

Abstract
Saccharomyces cerevisiae is an exceptional genetic system, with genetic crosses facilitated by its ability to be maintained in haploid and diploid forms. Such crosses are straightforward if the mating type/ploidy of the strains is known. Several techniques can determine mating type (or ploidy), but all have limitations. Here, we validate a simple, cheap and robust method to identify S. cerevisiae mating types. When cells of opposite mating type are mixed in liquid media, they ‘creep’ up the culture vessel sides, a phenotype that can be easily detected visually. In contrast, mixtures of the same mating type or with a diploid simply settle out. The phenotype is observable for several days under a range of routine growth conditions and with different media/strains. Microscopy suggests that cell aggregation during mating is responsible for the phenotype. Yeast knockout collection analysis identified 107 genes required for the creeping phenotype, with these being enriched for mating-specific genes. Surprisingly, the RIM101 signaling pathway was strongly represented. We propose that RIM101 signaling regulates aggregation as part of a wider, previously unrecognized role in mating. The simplicity and robustness of this method make it ideal for routine verification of S. cerevisiae mating type, with future studies required to verify its molecular basis.
Keywords: Saccharomyces cerevisiae, mating type assay, RIM101, mating phenotype, sexual aggregation, aggluntinin
Haploid baker's yeast exhibits a ‘creeping’ phenotype when mixed with cells of opposite mating type, forming a robust mating type assay that depends on sexual aggregation via agglutinins and the RIM101 pathway.
Introduction
The ability to maintain Saccharomyces cerevisiae in haploid and diploid forms as well as to perform genetic crosses is fundamental to the success of this yeast as a model organism for molecular genetics (Guthrie and Fink 1991, Botstein and Fink 2011). Saccharomyces cerevisiae has two mating types, a and α. In nature, S. cerevisiae is predominately inbreeding, as it routinely switches mating type to overcome mating type incompatibility (Haber 2012). However, in most laboratory S. cerevisiae strains, mating type is extremely stable and heritable due to deletion of the gene encoding the HO endonuclease that facilitates this mating type switching (Herskowitz and Jensen 1991). Mating type is determined by which of two idiomorphs (termed as such because of differences in length and sequence), MATa or MATα, is present at the MAT locus (Haber 2012). The MATα idiomorph contains two genes that specify the α-specific phenotype, while the MATa idiomorph contains a gene encoding a transcriptional repressor that, in a/α diploids, suppresses expression of haploid-specific genes. Haploid cells respond to the mating type pheromones produced by the opposite mating type, triggering a signaling cascade that eventually results in morphological changes that drive cell fusion and karyogamy (Bardwell 2005).
Mating between compatible S. cerevisiae cells in liquid cultures involves an aggregation process that physically holds cells together. This sexual aggregation is mediated by a- and α-agglutinin proteins that are expressed on the surface of the cell wall of haploid a and α strains, respectively (Banderas et al. 2016). The a-agglutinin is composed of two subunits encoded by the AGA1 and AGA2 genes, while the α-agglutinin is encoded by SAG1 (Lipke and Kurjan 1992). The agglutinin proteins adhere once they come into contact with each other like a sort of molecular Velcro that promotes cell–cell contact (Lipke and Kurjan 1992). Agglutinins are constitutively expressed at low levels, with their expression being upregulated in response to mating type pheromone to facilitate cell aggregation (Lipke and Kurjan 1992). The sexual aggregation mediated by these agglutinins is likely the first step for mating of yeast cells mixed in suspension (Banderas et al. 2016).
Performing genetic crosses in S. cerevisiae relies on knowing the mating types of the haploid strains involved. While the mating type and ploidy of established laboratory strains are generally known, there are a number of scenarios where they need to be determined. This includes when dissecting ascospores to produce haploid strains, when checking the mating of strains that lack convenient markers or when mutating the HO locus in wild-type strains. Currently, several methodologies are used to determine mating type and ploidy. Flow cytometry can be used to determine whether a cell is haploid or diploid, but does not distinguish mating type. A simple method to determine mating type or ploidy is to spread cells following mating on selective medium to look for cells containing markers from both parents, although this obviously requires appropriate markers (Sherman 1991). A PCR method utilizes MAT locus primers that produce different amplicon sizes depending on which mating type idiomorph is present (Bradbury et al. 2006). Microscopic observation of the ‘shmoo’ phenotype, which is a manifestation of morphological changes induced upon detection of the opposite mating type pheromone, is also used to identify mating type (Duntze et al. 1970, Sprague 1991, Merlini et al. 2013) following mixing of known MATa or MATα strains with the unknown strain(s). However, shmoo formation can be transient and is only observed in a small number of cells. Finally, the ‘halo’ method, which relies on inhibition of certain strains by pheromone production, can be used to determine mating type but requires tester strains with specific genotypes (Sprague 1991). Thus, although there is a diversity of methods, they are time-consuming, require specific components, and/or can produce inconsistent or transient results.
Here, we demonstrate a simple, cheap and robust method to enable the rapid identification of the mating type and ploidy of S. cerevisiae strains. The principle of the method is that when cells of opposite mating type are mixed, they ‘creep’ up the sides of the culture vessel in an easily observable manner, unlike mixtures involving the same mating type or a diploid strain, which simply settle out. An unknown strain can be mixed with known strains of each mating type in a culture medium, left overnight and simply observed to allow accurate distinction of both mating types as well as of diploid strains. The response is long-lasting, does not require specialized equipment or reagents, and requires minimal hands-on time. Investigation of the molecular basis for this ‘creeping’ phenotype using microscopy and the yeast knockout collection suggests that it is a consequence of cell aggregation, and indicates a surprising role of the RIM101 pathway in S. cerevisiae mating.
