

Abstract
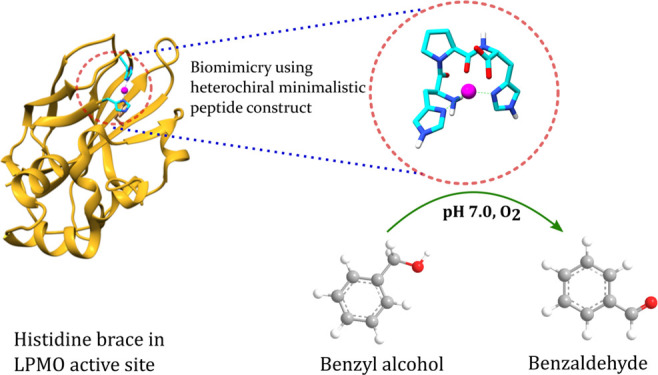
We employed a reductionist approach in designing the first heterochiral tripeptide that forms a robust heterogeneous short peptide catalyst similar to the “histidine brace” active site of lytic polysaccharide monooxygenases. The histidine brace is a conserved divalent copper ion-binding motif that comprises two histidine side chains and an amino group to create the T-shaped 3N geometry at the reaction center. The geometry parameters, including a large twist angle (73°) between the two imidazole rings of the model complex, are identical to those of native lytic polysaccharide monooxygenases (72.61°). The complex was synthesized and characterized as a structural and functional mimic of the histidine brace. UV–vis, vis-circular dichroism, Raman, and electron paramagnetic resonance spectroscopic analyses suggest a distorted square-pyramidal geometry with a 3N coordination at pH 7. Solution- and solid-state NMR results further confirm the 3N coordination in the copper center of the complex. The complex is pH-dependent and could catalyze the oxidation of benzyl alcohol in water to benzaldehyde with yields up to 82% in 3 h at pH 7 and above at 40 °C. The catalyst achieved 100% selectivity for benzaldehyde compared to conventional copper catalysis. The design of such a minimalist building block for functional soft materials with a pH switch can be a stepping stone in addressing needs for a cleaner and sustainable future catalyst.
Introduction
Mimicry of the enzyme active site through supramolecular assembly has been of great interest for the design and synthesis of various novel catalysts.1−3 Nature is a dominant source of inspiration in the area of supramolecular chemistry, and enzymes have served as natural prototypes for the design of supramolecular catalysts. In general, enzymes work by binding to their substrates and then use the action of two or more well-placed functional groups to achieve catalysis. Such an organization leads to substrate selectivity, reaction selectivity, and stereoselectivity. One way to achieve such natural enzyme-like precision in catalysis is by mimicking the active site of an enzyme. Biomimetic modeling of enzymes involves the design of compounds containing similar functional groups mimicking a specific enzyme’s active site.
Over the years, researchers have found that copper oxygenase enzymes possess His-Xaa-His (Xaa = amino acid) chelating sequences at their active sites.4,5 In addition, they exhibit δN versus εN tautomeric preferences in the imidazole group. Currently, efforts are directed toward trying to achieve a degree of control in the binding of transition metals in biologically common histidine-rich sites in a minimalistic construct. While histidine-containing linear peptides are reported as metalloenzyme mimetics with hydrolytic6,7 and oxidative activities,8−11 the metal chelation is highly unstable. As a consequence, the metal-binding ability of such peptides is rather limited, which directly affects their catalytic efficiency. A survey of the His imidazole group binding to copper proteins involved in redox chemistry, including O2 reactivity, indicates that the His-Xaa-His tripeptide motif is a frequently observed sequence. Some of the reported motifs include His-Thr-His in peptidylglycine α-hydroxylating monooxygenase12 and dopamine β-monooxygenase,13 His-Val-His in superoxide dismutases 5,14 and His-Gln-His in amyloid-like protein 2.15
Over the last decade, the introduction of amino acids of both chiralities in a peptide sequence has seen remarkable success in terms of novelty and creativity.16,17 Our laboratory reported the advantages of using d-amino acid in the sequence for the design of novel functional materials18 and enzymatically stable molecular constructs.19−21 The ability to tune the metal binding affinity of small peptides through the incorporation of unnatural d-amino acids holds great promise in designing ultra-short peptide motifs resembling the active site of an enzyme. The peptides which are diastereomerically different offer the possibility of altering the spatial orientation of amino acid side chains. This helps to modulate the local structure and interactions in the metal binding pockets without altering the side-chain chemistry.
Lytic polysaccharide monooxygenases (LPMOs) are metalloenzymes that activate molecular oxygen and cleave the C–H bond in polysaccharides.22 They are attracting considerable attention due to their industrial applications.23,24 LPMOs utilize copper as their functional active site metal for oxidizing recalcitrant polysaccharides in nature. The LPMO domains usually comprise 200–250 amino acids that drastically restrict their commercial application due to the excessive cost associated with their synthesis and stability at a range of temperatures and pH. Even the use of whole cell catalysis is limited in an industrial setup owing to the high cost of product purification. Interestingly, various spectroscopic and computational investigations have provided insights into the three-dimensional (3D) copper-containing active center featuring the “histidine brace”.22,25−27 The histidine brace is conserved and comprised a single divalent copper ion which is chelated with two nitrogen atoms of histidine at position 36 (one atom from the backbone and one from the side chain) and a nitrogen atom from a second histidine at position 135, forming an overall T-shaped 3N configuration.22,28 The main chain amino group of the N terminal histidine and imidazole side chain contributes two of its nitrogen, while the second conserved histidine is the source of the third nitrogen.
Conventionally, the aerobic oxidation of C–H bonds is performed using reducing agents and radical initiators under severe conditions.6 The last decade has seen significant research interest in developing catalysts that use readily available reagents at room temperature with molecular oxygen as the oxidant.29,30 This study focuses on developing a peptide-based, heterogeneous, environment-friendly, inexpensive molecular system with high catalytic efficiency. We have synthesized and characterized a copper-binding de novo-designed heterochiral tripeptide redox enzyme system that mimics the structurally conserved histidine brace found in LPMOs. The peptide–copper complex exhibits a very close chemical and structural resemblance to the active site geometry of the enzyme. It shows enhanced catalytic activity in the oxidation of benzyl alcohol in water. This heterochiral peptide–copper design may be an example of a structurally and functionally optimized biomimetic model for LPMOs.
Results
Characterization of the Active Site Geometry
The histidine brace is a result of the spatial interaction between two histidine residues at the 36th and 135th positions in a typical LPMO. To mimic the chemical and structural features of the LPMO active site, we have designed the peptide, His-Pro-DHis-NH2 (HPh), bearing two imidazole groups. A proline residue connects the two histidines, creating a T-shaped 3N geometry for copper coordination (Figure 1c). In our design, this spatial geometry was achieved by incorporating a d-histidine residue at the C-terminus of the sequence.
