Bartolec TK, Kafer GR, Pickett HA, Gaus K, Cesare AJ. Telomere Loop Dynamics in Chromosome End Protection. Molecular Cell. 2018;71:510–525. doi: 10.1016/j.molcel.2018.06.025. [DOI] [PubMed] [Google Scholar]
- Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, Choi AMK. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. The Journal of Experimental Medicine. 2006;203:2895–2906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. American Journal of Human Genetics. 2009;84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen T, Miller E, Bache S, Müller K, Ooms J, Robinson D, Seidel D, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H. Welcome to the Tidyverse. Journal of Open Source Software. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- Wiese KE, Nusse R, van Amerongen R. Wnt signalling: conquering complexity. Development (Cambridge, England) 2018;145:dev165902. doi: 10.1242/dev.165902. [DOI] [PubMed] [Google Scholar]
- Woo DH, Chen Q, Yang TLB, Glineburg MR, Hoge C, Leu NA, Johnson FB, Lengner CJ. Enhancing a Wnt-Telomere Feedback Loop Restores Intestinal Stem Cell Function in a Human Organotypic Model of Dyskeratosis Congenita. Cell Stem Cell. 2016;19:397–405. doi: 10.1016/j.stem.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T-LB, Chen Q, Deng JT, Jagannathan G, Tobias JW, Schultz DC, Wang S, Lengner CJ, Rustgi AK, Lynch JP, Johnson FB. Mutual reinforcement between telomere capping and canonical Wnt signalling in the intestinal stem cell niche. Nature Communications. 2017;8:14766. doi: 10.1038/ncomms14766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR. Senescence of Alveolar Stem Cells Drives Progressive Pulmonary Fibrosis. bioRxiv. 2019 doi: 10.1101/820175. [DOI] [PMC free article] [PubMed]
- Yihui Xie J. DT: A Wrapper of the JavaScript Library. DataTables 2020
- Yu G., Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, de Castro JPW, DeIuliis G, Ahangari F, Woolard T, Aurelien N, Arrojo E Drigo R, Gan Y, Graham M, Liu X, Homer RJ, Scanlan TS, Mannam P, Lee PJ, Herzog EL, Bianco AC, Kaminski N. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nature Medicine. 2018;24:39–49. doi: 10.1038/nm.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. enrichplot: Visualization of Functional Enrichment Result. Enrichplot 2020
- Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Toh L, Lau P, Wang X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. The Journal of Biological Chemistry. 2012;287:32494–32511. doi: 10.1074/jbc.M112.368282. [DOI] [PMC free article] [PubMed] [Google Scholar]