Methods
Yeast strains and standard growth conditions
All strains used in this study are listed in Table S1 (Supporting Information). Culture media (Formedium, Hunstanton UK) used were YPD (1% yeast extract, 2% Bacto Peptone and 2% glucose), YPGlycerol (1% yeast extract, 2% Bacto Peptone and 2% glycerol) and YNB (0.45% yeast nitrogen base with ammonium sulfate, appropriate amino acids and bases, and 2% glucose) (Treco and Lundblad 1993; Table S2, Supporting Information). Strains were stored in 15% glycerol at –80°C until use.
Mating type assay
Strains were revived from glycerol stocks on YPD medium and then grown overnight in appropriate liquid medium (YPD unless stated otherwise) at 30°C. Strains were diluted to OD600nm 0.2 in the same medium and added in equal volumes to a tube (2 mL per strain for a test tube, 250 µL for a microcentrifuge tube or 100 µL for a 96-well plate). Cultures were left with no shaking for ∼18 h at ∼22°C (room temperature) unless otherwise indicated, after which they were observed and photographed. Time-lapse photography was performed using a Nikon D850 camera with a 60-mm macro lens and a polarizing filter. Images were exposed at ISO200, f/16, for 10 s. Photographs were taken every minute from the start of the experiment for 20 h. Microscopic observation of cell aggregation was carried out for mating and nonmating cells using a LEICA ICC50 W microscope and the LAS (Leica Application Suite) EZ software (Leica Microsystems v3.2, Germany). Observations were performed on 10 μL of 200 μL mating and nonmating cell mixtures from a 96-well plate incubated on the bench with no shaking. Each 10 μL sample was gently collected from the bottom of the well where the creeping phenotype is seen and observed hourly from 2 to 7 h following mixing.
Multiplex PCR to confirm mating type assay
Primers MAT-a 5′-CAATGATTAAAATAGCATAGTCGG-3′, MAT-α 5′-CAGCACGGAATATGGGAC-3′ and MAT-R 5′-GGTGCATTTGTCATCCGTC-3′ (Bradbury et al. 2006) were used in a multiplex PCR reaction to amplify mating type-specific PCR products from 2 µL of overnight yeast culture (100 μL in YPD) in a final volume of 25 µL. PCR was performed with the KAPA2G Robust DNA polymerase in GC buffer (Custom Science, Auckland, New Zealand). Cycling parameters were initial incubation of 10 min at 94°C, then 35 cycles of 94°C for 25 s, 55°C for 25 s and 72°C for 90 s. The MATa amplicon (489 bp) and MATα amplicon (466 bp) were visualized on a 3% agarose gel run at 100 V for 4 h in sodium borate buffer (10 mM NaOH and 36 mM boric acid) and stained with ethidium bromide.
α-Factor testing
α-Factor (Zymo Research, Irvine, CA, US) was used at a final concentration of 50 µM following dilution in water. Details of the assays involving α-factor are provided in the legend to Fig. S5 (Supporting Information).
Genome-wide analysis of the mating type assay
The genetic basis for the ‘creeping’ phenotype was investigated via crosses of BY4742 (MATα) with the gene deletion library (MATa; BY4741 as parental strain) in round bottom 96-well plates. First, the gene deletion library (Open Biosystems, Huntsville, AL, US) was incubated overnight at 30°C in 200 µL YPD, and BY4742 was incubated overnight at 30°C in 5 mL YPD. The next day, 80 µL of BY4742 cells (1.26 × 107 cells/mL) were mixed with 120 µL of gene deletion library strains, incubated overnight at 30°C and imaged using a digital camera. Mating and nonmating phenotypes were identified through independent visual inspection by two people.
Phenotype enrichment
Genes required for the ‘creeping’ phenotype were analyzed for phenotype enrichment using YeastEnrichr (Chen et al. 2013, Kuleshov et al. 2016). Functional enrichment for phenotype was evaluated using text-mining associations of each gene with phenotypes reported in publications in PubMed. Significant enrichment was defined for categories with an adjusted P-value, a z-score (expected:observed ratio for each phenotype) and a combined score (the product of the z-score and negative logarithm of the P-value).
Spatial analysis of functional enrichment
Genes required for the ‘creeping’ phenotype were annotated for placement in a functionally annotated genetic interaction network (Costanzo et al. 2016) via spatial analysis of functional enrichment (SAFE; Baryshnikova 2016).
Network analysis
Genes required for the ‘creeping’ phenotype were annotated for function via network- and pathway-based community partitioning as previously described (Busby et al. 2019). Using NetworkAnalyst (Zhou et al. 2019), genes were mapped in a first-order network within an established yeast global interaction network (the STRING interactome; Szklarczyk et al. 2015) with a confidence cutoff of 900 and requirement for experimental evidence. Significant community modules (P < 0.05) were identified with the InfoMap algorithm via a comparison of network flow in a base network and our creeping phenotype network, and pathways enriched in these modules were determined using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Kanehisa and Goto 2000, Kanehisa et al. 2021) analysis (FDR < 0.05).
Analysis of publicly available RNA-seq data
We used the analyzed RNA-seq data in Table S4 (Supporting Information) from Read et al. (2016) to determine the effect of rim101 deletion on gene expression, with 1.5-fold difference used as our threshold for differential expression.
Results and discussion
A quick and reliable assay for determination of mating type in S. cerevisiae
Routine observations in our laboratory showed that S. cerevisiae cells of different mating types settled in non-shaking cultures differently to cells of the same mating type. The manifestation of this mating type phenotype is that cultures of S. cerevisiae cells of opposite mating types consistently ‘creep’ up the sides of the culture vessel when mixed and left overnight on the bench with no shaking (Fig. 1). This ‘creeping’ phenotype is not observed in cultures with a mixture of cells of the same mating type (a or α), or where one or both cultures are diploid—these instead simply settle to the bottom of the vessel (Fig. 1).