Figure 1.

(a) 3D structure of the lytic polysaccharide monooxygenase (PDB: 5FJQ) and active-site residues (cyan). (b) LigPlot showing the ligand interactions with the histidine residues in the LPMO active site. (c) Chemical structure of the designed peptide mimicking the histidine brace. (d,e) Comparison between the structural parameters of the copper-chelated modeled peptide and enzyme active site, respectively (Table S1, Supporting Information).
The Cu–HPh complex was modeled using Avogadro 1.2.0.31,32 Interestingly, we have observed a large twist angle (73°) between the two imidazole groups (Figure 1e), which is in agreement with the reported LPMO (Protein Data Bank, 5FJQ.pdb). This endorses the observation that the Cu–HPh complex successfully mimics the structural motif of the Histidine brace.
In the next phase, we have synthesized the peptide molecule and characterized the physiochemical properties of the complex using various spectroscopic techniques.
Cu Binding to HPh
The pH-dependent coordination of Cu(II) with the designed peptide was verified in a wide range of pH values ranging from 3 to 9 using UV–vis spectroscopy. A 0.9:1 Cu(II) to peptide stoichiometry is considered for the experiments to ensure that there is no excess of copper, thus preventing a copper hydroxide precipitation in neutral and alkaline pH. At low pH, λmax of the d–d bands are around 800 nm, which correlates with a band for a Cu(II) aqueous ion (Figures 2a and S4a, Supporting Information). With the increase in pH, a blue shift is observed. The absorption peak at 620 nm is reported to be specific to the 3N Cu coordination.33 The position of these bands indicates a square-pyramidal geometry around the Cu(II) coordination sphere with the dx2–y2 ground state4,34,35 and is close to the d–d transition seen in LPMO (655 nm).35,36 The absence of any absorption band at 520 nm further confirms the 3N geometry.37 Furthermore, we have employed UV–vis spectroscopy to verify the stability of the complex at pH 7 with respect to time (Figure S5, Supporting Information). The absence of any difference in the spectral parameters of the complex up to 180 min suggests that there is no leaching of the Cu(II) ions.
Figure 2.

Analysis of the Cu–HPh complex formation: (a) UV–vis, (b) CD, and (c) Raman spectra at different pH values. Experimental conditions: final concentrations of 5 mM HPh and 4.5 mM CuCl2 were titrated with 1 μL aliquots of 1 M HCl or NaOH solution, and the reaction was left to equilibrate for 10 min after each addition.
The circular dichroism (CD) data are in good agreement with the pH-dependent UV–vis spectra. Major changes in visible-CD spectra are observed at pH 7 and above, suggesting that at this pH, a change in the coordinating ligands around the copper ion occurs. At pH 7, two negative bands around 550 and 310 nm (Figures 2b and S2, Supporting Information) are accompanied by a strong negative peak at 280 nm, validating an N-imidazole-to-Cu(II) charge transfer.38 At pH 9, however, the negative band at 550 nm broadens and, together with the strong negative band at 310 nm, indicates the presence of mixed species. Furthermore, the positive bands at 650 and 750 nm suggest coordination of Cu(II) with proposed donors 2Nim and N–, as reported in different Cu(II)–histidine complexes.6,39 In the UV-CD analysis, we have observed a negative maximum at 330 nm at pH 7 and above (Figure S3, Supporting Information). This typically suggests the characteristic charge transfer transitions between Nim and Cu2+.40,41 Besides, the electronic transition of the amide group is affected by the solvent environment.
This may explain the observed increase in the negative intensity of the UV-CD spectrum at 230 nm at higher pH. Further characterization of the Cu–HPh complex formation was obtained by measuring Raman spectra at pH 3, 7, and 9 (Figure 2c). The Raman signal at 1543 cm–1 for pH 3 corresponds to the C‡C stretching vibration and is similar to that of the control (Figure 2c). The shift to the higher frequency (Δϑ = 11 cm–1) upon increasing the pH to 7 and above signifies the chelation of the imidazole group.42 The Raman spectra of the control and the complex at pH 3 are dominated by a single strong band at 1426 cm–1 that is due to the Nτ–C2–Nπ symmetric stretch (Figure 2c).43,44 In case of the complexes formed at pH 7 and above, we have observed a higher vC4–C5 (1590–1568 cm–1) and a band at around 1290 cm–1, attributed to a stretch mode of the C2–Nπ–C4 linkage,45,46 suggesting Nτ–H and Nπ–M forms.47 The vC4–C5 (1550–1530 cm–1) at pH 3 suggests the absence of any metal ion coordination and is similar to the control. In addition, the Raman band at around 1275 cm–1 at pH 7 and above is useful as supporting evidence for the metal binding to Nπ.47 The increase in pH resulted in the increased peak intensity at 1672 cm–1, ascribed to the C=N stretching vibration.
EPR Characterization
The coordination of the Cu–HPh complex was investigated at pH 3 and 7 using low-temperature electron paramagnetic resonance (EPR) spectroscopy (100 K). The hyperfine features have been resolved into parallel (g∥) and perpendicular (g⊥) regions (Figure 3a). The axial EPR spectra for pH 7 with g∥ = 2.32 > g⊥ = 2.03 suggest the presence of the 1B2g ground state with the unpaired electron in the dx2–y2 orbital.35,37 The complex exhibits EPR parameters very close to those of the LPMOs (g∥ = 2.23–2.28; g⊥ = 2.06–2.09).35 A simulation of the EPR spectrum was obtained to validate the spectral features of the complex formed at pH 7 (Figure S6, Supporting Information). Comparison of the EPR spectra of the complex and copper salt at pH 7 (g∥, 2.50 > g 2.11, Figure S8, Supporting Information) suggests that there is no leaching of Cu(II) from the complex, which is in line with our observation in UV–vis analysis. Interestingly, no g∥ component was observed in the case of pH 3 (g = 2.09), suggesting isotropy in the applied field.48 In agreement with the UV–vis and CD spectra, the EPR fingerprints at pH 7 show the presence of the 3N geometry.35,37
Figure 3.

(a) Frozen-solution EPR of the Cu–HPh complex at pH 3 (blue) and pH 7(red) in 30% glycerol (v/v). (b) Solution-state 1H NMR of the Cu–HPh complex at pH 3 and pH 7. (c) Solid-state 1H NMR of the Cu–HPh complex at pH 3 and pH 7; * the peak at −5 ppm is due to the spectrometer artifact. (d) Solid-state 13C NMR of Cu–HPh complex at pH 3 and 7. The peaks in the 13C spectra correspond to the peptide backbone shown in Figure S9, Supporting Information.