Figure 1.

A 50:50 mixture of a + α cells reveals a creeping phenotype. YPD cultures (OD600nm 0.2) were mixed in equal volumes of 2 mL each in 20-mL glass tubes (A), 250 μL each in microcentrifuge tubes (B) and 100 μL each in a round-bottomed 96-well plate (C). Mixtures were allowed to settle on the bench for 18 h and photographed. The creeping phenotype is only observed for the mixtures of opposite mating types (a + α). Mating types a (a) and α (α), and diploid cells (2n) are indicated below the tubes. The phenotype was not observed in flat-bottomed 96-well plates. (D) Time-lapse photographs of an a + α assay at 0, 4, 8 and 12 h show the formation of the creeping phenotype compared with a tight pellet for nonmating cells. For full time lapse over 20 h, see Fig. S1 (Supporting Information), and for a video, see Movie S1 (Supporting Information).
We decided to investigate this observation further using a variety of vessels to determine whether it could form a robust mating type assay. We found that the creeping phenotype is observed in glass test tubes, plastic microfuge tubes and round bottom microwell plates (Fig. 1A–C), demonstrating its scalability and flexibility, although we did not observe the phenotype in flat-bottomed microwell plates. Time-lapse photography revealed that the formation of the creeping phenotype is visible ∼4 h after the mixing of YPD cultures of a and α cells at room temperature, and intensifies between ∼4 and 8 h (Fig. 1D; see Movie S1, Supporting Information, for a complete video of the formation of the creeping phenotype over 20 h). Moreover, the phenotype is visible for several days. These results show that this assay is a simple way to distinguish MATa and MATα strains (formation of the creeping phenotype with one or other of the known mating type tester strains), as well as diploid strains (no creeping phenotype with either known mating type tester strain), and that the assay can be performed in a variety of culture vessel formats.
Determination of conditions compatible with the mating type assay
We next wanted to characterize how robust the mating type phenotype is under different conditions to assess its practicality as a mating type assay. We first tried different media, including rich and synthetic media, and water. The creeping phenotype forms reliably in both rich and synthetic media containing glucose as the carbon source, but is less obvious when glycerol is used and does not manifest in water (Fig. 2A). Next we tested the effects of cell density and partner ratio on the phenotype for cells grown in glucose rich medium. Cell density can be varied from 0.2 to 1.8 (OD600nm) without affecting the formation of the creeping phenotype (Fig. 2B), while increasing the proportion of one partner over 60% impedes the creeping phenotype (Fig. 2C). The assay is symmetric, in that disruption of the creeping phenotype is observed equally regardless of which partner (a or α) is present in excess (Fig. S2, Supporting Information). Interestingly, detection of the creeping phenotype is improved in 96-well plates compared to tubes (compare Fig. S2, Supporting Information, with Fig. 2C). Finally, the creeping phenotype is obvious at temperatures in the optimal growth range for S. cerevisiae (∼22 and 30°C), but is nevertheless still visible when the cultures are incubated at 37°C (Fig. 2D). However, as for cells in water, formation of the creeping phenotype does not occur at 4°C (Fig. 2D).
Figure 2.

Establishing working parameters for the mating assay. Four parameters were varied: medium (A), cell density (B), partner ratio (C) and temperature (D). Strains were diluted to OD600 0.2 in YPD, mixed equally (50:50) and incubated at room temperature (∼22°C) unless otherwise indicated. Mating type a (a) and mating type α (α) cells are indicated below the tubes. All mixtures were photographed at 18 h. (A) Overnight YPD cultures were diluted in the indicated media or water prior to the mating assay. Note that the YNB and water images appear black and white because of medium composition, not photography. (B) The same overnight YPD cultures from panel (A) were diluted to OD600nm 0.2, 0.6, 1.0, 1.4 or 1.8 to perform the assay. (C) Opposite mating type cells were mixed at the different ratios indicated in a final volume of 4 mL (ratios represent α:a; see Fig. S2, Supporting Information, for the reciprocal ratios). (D) Mixtures were incubated at 4°C, room temperature, 30 or 37°C for 18 h. Note that the first panel in (A) and in (B), the last panel in (C) and the second panel in (D) are all the same.
Mating type determination of different strains of S. cerevisiae
To assess how robust the assay is to strain type, 14 S. cerevisiae strains derived from different wild isolates (Cubillos et al. 2009) that include representatives of each of the five major, non-mosaic S. cerevisiae clades described by Liti et al. (2009) were selected (Table S1, Supporting Information). To test whether the assay is able to determine their mating types, each strain was individually mixed with known tester a and α strains using standard conditions (YPD medium, OD600nm 0.2, overnight incubation on the bench without shaking). The length of incubation time necessary to unambiguously visualize the creeping phenotype varied depending on the growth rate of the strain. The mating type determinations made using this assay matched that recorded for each strain, with one exception (Fig. 3A; Fig. S3, Supporting Information). The one exception is illuminating: we found a discrepancy between the mating type determined by us and that expected (strain 1; MATα by our assay but expected to be MATa; Table S1, Supporting Information). To check this strain and validate the mating type determinations, we carried out multiplex PCR on seven strains with primers previously designed to determine the presence of the a or α idiomorph within the active MAT locus (Bradbury et al. 2006). Each strain gave the 489 bp a or 466 bp α band expected from the creeping phenotype assay (Fig. 3B), including the discrepant strain. Therefore, this discrepant strain is MATα as the creeping phenotype suggests, with the discrepancy likely a strain designation error rather than a problem with the assay. Together, these results show that the assay can accurately determine mating type for a variety of S. cerevisiae strains under a variety of conditions.