NMR Characterization
In order to further verify the 3N binding modes of the complex, we recorded the 1H and 13C NMR spectra of HPh and HPh in the presence of Cu(II) at pH 3 and 7. We have employed both solid-state and solution-state NMR for the analysis. The UV–vis spectrum shows no complex formation at pH 3 and is therefore considered as a reference here. The 1H NMR spectrum in the solution state at pH 3 further validates this observation (Figure 3b). Moreover, the solution-state 1H spectra of the sample at pH 7 show broad featureless lines due to fast relaxation, indicating the Cu–HPh complex formation. In the solid state, the 1H and 13C NMR spectra of the nascent peptide showed resolved resonances. In contrast, for the Cu(II) complex, 1H and 13C solid-state NMR spectra showed broadening of the resonances (Figure 3c,d). The 1H NMR spectrum of the complex shows a nonselective broadening of the signals over a spectral range of 20 ppm due to the binding of the paramagnetic Cu(II). In the nascent peptide 1H spectrum, a clear peak at 14.5 ppm can be attributed to the ring HN proton. In contrast, the proton resonance in the complex is either broadened out or shifted to a new position. Similar nonselective broadening of the peaks was also observed in the carbon spectrum of the Cu(II) complex, and a new peak appears at 0–5 ppm. This peak broadening and change in shifts may be attributed to the paramagnetic center of Cu2+ and are absent for the nascent peptide.
Longitudinal relaxation (T1) values for protons of the nascent peptide were estimated to be 600–700 ms by using a saturation recovery experiment. The proton signal of the peptide with the complex was collected at 0.1, 0.2, and 1 s. We observed that upon complex formation, the proton signal recovered to equilibrium magnetization with less than 0.1 s delay after saturation. We expect the actual T1 values to be much less than 0.1 s, which is an order of magnitude smaller in comparison to that of the nascent peptide (pH 3). The observed large difference in the T1 values of the nascent peptide and the complex is a good indicator of a complex with a paramagnetic center.
Morphological and Elemental Characterization of the Complex
Field emission scanning electron microscopy (FESEM) was employed to determine the morphology of the Cu–HPh complex. It shows flower-like structures that are formed by the agglomeration of uniform microflakes stacked in different orientations (Figure 4a). Transmission electron microscopy (TEM) of the complex after negative staining confirms solid flake-like structures of around 214 nm (Figure 4b). The energy-dispersive X-ray spectroscopy (EDS) line mapping of the structure was performed to confirm the presence of copper ions (Figure 4c). The intensity of Cu signals shown in the red line reveals its abundance. The signals from other elements like phosphorus may be attributed to the phosphate buffer used for the experiment.
Figure 4.

Morphology characterization of the complex. (a) FESEM image showing the flake-like morphology (inset) that is clustered to form a flower of around 214 nm in diameter; (b) negatively stained TEM image of the “flower”; and (c) corresponding SEM-EDS line map spectrum of the complex showing carbon (magenta), nitrogen (purple), oxygen (pink), sulfur (blue), phosphorus (cyan), and copper (red).
Catalytic Efficiency of the Designed Tripeptide Catalyst
We assessed the catalytic activity of the peptide complex for the selective aerobic oxidation of benzyl alcohol to its corresponding aldehyde (Scheme 1).
Scheme 1. Selective Oxidation of Benzyl Alcohol to Benzaldehyde in the Presence of the Cu–HPh Complex at 40 °C.

The oxidation of benzyl alcohol was carried out in a round-bottom flask (25 mL) fitted with an O2 balloon atop a magnetic stirrer at 40 °C. We screened different catalyst loadings (10, 15, and 30 mol %) and monitored the rate of conversion of benzyl alcohol at different time intervals for 3 h. At low catalytic loads (10 and 15 mol %), the percent conversion of benzaldehyde after 3 h was also low at 10 and 27%, respectively [Figure 5b (olive), Figure S11a,b, Supporting Information].
Figure 5.

Conversion of benzyl alcohol into benzaldehyde in water: pH = 7 at 40 °C is the standard reaction conditions used except in the pH titration experiment. (a) Conversion comparison between copper alone and the complex at 30% loading as a function of time. (b) Histogram comparing percentage conversion by nascent HPh (red), copper (black), and the Cu–HPh complex (olive) after 3 h. (c) Conversion of benzyl alcohol into benzaldehyde at 40 °C in 6 h under aerobic conditions. (d) Histogram showing the catalytic efficiency of the Cu–HPh complex at different pH (mean ± standard deviation, n = 3).
In general, the trend suggests that by doubling the catalyst loading from 15 to 30 mol %, we could achieve an impressive 300% increase in the catalytic efficiency. Next, we compared the catalytic efficiency of the catalyst and Cu(II) at 30 mol % catalyst loading (Figures 5a, S11c, and S13c, Supporting Information). After 3 h of the reaction, we have observed 82% conversion of benzyl alcohol in the case of the complex with 100% selectivity. For Cu(II), however, the conversion was low (62%) and is comparable to previous reports that suggest a moderate conversion by Cu(II) alone (72–83% conversion after 24 h at 70 °C).49 Furthermore, we have then compared the catalytic efficiency of the complex with an equimolar concentration of individual entities, that is, peptide and copper alone as control (Figure 5b, red and black, respectively, and Figure S10, Supporting Information). While HPh alone did not show any significant conversion at even 30 mol % loading, we have observed an increased catalysis by Cu(II) after 90 min of the reaction(Figures S10a,b and S12–S13, Supporting Information). We have considered 30 mol % catalyst loading for all subsequent experiments unless mentioned otherwise.
Next, we explored the scope of achieving complete conversion of benzyl alcohol by extending the reaction time to 12 h (Figure 5c). While no significant increase in the conversion (in percentage) was observed, the selectivity remained 100%, and no trace of benzoic acid was observed in the high-performance liquid chromatography (HPLC) spectra. This may be attributed to the application of low temperature in the reaction, which prevents auto-oxidation of the aldehyde.
Dependency of pH in Modulating Catalysis
The measurement of the reaction rate as a function of pH provides essential information about the underlying correlation between the protonation states of the imidazole nitrogen and catalysis. The protonation state affects the imidazole donor–acceptor capabilities, which are key determinants for redox properties and reactivity. In addition, metal-induced histidine deprotonation was also suggested for different metalloenzymes at varied pH.50 Therefore, we have tested the catalytic efficiency of our complex across pH 6, the effective pKa of histidine. Extrapolation from Figure 5d suggests that the catalytic efficiency of the peptide increases with increase in pH. At pH 3, no product formation was detected in the HPLC spectra, but the conversion rate increased to 35% at pH 5. In the case of higher pH (above pH 6), however, the conversion has reached 80%, demonstrating the formation of a tailor-made catalytic active site at a physiological pH. Morphological verification of the catalyst at different pH reveals the gradual transition from the disordered to the more ordered nanoassembly at higher pH (Figure 6).
Figure 6.

FESEM characterization of the assemblies formed by the Cu–His complex at different pH. Scale 10 μm.
Discussion
Mimicking the active site of metalloenzymes in artificial systems is the key to understand the selectivity and efficiency of enzymatic reactions. Specifically, one must design structural sites where the metal can impart thermodynamic stability, direct the protein fold, and overcome nonpreferred metal-binding geometries. Recent advancements in metal–peptide supramolecular chemistry led to the development of artificial macromolecular structures that operate as a geometric template functioning as an electron reservoir.51,52 However, the designed metallopeptides show significant lack of catalytic efficiency relative to their natural counterparts.53 Over the years, the advancements in understanding the balance between protein scaffold stability and metal ion coordination preferences lead to exploring the unnatural d-amino acids.54 The ability to tune the metal-binding affinity of small peptides by incorporating d-amino acids and the preorganization of the peptide structure holds great potential in designing ultra-short catalytic domains derived from the active site of a metalloenzyme.
In the present study, we have designed a tripeptide to mimic the histidine brace motif found in the active site of an industrially important enzyme LPMO. Our goal is to generate a structural mimic through a minimalistic design approach. The otherwise difficult mimicry of the LPMO active site was achieved by incorporating d-histidine at the C-terminal. The modeling studies suggest that copper coordinates with the peptide through 3N coordination, which results in a distorted square-pyramidal geometry, similar to that observed in LPMO active sites.
Interestingly, one can modulate the protonation state of histidine across the physiological pH (pKa ≈ 6–7). The imidazole group of histidine ligands can bind to the Cu ion through either the δN or εN site, and the tautomeric preference varies with pH. The spectroscopic characterization at various pH suggests that the complex formation occurs only at pH 7 or above. Increasing the pH from 3 to 9 resulted in an increase in the intensity of the d–d transition band in the UV and vis-CD spectra. A plausible explanation for this phenomenon is the participation of one amide nitrogen (Nam) atom and two imidazole nitrogen (Nim) atoms to form an equatorial plane coordination, while the fourth coordination site is occupied by the O5 atom of the water molecule. The remaining apical position was occupied by the O4 atom of another water molecule completing the square-pyramidal geometry. Relatively strong CD bands are often observed for the d–d transitions of tetragonal complexes, involving backbone amide and histidine coordinations through the imidazole ring.37 Additionally, it causes a distortion in the tetragonally elongated symmetry, causing large transition and hence the enhanced d–d transition with increasing pH.55,56 The spectral changes in UV–vis and vis-CD spectra between the pH range of 7 and 9 are relatively small, suggesting the formation of a predominant species at pH 7. The coordination of Nam and Nim is also confirmed from the vibrational Raman spectra. The Nim coordination can be extracted from the drop in intensity of the Raman-active N–H deformation bands at 1440 cm–1. Furthermore, the signal corresponding to the Raman C=C or C=N stretching band (1543 cm–1) observed at pH 3 also red-shifts to 1554 cm–1 at pH 7 and 9, suggesting formation of a complex with Nτ coordination.37,42 The presence of the Nτ coordination form is further evidenced by the small bandwidth of the Raman-active ring-breathing vibration at 1278 cm–1.42,57,58 The EPR spectrum of the complex at pH 7 and 9 is typical for a Cu(II) complex with a distorted square-pyramidal geometry.35 Based on the NMR experiments, we can conclude that the predominant species is in the 3N form as it is able to considerably accelerate the Cu(II) exchange (within milliseconds).37,59 Overall, our model Cu–HPh complex can reproduce the physicochemical features of the active site of LPMOs.
Catalytic activity of our model complex was tested for the oxidation of benzyl alcohol in water. While His alone does have very low to no catalytic activity, it is the most important amino acid playing the role of a proton donor or acceptor in a catalytic reaction.60 The results above clearly demonstrate that the binding of a copper ion to the structure is through the creation of multivalency of the histidine residues at pH 7. Based on previous reports,61,62 we postulate that the protonation of one of the imidazole near pH 7 facilitates H-atom transfer in the presence of oxygen. Stahl and co-workers have earlier demonstrated this to be the most energetically favorable route for the oxidation of a Cu-alkoxide.63
Conclusions
The work presented here describes the design, synthesis, and characterization of a minimalistic de novo designed tri-peptide (HPh) as a model for the histidine brace active site of LPMO. The complex is characterized by various spectroscopic techniques, and the catalytic function was tested for the model benzyl alcohol oxidation. The molecular structure of the Cu–HPh complex adopts very similar distorted square-pyramidal coordination geometry, as seen in the LPMO active site. The position of d–d transitions as observed in UV–vis CD and Raman spectroscopies supports the existence of the square-pyramidal geometry of the complex through 3N coordination at pH 7, identical to the histidine brace. The EPR fingerprints suggest the 1B2g ground state with the unpaired electron on the dx2–y2 orbital. The pH dependence is explained by the absence of any complex at pH below 7. The 3N coordination is further validated by the solid-state NMR analysis that shows an accelerated Cu(II) exchange (milliseconds or faster) at pH 7. The heterogeneous Cu–HPh complex showed a high yield of benzaldehyde, up to 82% through selective oxidation of benzyl alcohol in water, in the presence of molecular oxygen at 40 °C. This study presents a minimalistic design strategy using d-amino acid for structural and functional mimicry of the LPMO active site, which can be a stepping stone for generating sustainable and environment-friendly, alternative catalytic solutions.
Experimental Section
Materials
All reagents were of analytical grade and used without further purification. All the amino acids and resin were purchased from Novabiochem (Merck, Germany). Copper chloride dihydrate (CuCl2·2H2O) was procured from Sigma-Aldrich, India. All solutions were prepared in Milli-Q (18 MW) water.