Figure 3.
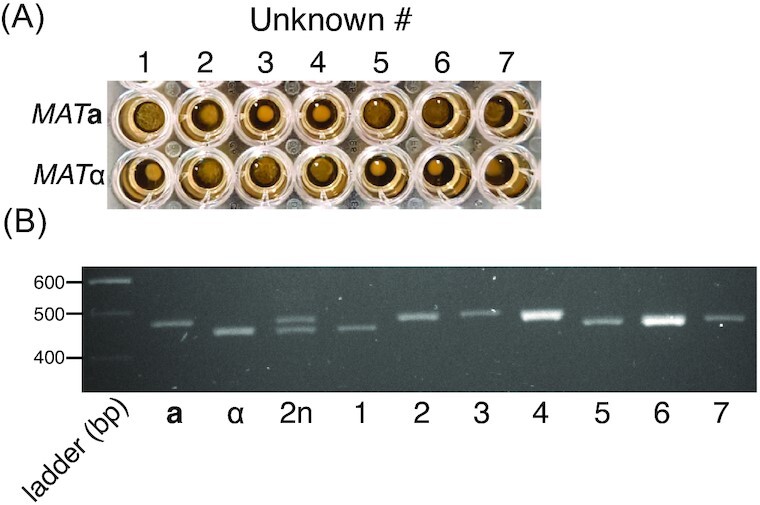
Determination of mating type of wild S. cerevisiae strains.(A) Seven strains from the Mosaic and Wine/European clades identified in Liti et al. (2009) were mixed with MATa and MATα tester strains using standard assay conditions (YPD medium, OD600nm 0.2, room temperature incubation). Mixtures were photographed after 18 h. The creeping phenotype is visible for strains 1, 5, 6 and 7 crossed with MATa tester but not with MATα tester. The opposite result is visible for strains 2, 3 and 4. (B) Mating assay results were confirmed by multiplex PCR carried out on cells from the seven strains and tester a, α and 2n strains. Expected MATa and MATα specific amplicons are 489 and 466 bp, respectively. As expected, both amplicons are observed in the diploid strain. Seven additional strains are shown in Fig. S3 (Supporting Information).
Sexual aggregation is implicated in the creeping phenotype
To shed light on the basis of the creeping phenotype, we made microscopic observations of the cells as the cultures began to creep. These observations show increased aggregation of cells with time when both mating types are present, but not when only one mating type is present (Fig. 4; Fig. S4, Supporting Information). Therefore, these results implicate aggregation as the basis for the creeping phenotype. This is consistent with the nature of the phenotype (cells ‘moving’ up the vessel sides through adherence and growth), the requirement for a medium supporting growth, the poor performance on glycerol (which is not a favored carbon source for S. cerevisiae), the absence of the phenotype at 4°C (sexual aggregation has previously been reported to be cold sensitive; Zhao et al. 2001) and the phenotype becoming evident from ∼4 h (around the time of mating initiation; Duntze et al. 1970, Sena et al. 1973). To determine whether response to the opposite mating type is sufficient to generate the creeping phenotype, we treated cells with α−factor. However, addition of α−factor did not result in the creeping phenotype in MATa cells alone (Fig. S5, Supporting Information), suggesting that mating response (e.g. shmoo formation) is not sufficient to realize the phenotype. Instead, it is likely that growth (which is inhibited by α−factor via cell cycle arrest) and/or the presence of the opposite mating partner is necessary.
Figure 4.
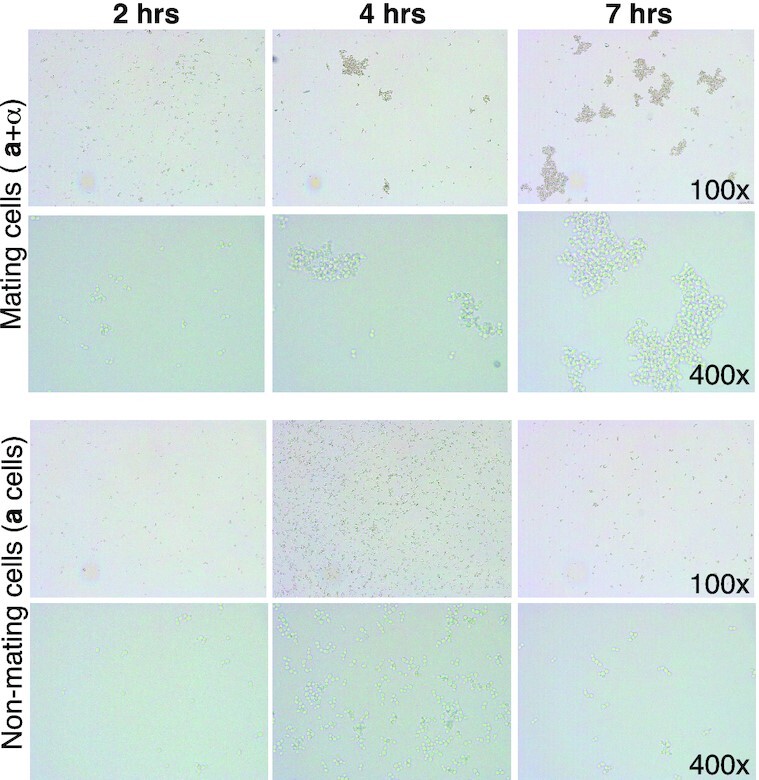
Creeping cells aggregate over time. Mating and nonmating samples of a+ α cells or haploid a cells were observed under the microscope. Images were taken from 2 to 7 h after cells were mixed. Representative images at ×100 and ×400 magnification are shown. Extensive cell aggregation is observed for mating cells from 4 h following mixing, while no appreciable aggregation is observed for nonmating cells over the same time period. See Fig. S4 (Supporting Information) for the full time course.