Modeling of the Complex
The [CuII(LH3)(H2O)2]+ complex was modeled and energy-minimized using the MMFF94 force field with a convergence of 10 × 10–7 Avogadro 1.2.0 (http://avogadro.cc/),32 with frequency calculations carried out using Gaussian 09W using the B3LYP/3-21g level of theory.31
Peptide Synthesis
The peptide was synthesized manually by solid-phase peptide synthesis using Fmoc chemistry on a 4-(hydroxymethyl) phenoxyacetic acid (HMPA) resin (0.74 mmol/g, Novabiochem). The peptide was precipitated in cold ether and purified by reverse-phase HPLC (Shimadzu Ltd, Japan) using an analytical C-18 column (Figure S1a). The gradient elution of 10–100% acetonitrile in water with 0.1% tetrafluoroacetic acid (TFA) at 0.5 mL min–1 was used. The HPLC chromatogram was recorded at 210 nm for fraction collection. The molecular weight of the peptide was evaluated using high-resolution (HR) mass spectroscopy (MS) analysis (Figure S1b). The peptide was stored at 4 °C for further use.
Synthesis of the Cu–HPh Complex
Metal to ligand ratios were maintained at 0.9:1(4.5 mM CuCl2/5 mM HPh) to ensure total binding of Cu(II) to the peptide in phosphate buffer. The mixtures were stirred for 30 min at room temperature before UV–vis absorption measurements. The precipitates were filtered through a 0.2 μm filter, washed several times with water to remove excess Cu(II) followed by hexane, and dried in vacuum. The mass of the complex was verified using electrospray ionization (ESI)-MS (Figure S14, Supporting Information).
UV–Vis
UV–vis absorption measurements in the range of pH 3–9 were performed. The Cu/HPh ratio was maintained at 0.9:1 for all the experiments. The spectra were recorded in the 400–800 nm spectral range using a dual-beam spectrometer (Agilent Cary 60) in a quartz cuvette with a 1 cm path length. pH titrations were carried out by adding drops of 1 M NaOH directly to the samples. Solutions were allowed to saturate for at least 5 min before recording the spectra. The extinction coefficient for the absorption at 620 nm is presented in Table S2, Supporting Information.
Circular Dichroism
CD spectra were recorded on a JASCO J-1700 (JASCO) spectropolarimeter at room temperature. Measurements were performed in the 800–190 nm range with a 3 mm path quartz cuvette with a final volume of 0.5 μL. The CD experiments were performed at a low concentration, maintaining the 0.9:1 ratio of Cu and peptide (1.8 mM CuCl2 and 2 mM peptide). The pH titration was performed by adding 1 M NaOH.
Raman Spectroscopy
Raman spectroscopy was performed using a laser micro-Raman system (Horiba Scientific, LabRam HR Evolution Raman Spectrometer) equipped with a charge coupled device (CCD) detector. A 532 nm laser (argon) was used to excite the sample (output power of 16.5 mW). The laser was focused using a 20× objective. Ten microliters of the sample was deposited on a Si window and air-dried before taking measurements. For each sample, 10 accumulations were averaged with an exposure time of 20 s. The spectral range for all Raman spectra is 1750–1200 cm–1.
EPR Analysis
EPR spectra of the sample were recorded at the X-band frequency (9.5 GHz) using a JEOL (model JES FA200) spectrometer. The following parameters were used: modulation frequency, 100.00 kHz; modulation amplitude, 10.00 G; microwave power, 1.002 mW; time constant, 2.560 ms; receivers gain, 1.00 × 103. Ethylene glycol was added as a cryo-protectant to each sample (30% v/v). Samples were freeze-quenched in liquid nitrogen before carrying out the measurements at 77 K.
1H and 13C Solid-State NMR Analyses
Solid-State NMR
1H and 13C solid-state NMR spectra of the HPh peptide and the complex were acquired at 60 kHz magic angle spinning (MAS) frequency on a Bruker 700 MHz Avance-III spectrometer using 1.3 mm rotors and HXY triple resonance probe. For proton decoupling during acquisition, a rCWApA decoupling sequence with an rf amplitude of 12 kHz was used. One pulse proton spectrum was obtained with a pulse length of 2.7 μs. For the complex, 13C spectra were obtained using one pulse (2.5 μs) with 1H heteronuclear decoupling applied during acquisition. The proton spectrum of the complex was acquired with 32 transients and a recycle delay of 0.2 s. For the 13C spectrum of peptide, 20,480 transients with a recycle delay of 0.1 s were collected.
For the peptide, 13C spectra were acquired with one pulse and 1H–13C cross-polarization with a 1 ms contact time and heteronuclear decoupling of protons during the acquisition. The 1H and 13C spectra of the peptide were acquired with 8 transients, 2 s recycle delay and 12,288 transients, and 2 s recycle delay, respectively. 1H spectra were referenced with respect to the HN-Leu peak of the N-formyl-l-methionyl-l-leucyl-l- phenylalanine-OMe (MLF) tripeptide at 9 ppm as an external reference. The 13C spectra were calibrated with reference to the proton spectrum using the gyromagnetic ratio of proton to the carbon nuclei.
Longitudinal relaxation (T1) values for protons of nascent peptide were measured using a saturation recovery experiment. In the saturation recovery experiment, the delay values used were 1, 50, 100, 200, 300, 500, 800 ms, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 9, and 12 s. Each experimental point is obtained with eight transients. Initial saturation of the proton signal is achieved by a train of 50 pulses with a length of 3.15 μs and an inter pulse delay of 8 μs.
Solution-State NMR
Solution-state proton NMR spectra of the peptide and the complex have been acquired in a 90%·H2O + 10% D2O solvent at pH 3 and 7 using a Bruker 300 Avance III spectrometer with a 5 mm H/X probe. Water suppression was carried out using the pre-saturation technique. All the spectra were calibrated with respect to the D2O signal (4.79 ppm). The pulse length for 1H was 14 μs.
FESEM Analysis
The complex formed at pH 7 and 9 was directly loaded on a glass cover slide. Samples at pH below 7 were prepared by drop-casting on a clean coverslip, followed by vacuum drying. Samples were sputtered with platinum in a JEOL JFC-1600 high-resolution sputter coater at 30 mA for 120 s. The surface of interest was examined using a JEOL JSM-7400F field-emission scanning electron microscopy system at an acceleration voltage of 15 kV. SEM–EDS mapping was performed at 2 kV.
FETEM Analysis
The complex was loaded on a carbon-coated copper mesh grid (300 mess) and was negatively stained using 2% (w/v) saturated uranyl acetate. After 30 s, the excess stain was wicked away, and the grids were allowed to air-dry. Images were captured using a JEM-2100F (Joel) field emission transmission electron microscope at 200 kV.