Functional genomics confirms the creeping phenotype is associated with mating
To gain further insight into the mechanism behind the creeping phenotype, we used a yeast deletion library (Giaever and Nislow 2014) to examine the impact of nonessential gene deletions on the ability to produce the creeping phenotype. To do this, we mated 4294 haploid gene deletion strains in a MATa background with a wild-type MATα strain, performing two biological replicate experiments. From this we identified 107 gene deletion strains that failed to produce the creeping phenotype in either replicate (Table S3, Supporting Information). These 107 genes comprised 84 characterized genes, and 23 uncharacterized open reading frames (ORFs) that encode putative proteins, dubious ORFs or proteins of unknown function.
As expected, the 107 genes required for the creeping phenotype are strongly enriched for genes involved in yeast mating. This conclusion is supported by the results of an unbiased assessment of the genetics underlying the creeping phenotype. This was performed by using YeastEnrichr (Kuleshov et al. 2016) to identify overrepresented phenotypes associated with the 107 genes compared to random gene sets. The top four functions (by adjusted P-value < 0.05) are all involved in mating (mating efficiency, mating response, pheromone production and pheromone sensitivity), as are other highly enriched functions such as nuclear fusion during mating and shmoo formation (Table S4, Supporting Information). Moreover, most of the genes reported to be involved in mating signaling (Merlini et al. 2013) were among our 107 gene set, although none of the genes involved in cell–cell fusion were (Table 1). This enrichment in mating signaling but not cell fusion is consistent with the creeping phenotype resulting from aggregation, which occurs before fusion. Surprisingly given the deletion collection is MATa, STE2 that encodes the receptor for pheromone α (Burkholder and Hartwell 1985) was not among our 107 genes (Table 1). However, it has been reported that strains deleted for STE2 display a low background of mating (Jahng et al. 1988), which might explain the persistence of the creeping phenotype in absence of STE2. Conversely, the inclusion of MFA1 among our 107 genes (Table 1) is surprising considering that MFA1 and MFA2 both encode the a pheromone and were shown to be functionally redundant (Michaelis and Herskowitz 1988). This might suggest that MFA2 is not able to compensate for the absence of MFA1 whereas MFA1 can compensate for the absence of MFA2 to produce the creeping phenotype, which is consistent with MFA2 producing 3-fold less a pheromone than MFA1 (Chen et al. 1997). Finally, consistent with the evidence implicating aggregation, we found that the creeping phenotype was not detectable in the absence of the AGA1 or AGA2 agglutinin genes (Table S3, Supporting Information). Thus, as expected, the creeping phenotype seems to depend on yeast mating genetic pathways and the genetic results are consistent with the involvement of mating aggregation.
Table 1.
Overlap between mating signaling/cell–cell fusion genes and genes required for the creeping phenotype.
| Function | Gene | Note |
|---|---|---|
| Signaling components | ||
| Pheromone a | MFA1 a , MFA2*b | |
| Pheromone α | MF(ALPHA)1 | α-Specific genec |
| MF(ALPHA)2 | α-Specific gene | |
| G-protein coupled receptors | ||
| Receptor for pheromone a | STE3 | α-Specific gene |
| Receptor for pheromone α | STE2* | |
| G-protein α subunit | GPA1 | |
| G-protein β subunit | STE4 | |
| G-protein γ subunit | STE18 | Not in the YKO libraryd |
| PAK kinase | STE20 | |
| MAPK scaffold | STE5 | |
| MAPK scaffold | STE50 | |
| MAPKKK | STE11 | |
| MAPKK | STE7 | |
| MAPK | FUS3, KSS1 | |
| Transcription factor | STE12 | Not in the YKO library |
| Scaffold for shmoo orientation | FAR1 | |
| Cdc42 GTPase | CDC42 | Essential gene |
| Cdc42-GEF | CDC24 | Essential gene |
| Cdc42, Ste5 & Ste20-scaffold | BEM1 | |
| Ras GTPase | RAS1 | |
| Formins | BNI1 , BNR1 | |
| Fusion components | ||
| Transmembrane proteins | PRM1, FIG1, KEX2 | |
| Cell wall remodeling | FUS1, FUS2 | |
| Lipid raft protein | RVS161 | |
| Type V myosin | MYO2 | Not in the YKO library |
| Tropomyosin | TPM1 |
Genes required for the creeping phenotype are shown in bold.
Asterices indicate genes whose deletion unexpectedly does not affect the creeping phenotype; see text for discussion.
α-Specific genes are not expected to affect the creeping phenotype in the MATa gene deletion YKO library context.
Genes that were not present (either missing or essential) in the deletion library are indicated.