Catalysis
0.5 mmol of benzyl alcohol was added along with different amounts of the peptide complex (10, 15, and 30 mol %, respectively) to a round-bottom flask, capped using an O2 balloon and left to react at 40 °C in phosphate buffer (pH 7) for 3 h. The reaction was stirred continuously using a magnetic stirrer. Aliquots (25 μL) were collected at regular time intervals and filtered using a 0.2 μm filter. The filtrates were then analyzed for product formation using a reverse-phase high-performance liquid chromatograph (Shimadzu Prominence, Shimadzu Ltd, Japan) with a C18 column at 283 nm.
Acknowledgments
This work was supported by the Department of Biotechnology; project number BT/PR25526/NER/95/1238/2017. J.S. thanks CSIR-UGC for financial assistance during his Ph.D. through the NET JRF program. The authors acknowledge the Central Instrumentation Facility, IIT Guwahati and the Center for Nanotechnology, IIT Guwahati for analytical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c07075.
Summary of geometric parameters of the designed complex; HPLC and mass characterization results of the synthesized peptide; CD, EPR, and NMR characterization results of the complex at different pH; and catalytic characterization of the complex, peptide, and Cu(II) at different pH (PDF)
Author Contributions
V.R. conceived and directed the ideas and planned and performed the overall execution. V.T.B. designed the catalysis experiments. J.S. performed the experiments. V.R., V.T.B., and J.S. analyzed and interpreted data and contributed to the editing of the manuscript. L.R.P. and V.A. analyzed and interpreted NMR experiment results. A.S.R. did modeling experiments. J.S. and V.R. analyzed the results and wrote the paper in close collaboration with all the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Kuah E.; Toh S.; Yee J.; Ma Q.; Gao Z. Enzyme mimics: advances and applications. Chem. - Eur. J. 2016, 22, 8404–8430. 10.1002/chem.201504394. [DOI] [PubMed] [Google Scholar]
- Garg M.; Vishwakarma N.; Sharma A. L.; Mizaikoff B.; Singh S. Lysine-Functionalized Tungsten Disulfide Quantum Dots as Artificial Enzyme Mimics for Oxidative Stress Biomarker Sensing. ACS Omega 2020, 5, 1927–1937. 10.1021/acsomega.9b03655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putti M.; Stassen O. M. J. A.; Schotman M. J. G.; Sahlgren C. M.; Dankers P. Y. W. Influence of the assembly state on the functionality of a supramolecular jagged1-mimicking peptide additive. ACS Omega 2019, 4, 8178–8187. 10.1021/acsomega.9b00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa K.; Korabik M.; Kowalik-Jankowska T. Coordination properties of Cu (II) ions towards the peptides based on the His-Xaa-His motif from Fusobacterium nucleatum P1 protein. J. Inorg. Biochem. 2019, 201, 110819. 10.1016/j.jinorgbio.2019.110819. [DOI] [PubMed] [Google Scholar]
- Park G. Y.; Lee J. Y.; Himes R. A.; Thomas G. S.; Blackburn N. J.; Karlin K. D. Copper–peptide complex structure and reactivity when found in conserved his-xaa-his sequences. J. Am. Chem. Soc. 2014, 136, 12532–12535. 10.1021/ja505098v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancs Á.; Selmeczi K.; May N. V.; Gajda T. On the copper (ii) binding of asymmetrically functionalized tripodal peptides: solution equilibrium, structure, and enzyme mimicking. New J. Chem. 2018, 42, 7746–7757. 10.1039/c7nj04716a. [DOI] [Google Scholar]
- Kopera E.; Krȩżel A.; Protas A. M.; Belczyk A.; Bonna A.; Wysłouch-Cieszyńska A.; Poznański J.; Bal W. Inorg. Chem. 2010, 49, 6636–6645. 10.1021/ic1005709. [DOI] [PubMed] [Google Scholar]
- Árus D.; Jancsó A.; Szunyogh D.; Matyuska F.; Nagy N. V.; Hoffmann E.; Körtvélyesi T.; Gajda T. On the possible roles of N-terminal His-rich domains of Cu, Zn SODs of some Gram-negative bacteria. J. Inorg. Biochem. 2012, 106, 10–18. 10.1016/j.jinorgbio.2011.09.029. [DOI] [PubMed] [Google Scholar]
- Jakab N. I.; Jancsó A.; Gajda T.; Gyurcsik B.; Rockenbauer A. Copper (II), nickel (II) and zinc (II) complexes of N-acetyl-His-Pro-His-His-NH2: Equilibria, solution structure and enzyme mimicking. J. Inorg. Biochem. 2008, 102, 1438–1448. 10.1016/j.jinorgbio.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Jancsó A.; Paksi Z.; Jakab N.; Gyurcsik B.; Rockenbauer A.; Gajda T. Solution chemical properties and catecholase-like activity of the copper (II)–Ac-His-His-Gly-His-OH system, a relevant functional model for copper containing oxidases. Dalton Trans. 2005, 3187–3194. 10.1039/b507655b. [DOI] [PubMed] [Google Scholar]
- Timári S.; Cerea R.; Várnagy K. Characterization of CuZnSOD model complexes from a redox point of view: Redox properties of copper (II) complexes of imidazole containing ligands. J. Inorg. Biochem. 2011, 105, 1009–1017. 10.1016/j.jinorgbio.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Prigge S. T.; Kolhekar A. S.; Eipper B. A.; Mains R. E.; Amzel L. M. Amidation of bioactive peptides: the structure of peptidylglycine α-hydroxylating monooxygenase. Science 1997, 278, 1300–1305. 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- Prigge S. T.; Mains R. E.; Eipper B. A.; Amzel L. M. New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cell. Mol. Life Sci. 2000, 57, 1236–1259. 10.1007/pl00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason J. E.; Galaleldeen A.; Peterson R. L.; Taylor A. B.; Holloway S. P.; Waninger-Saroni J.; Cormack B. P.; Cabelli D. E.; Hart P. J.; Culotta V. C. Candida albicans SOD5 represents the prototype of an unprecedented class of Cu-only superoxide dismutases required for pathogen defense. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 5866–5871. 10.1073/pnas.1400137111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse L.; Beher D.; Masters C. L.; Multhaup G. The βA4 amyloid precursor protein binding to copper. FEBS Lett. 1994, 349, 109–116. 10.1016/0014-5793(94)00658-x. [DOI] [PubMed] [Google Scholar]