Functional enrichment and network analyses implicate RIM101 signaling in the creeping phenotype
To shed more light on the genetic basis for the creeping phenotype, we performed two further enrichment analyses on our set of 107 genes to determine whether they are enriched in specific genetic interaction pathways. SAFE calculates whether any of the 19 previously identified genetic interaction domains in yeast are enriched among a list of provided genes (Baryshnikova 2016, Costanzo et al. 2016), while NetworkAnalyst (Zhou et al. 2019) shows how a list of provided genes maps onto an established global network composed of all genetic interactions from the STRING interactome (Szklarczyk et al. 2015). Application of SAFE to the 107 genes revealed a concentration of genetic interactions in six functional domains: multivesicular body (MVB) sorting/RIM signaling, mitochondria, chromatin, transcription, metabolism and cytokinesis (Fig. 5A). Moreover, NetworkAnalyst classified the 107 genes into an interactive network connecting 65 of the 107 genes with eight statistically significant modules (P < 0.05) representative of subnetworks that are more interactive than expected at random (Fig. 5B). To evaluate overrepresentation of pathways in these modules, an enrichment analysis of interactions for association to the KEGG pathway database was conducted. Five pathways were significantly enriched for five of the modules (Fig. 5B): MAPK signaling (P = 1.9 × 10–20), ribosome (P = 5.4 × 10–7), endocytosis (P = 5.0 × 10–4), longevity regulating (P = 7.3 × 10–3) and oxidative phosphorylation (P = 1.6 × 10–5). These analyses also show enrichment of mating pathways. For example, the MAPK signaling module (Fig. 5B) contains many hallmark mating genes (e.g.STE4, STE5, STE7, STE11, STE20, STE50, GPA1, BNI1, BNR1, BEM1 and RAS1) and is integral to the mating pathway (Bardwell 2005; Table 1). Thus, these modules can provide functional insight into the creeping phenotype.
Figure 5.

Enrichment analyses of genes required for the creeping phenotype.(A) SAFE of the 107 deletion strains required for the creeping phenotype. All 107 genes were mapped in an established genetic interaction network to identify overrepresented regions. The 107 genes show significant membership to 6 of the previously defined 19 functional regions (Baryshnikova 2016, Costanzo et al. 2016), illustrated by dotted ellipses, with the intensity of color proportional to the number of genes. (B) Pathway enrichment for first-order minimum network analysis of the genes required for the creeping phenotype. Sixty-five of the 107 genes appear in this network. Genes required for creeping phenotype are shown as square nodes and interacting genes as circular nodes. The first-order network was generated in NetworkAnalyst (Zhou et al. 2019) with the STRING interactome database where nodes and edges represent genes and genetic/physical interactions, respectively. Node size is based on betweenness score, which is indicative of node importance for overall network connectivity. Nodes are colored by module analysis representing tightly clustered subnetworks with more internal connections than randomly expected in the whole network. Nodes not assigned to a module are without color. The most significantly enriched pathway (lowest P-value < 0.05) in the KEGG database was identified for each module and are listed in the legend; modules without significant enrichment are also listed for clarity.
The strongest and most surprising enrichment in both enrichment analyses was for RIM101 signaling (auto-annotated in NetworkAnalyst as ‘endocytosis’, presumably because the RIM101 pathway contains Endosomal Sorting Complex Required for Transport [ESCRT] proteins; Fig. 5). This enrichment is striking: of the twelve genes in the RIM101 pathway, nine are among our 107 gene set (Fig. 6). Eight of these appear in the NetworkAnalyst analysis (Fig. 5B). The exception is YGR122W, which is required for the creeping phenotype but fell below the NetworkAnalyst protein-protein interaction confidence threshold. IME1 does not show as required for the creeping phenotype, but, although appearing in NetworkAnalyst endocytosis module, is not considered part of the core RIM101 pathway (Lorenz and Cohen 2014; Fig. 6). The remaining two genes, SNF7 and STP22, were not present in the gene deletion library. Thus, all the core RIM101 pathway genes we could assess are required for the creeping phenotype.
Figure 6.
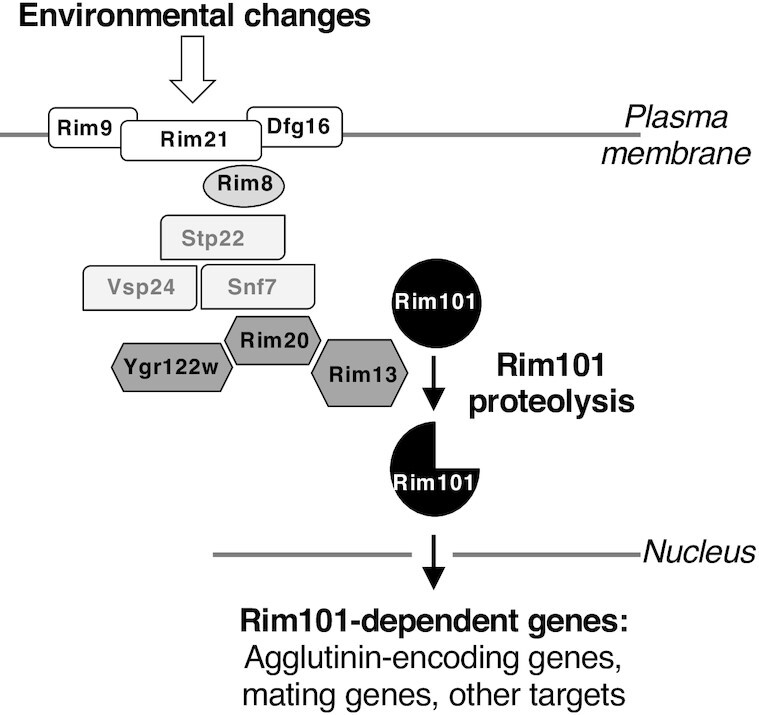
Schematic representation of the RIM101 signaling pathway and its potential involvement in S. cerevisiae mating. The fungal-specific RIM101 signaling pathway is based on the proteolytic processing of the Rim101 transcription factor. The integral membrane proteins Rim21, Dfg6 and Rim9 constitute a surface response complex that recruits the arrestin-like Rim8 at the plasma membrane. Downstream ESCRT proteins Stp22, Vsp24 and Snf7 are recruited in a Rim8-dependent manner to constitute a platform on which the proteolytic complex (Ygr122w, Rim20 and Rim13) forms. The calpain-like cysteine protease Rim13 then cleaves Rim101, inducing its nuclear localization and cellular transcriptional response. We propose that Rim101 targets include genes responsible for the sexual agglutination response among other genes that mediate mating. Apart from SNF7 and STP22, which were not present in our gene deletion library screen, all genes in the RIM101 pathway were found to be required for the creeping phenotype (Fig. 5B; Table S3, Supporting Information). Adapted from Ost et al. (2015).