- Peacock A. F. A.; Hemmingsen L.; Pecoraro V. L. Using diastereopeptides to control metal ion coordination in proteins. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16566–16571. 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionna M.; Styan K. E.; Marchesan S. The unexpected advantages of using D-amino acids for peptide self-assembly into nanostructured hydrogels for medicine. Curr. Top. Med. Chem. 2016, 16, 2009–2018. 10.2174/1568026616999160212120302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan S.; Hazam P. K.; Ramakrishnan V. Symmetry-directed self-organization in peptide nanoassemblies through aromatic π–π interactions. J. Phys. Chem. B 2017, 121, 404–411. 10.1021/acs.jpcb.6b09474. [DOI] [PubMed] [Google Scholar]
- Hazam P. K.; Jerath G.; Chaudhary N.; Ramakrishnan V. Peptido-mimetic approach in the design of syndiotactic antimicrobial peptides. Int. J. Pept. Res. Ther. 2018, 24, 299–307. 10.1007/s10989-017-9615-3. [DOI] [Google Scholar]
- Hazam P. K.; Jerath G.; Kumar A.; Chaudhary N.; Ramakrishnan V. Effect of tacticity-derived topological constraints in bactericidal peptides. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1388–1395. 10.1016/j.bbamem.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Jerath G.; Goyal R.; Trivedi V.; Santhoshkumar T. R.; Ramakrishnan V. Syndiotactic peptides for targeted delivery. Acta Biomater. 2019, 87, 130–139. 10.1016/j.actbio.2019.01.036. [DOI] [PubMed] [Google Scholar]
- Ciano L.; Davies G. J.; Tolman W. B.; Walton P. H. Bracing copper for the catalytic oxidation of C–H bonds. Natl. Catal. 2018, 1, 571–577. 10.1038/s41929-018-0110-9. [DOI] [Google Scholar]
- Arora R.; Bharval P.; Sarswati S.; Sen T. Z.; Yennamalli R. M. Structural dynamics of lytic polysaccharide monoxygenases reveals a highly flexible substrate binding region. J. Mol. Graphics Modell. 2019, 88, 1–10. 10.1016/j.jmgm.2018.12.012. [DOI] [PubMed] [Google Scholar]
- Horn S. J.; Vaaje-Kolstad G.; Westereng B.; Eijsink V. G. Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 2012, 5, 45. 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Span E. A.; Marletta M. A. The framework of polysaccharide monooxygenase structure and chemistry. Curr. Opin. Struct. Biol. 2015, 35, 93–99. 10.1016/j.sbi.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Walton P. H.; Davies G. J. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Curr. Opin. Chem. Biol. 2016, 31, 195–207. 10.1016/j.cbpa.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Kjaergaard C. H.; Qayyum M. F.; Wong S. D.; Xu F.; Hemsworth G. R.; Walton D. J.; Young N. A.; Davies G. J.; Walton P. H.; Johansen K. S.; Hodgson K. O.; Hedman B.; Solomon E. I. Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 8797–8802. 10.1073/pnas.1408115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaaje-Kolstad G.; Forsberg Z.; Loose J. S.; Bissaro B.; Eijsink V. G. Structural diversity of lytic polysaccharide monooxygenases. Curr. Opin. Struct. Biol. 2017, 44, 67–76. 10.1016/j.sbi.2016.12.012. [DOI] [PubMed] [Google Scholar]
- Hoover J. M.; Stahl S. S. Highly practical copper (I)/TEMPO catalyst system for chemoselective aerobic oxidation of primary alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. 10.1021/ja206230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell R.; Garcia-Bosch I.. Synthetic Copper Complexes as Cu-Dependent Monooxygenase Model Systems and Catalysts for Dioxygen Reduction and Water Oxidation, 2021; pp 436–473.
- Frisch M.; Trucks G.; Schlegel H.; Scuseria G.; Robb M.; Cheeseman J.; Scalmani G.; Barone V.; Petersson G.; Nakatsuji H.. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016.
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozłowski H.; Bal W.; Dyba M.; Kowalik-Jankowska T. Specific structure–stability relations in metallopeptides. Coord. Chem. Rev. 1999, 184, 319–346. 10.1016/s0010-8545(98)00261-6. [DOI] [Google Scholar]
- Cai D.; Klinman J. P. Copper amine oxidase: heterologous expression, purification, and characterization of an active enzyme in Saccharomyces cerevisiae. Biochemistry 1994, 33, 7647–7653. 10.1021/bi00190a019. [DOI] [PubMed] [Google Scholar]
- Muthuramalingam S.; Maheshwaran D.; Velusamy M.; Mayilmurugan R. Regioselective oxidative carbon-oxygen bond cleavage catalysed by copper (II) complexes: A relevant model study for lytic polysaccharides monooxygenases activity. J. Catal. 2019, 372, 352–361. 10.1016/j.jcat.2019.03.019. [DOI] [Google Scholar]
- Garajova S.; Mathieu Y.; Beccia M. R.; Bennati-Granier C.; Biaso F.; Fanuel M.; Ropartz D.; Guigliarelli B.; Record E.; Rogniaux H.; Henrissat B.; Berrin J. G. Single-domain flavoenzymes trigger lytic polysaccharide monooxygenases for oxidative degradation of cellulose. Sci. Rep. 2016, 6, 28276. 10.1038/srep28276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez P.; Vileno B.; Bossak K.; El Khoury Y.; Hellwig P.; Bal W.; Hureau C.; Faller P. Cu (II) binding to the peptide Ala-His-His, a chimera of the canonical Cu (II)-binding motifs Xxx-His and Xxx-Zzz-His. Inorg. Chem. 2017, 56, 14870–14879. 10.1021/acs.inorgchem.7b01996. [DOI] [PubMed] [Google Scholar]
- Fawcett T. G.; Bernarducci E. E.; Krogh-Jespersen K.; Schugar H. J. Charge-transfer absorptions of copper (II)-imidazole and copper (II)-imidazolate chromophores. J. Am. Chem. Soc. 1980, 102, 2598–2604. 10.1021/ja00528a013. [DOI] [Google Scholar]
- Myari A.; Malandrinos G.; Deligiannakis Y.; Plakatouras J. C.; Hadjiliadis N.; Nagy Z.; Sòvágó I. Interaction of Cu2+ with His–Val–His and of Zn2+ with His–Val–Gly–Asp, two peptides surrounding metal ions in Cu, Zn-superoxide dismutase enzyme. J. Inorg. Biochem. 2001, 85, 253–261. 10.1016/s0162-0134(01)00204-5. [DOI] [PubMed] [Google Scholar]