The RIM101 pathway is a conserved fungal-specific signaling pathway that mediates adaptation to external pH (Penalva and Arst 2004). It is named after the pH-responsive transcription factor Rim101 whose activation by C-terminal proteolytic cleavage is the key output of the pathway (Orejas et al. 1995, Li and Mitchell 1997; Fig. 6). Besides its role in environmental pH response, the pathway is involved in diverse cellular behaviors that include sporulation and haploid invasive growth in S. cerevisiae (Su and Mitchell 1993, Li and Mitchell 1997), salt tolerance in S. cerevisiae and Candida albicans (Lamb and Mitchell 2003), pathogenicity in C. albicans, Aspergillus fumigatus and Cryptococcus neoformans (Davis 2009), and cell wall assembly in S. cerevisiae and C. neoformans (Castrejon et al. 2006, O'Meara et al. 2013, Zhao et al. 2019).
Involvement of the RIM101 pathway in the creeping phenotype of S. cerevisiae mating cells strongly suggests that this signaling pathway also plays an important role in the first step(s) of mating, particularly in mating partner aggregation (Fig. 4; Fig. S4, Supporting Information). We hypothesize that the RIM101 pathway mediates this process by regulating expression of a- and α-agglutinin encoding genes, given Rim101 is a transcription factor. While we are not aware of the RIM101 pathway having previously been proposed to mediate mating in S. cerevisiae, RIM101 pathway mutants have been reported to display mating defects (Castrejon et al. 2006). Direct support for our hypothesis comes from the results showing that deletion of both RIM101 and RIM13 (encoding the protease responsible for Rim101 activation; Fig. 6) results in downregulated expression of AGA2 and MFA1 (which encode the a-agglutinin adhesive subunit and pheromone a, respectively) in a MATa strain (Lamb and Mitchell 2003; Table S5, Supporting Information). In addition, when we examined a publicly available genome-wide RNA-seq dataset where RIM101 was deleted in a MATα strain (Read et al. 2016), we found that the α-agglutinin and pheromone α encoding genes (SAG1, MF(ALPHA)1, MF(ALPHA)2) were significantly downregulated (Table S5, Supporting Information). These data also show downregulation of AGA1, encoding the a-agglutinin anchor subunit, in the MATα context (Table S5, Supporting Information), which is likely an outcome of AGA1 being expressed in both MATa and MATα cells (Lipke and Kurjan 1992). While neither study was conducted under active mating conditions and so may not reflect expression during mating, the fact that these RNA-seq data mirror the impacts on agglutinin and pheromone seen with RIM101 deletion in a MATa strain implies that Rim101 regulates expression of mating aggregation genes in both mating types. Moreover, expression of RIM101 is absolutely required in both partners for mating in the yeast Yarrowia lipolytica (Lambert et al. 1997), although the mating was assessed on solid rather than liquid medium. Finally, Table S5 (Supporting Information) shows that around two-thirds (19 out of 30) of the genes reported to be involved in mating signaling (Merlini et al. 2013) have their expression significantly affected by RIM101 deletion, as do 43 of the 95 genes required for the creeping phenotype. These results suggest a wider involvement of the RIM101 pathway in mating, although the fact that 30 of the 43 RIM101 deletion-affected genes required for the creeping phenotype are upregulated rather than downregulated upon RIM101 deletion shows that this regulation is not simply Rim101 generally activating mating pathway genes. Altogether, our data and existing evidence suggest a novel and direct role for the RIM101 signaling pathway in sexual agglutinin expression as part of a more general role in mating signaling, thereby explaining the involvement of this pathway in the creeping phenotype.
Conclusions
Here, we describe a rapid, inexpensive and robust method to determine not only the mating type of an unknown S. cerevisiae strain but also whether the strain is haploid or diploid. While we serendipitously discovered the creeping phenotype in our laboratory, anecdotally it seems to be used as a mating type assay in some laboratories, but we are not aware of it having been previously published. Thus, here we focused on evaluating and optimizing the phenotype to provide it as an alternative mating type assay for the yeast community. The assay is as simple as mixing an ‘unknown’ sample with known MATa and MATα strains at roughly equal cell densities and observing the results after 18 h; thus, it is cheap and robust, and requires minimal hands-on time. So far, all S. cerevisiae strains we have tried in our laboratory are amenable to the assay, but we do not know whether it will work for all S. cerevisiae strains or for other Saccharomyces species.
Our microscopy data suggest that the phenotype results from cell aggregation that occurs during yeast mating. This conclusion is supported by the timing of the creeping phenotype, which occurs around the start of mating following mixing of compatible strains and is concomitant with visible signs of aggregation. It is also consistent with the requirement for conditions facilitating growth, and in particular with the longevity of the phenotype—well past when mating is likely to have completed. Moreover, demonstration that the creeping phenotype depends on many characterized mating genes, and in particular on the AGA1 and AGA2 agglutinin genes, is also consistent with aggregation underlying the phenotype. The aggregation appears to allow cells to position themselves for long enough in close proximity so that cell–cell recognition pathways are able to facilitate cell fusion (Banderas et al. 2016).