- Watly J.; Simonovsky E.; Wieczorek R.; Barbosa N.; Miller Y.; Kozlowski H. Insight into the Coordination and the Binding Sites of Cu2+ by the Histidyl-6-Tag using Experimental and Computational Tools. Inorg. Chem. 2014, 53, 6675–6683. 10.1021/ic500387u. [DOI] [PubMed] [Google Scholar]
- Kowalik-Jankowska T.; Ruta-Dolejsz M.; Wiśniewska K.; Łankiewicz L.; Kozłowski H. Copper (II) complexation by human and mouse fragments (11–16) of β-amyloid peptide. J. Chem. Soc., Dalton Trans. 2000, 24, 4511–4519. 10.1039/b006125p. [DOI] [Google Scholar]
- Itabashi M.; Itoh K. Raman Scattering Study on Coordination Structures of Cu (II)–L-Histidine (1: 2) in Aqueous Solutions. Bull. Chem. Soc. Jpn. 1980, 53, 3131–3137. 10.1246/bcsj.53.3131. [DOI] [Google Scholar]
- Lord R. C.; Yu N.-T. Laser-excited Raman spectroscopy of biomolecules: II. Native ribonuclease and α-chymotrypsin. J. Mol. Struct. 1970, 51, 203–213. 10.1016/0022-2836(70)90137-3. [DOI] [PubMed] [Google Scholar]
- Tasumi M.; Harada I.; Takamatsu T.; Takahashi S. Raman studies of L-histidine and related compounds in aqueous solutions. J. Raman Spectrosc. 1982, 12, 149–151. 10.1002/jrs.1250120212. [DOI] [Google Scholar]
- Hashimoto S.; Ono K.; Takeuchi H. UV resonance Raman scattering from metal-coordinating histidine residues in Cu, Zn-superoxide dismutase. J. Raman Spectrosc. 1998, 29, 969–975. . [DOI] [Google Scholar]
- Hasegawa K.; Ono T.-a.; Noguchi T. Ab initio density functional theory calculations and vibrational analysis of zinc-bound 4-methylimidazole as a model of a histidine ligand in metalloenzymes. J. Phys. Chem. A 2002, 106, 3377–3390. 10.1021/jp012251f. [DOI] [Google Scholar]
- Takeuchi H. Raman structural markers of tryptophan and histidine side chains in proteins. Biopolymers 2003, 72, 305–317. 10.1002/bip.10440. [DOI] [PubMed] [Google Scholar]
- Stanila A.; Marcu A.; Rusu D.; Rusu M.; David L. Spectroscopic studies of some copper (II) complexes with amino acids. J. Mol. Struct. 2007, 834–836, 364–368. 10.1016/j.molstruc.2006.11.048. [DOI] [Google Scholar]
- Marti E. M.; Methivier C.; Dubot P.; Pradier C. M. Adsorption of (S)-histidine on Cu (110) and oxygen-covered Cu (110), a combined Fourier transform reflection absorption infrared spectroscopy and force field calculation study. J. Phys. Chem. B 2003, 107, 10785–10792. 10.1021/jp035355u. [DOI] [Google Scholar]
- Smulevich G.; Mauro J. M.; Fishel L. A.; English A. M.; Kraut J.; Spiro T. G. Heme pocket interactions in cytochrome c peroxidase studied by site-directed mutagenesis and resonance Raman spectroscopy. Biochemistry 1988, 27, 5477–5485. 10.1021/bi00415a014. [DOI] [PubMed] [Google Scholar]
- Escobar S.; Velasco-Lozano S.; Lu C.-H.; Lin Y.-F.; Mesa M.; Bernal C.; López-Gallego F. Understanding the functional properties of bio-inorganic nanoflowers as biocatalysts by deciphering the metal-binding sites of enzymes. J. Mater. Chem. B 2017, 5, 4478–4486. 10.1039/c6tb03295h. [DOI] [PubMed] [Google Scholar]
- Gulseren G.; Khalily M. A.; Tekinay A. B.; Guler M. O. Catalytic supramolecular self-assembled peptide nanostructures for ester hydrolysis. J. Mater. Chem. B 2016, 4, 4605–4611. 10.1039/c6tb00795c. [DOI] [PubMed] [Google Scholar]
- Kaplan J.; DeGrado W. F. De novo design of catalytic proteins. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11566–11570. 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalakshmi R.; Balaram P.. The use of D-amino acids in peptide design, D-Amino Acids. A New Frontier in Amino Acid and Protein Research—Practical Methods and Protocols; Nova Science Publishers Inc, 2007; pp 415–430. [Google Scholar]
- Tishmack P. A.; Bashford D.; Harms E.; Van Etten R. L. Use of 1H NMR Spectroscopy and Computer Simulations To Analyze Histidine p K a Changes in a Protein Tyrosine Phosphatase: Experimental and Theoretical Determination of Electrostatic Properties in a Small Protein. Biochemistry 1997, 36, 11984–11994. 10.1021/bi9712448. [DOI] [PubMed] [Google Scholar]
- Schipper P. E. Use of dispersion-induced circular dichroism (DICD) in spectroscopic assignment: applications to copper (II) complexes. J. Am. Chem. Soc. 1976, 98, 7938–7944. 10.1021/ja00441a009. [DOI] [Google Scholar]
- Mesu J. G.; Visser T.; Soulimani F.; van Faassen E. E.; de Peinder P.; Beale A. M.; Weckhuysen B. M. New insights into the coordination chemistry and molecular structure of copper (II) histidine complexes in aqueous solutions. Inorg. Chem. 2006, 45, 1960–1971. 10.1021/ic051305n. [DOI] [PubMed] [Google Scholar]
- Torreggiani A.; Tamba M.; Bonora S.; Fini G. Raman and IR study on copper binding of histamine. Biopolymers 2003, 72, 290–298. 10.1002/bip.10406. [DOI] [PubMed] [Google Scholar]
- Zoroddu M. A.; Medici S.; Peana M. Copper and nickel binding in multi-histidinic peptide fragments. J. Inorg. Biochem. 2009, 103, 1214–1220. 10.1016/j.jinorgbio.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Duncan K. L.; Ulijn R. V. Short peptides in minimalistic biocatalyst design. Biocatalysis 2015, 1, 67–81. 10.1515/boca-2015-0005. [DOI] [Google Scholar]
- Li S.; Hong M. Protonation, tautomerization, and rotameric structure of histidine: a comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. 10.1021/ja108943n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer G.; Okon M.; McIntosh L. P. pH-dependent random coil 1 H, 13 C, and 15 N chemical shifts of the ionizable amino acids: a guide for protein pK a measurements. J. Biomol. NMR 2014, 60, 109–129. 10.1007/s10858-014-9862-y. [DOI] [PubMed] [Google Scholar]
- McCann S. D.; Stahl S. S. Copper-catalyzed aerobic oxidations of organic molecules: pathways for two-electron oxidation with a four-electron oxidant and a one-electron redox-active catalyst. Acc. Chem. Res. 2015, 48, 1756–1766. 10.1021/acs.accounts.5b00060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.