Surprisingly, our functional genomics screen identified the RIM101 pathway as being necessary for the creeping phenotype. All components of this pathway we could assess (all three surface response complex genes RIM9, RIM21 and DFG16; two of the four of Rim8-dependent platform component genes (RIM8 and VSP24); and all three proteolytic complex genes RIM13, RIM20 and YGR122w) are required for the creeping phenotype. The gene encoding the transcription factor Rim101 is also required, proteolytic activation of which is the key output of the pathway. We suggest that Rim101 activates the agglutinin genes; thus, dependence of the creeping phenotype on the RIM101 pathway can be explained simply by a decrease of the expression of these genes in the absence of Rim101 activation. Our examination of published data supports this contention, and a more general, previously unrecognized role for RIM101 signaling in S. cerevisiae mating, albeit one that appears to involve both upregulation and downregulation of different mating genes by Rim101. While Rim101-dependent regulation of the agglutinin genes can explain the dependence of the creeping phenotype on the RIM101 pathway, the trigger that leads to Rim101 activation during mating is unknown. Thus, future work will be required to confirm whether Rim101 directly regulates the agglutinin genes and other mating genes, and how the RIM101 pathway is activated during mating. The simplicity of assaying the creeping phenotype will aid these future approaches, as well as further investigations into the phenotype.
Acknowledgments
We thank Ganley Lab members for comments on the manuscript.
Supplementary Material
Contributor Information
Samantha D M Arras, School of Biological Sciences, University of Auckland, Auckland 1142, New Zealand.
Taylor R Hibbard, School of Biological Sciences, Victoria University of Wellington, Wellington 6012, New Zealand.
Lucy Mitsugi-McHattie, School of Biological Sciences, University of Auckland, Auckland 1142, New Zealand.
Matthew A Woods, Digital Life Institute, University of Auckland, Auckland 0632, New Zealand.
Charlotte E Johnson, School of Biological Sciences, University of Auckland, Auckland 1142, New Zealand.
Andrew Munkacsi, School of Biological Sciences, Victoria University of Wellington, Wellington 6012, New Zealand.
Sylvie Hermann-Le Denmat, School of Biological Sciences, University of Auckland, Auckland 1142, New Zealand.
Austen R D Ganley, School of Biological Sciences, University of Auckland, Auckland 1142, New Zealand; Digital Life Institute, University of Auckland, Auckland 0632, New Zealand; Institute of Natural and Mathematical Sciences, Massey University, Auckland 0632, New Zealand.
Funding
This work was supported by a grant from the New Zealand Marsden Fund (10-MAU-072) to Austen R. D. Ganley.
Conflict of interest statement. None declared.
References
- Banderas A, Koltai M, Anders Aet al. . Sensory input attenuation allows predictive sexual response in yeast. Nat Commun. 2016;7:12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2005;26:339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A. Systematic functional annotation and visualization of biological networks. Cell Syst. 2016;2:412–21. [DOI] [PubMed] [Google Scholar]
- Botstein D, Fink GR. Yeast: an experimental organism for 21st century biology. Genetics. 2011;189:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury JE, Richards KD, Niederer HAet al. . A homozygous diploid subset of commercial wine yeast strains. Antonie Van Leeuwenhoek. 2006;89:27–37. [DOI] [PubMed] [Google Scholar]
- Burkholder AC, Hartwell LH. The yeast alpha-factor receptor: structural properties deduced from the sequence of the STE2 gene. Nucleic Acids Res. 1985;13:8463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby BP, Niktab E, Roberts CAet al. . Genetic interaction networks mediate individual statin drug response in Saccharomycescerevisiae. NPJ Syst Biol Appl. 2019;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrejon F, Gomez A, Sanz Met al. . The RIM101 pathway contributes to yeast cell wall assembly and its function becomes essential in the absence of mitogen-activated protein kinase Slt2p. Eukaryot Cell. 2006;5:507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Yet al. . Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Sapperstein SK, Choi JDet al. . Biogenesis of the Saccharomycescerevisiae mating pheromone a-factor. J Cell Biol. 1997;136:251–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch ENet al. . A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353:aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos FA, Louis EJ, Liti G. Generation of a large set of genetically tractable haploid and diploid Saccharomyces strains. FEMS Yeast Res. 2009;9:1217–25. [DOI] [PubMed] [Google Scholar]
- Davis DA. How human pathogenic fungi sense and adapt to pH: the link to virulence. Curr Opin Microbiol. 2009;12:365–70. [DOI] [PubMed] [Google Scholar]
- Duntze W, MacKay V, Manney TR. Saccharomyces cerevisiae: a diffusible sex factor. Science. 1970;168:1472–3. [DOI] [PubMed] [Google Scholar]
- Giaever G, Nislow C. The yeast deletion collection: a decade of functional genomics. Genetics. 2014;197:451–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular Biology. Vol. 194. San Diego, CA: Academic Press, 1991. [Google Scholar]
- Haber JE. Mating-type genes and MAT switching in Saccharomycescerevisiae. Genetics. 2012;191:33–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I, Jensen RE. Putting the HO gene to work: practical uses for mating-type switching. Methods Enzymol. 1991;194:132–46. [DOI] [PubMed] [Google Scholar]
- Jahng KY, Ferguson J, Reed SI. Mutations in a gene encoding the alpha subunit of a Saccharomycescerevisiae G protein indicate a role in mating pheromone signaling. Mol Cell Biol. 1988;8:2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Sato Yet al. . KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard ADet al. . Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TM, Mitchell AP. The transcription factor Rim101p governs ion tolerance and cell differentiation by direct repression of the regulatory genes NRG1 and SMP1 in Saccharomycescerevisiae. Mol Cell Biol. 2003;23:677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M, Blanchin-Roland S, Le Louedec Fet al. . Genetic analysis of regulatory mutants affecting synthesis of extracellular proteinases in the yeast Yarrowialipolytica: identification of a RIM101/pacC homolog. Mol Cell Biol. 1997;17:3966–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Mitchell AP. Proteolytic activation of Rim1p, a positive regulator of yeast sporulation and invasive growth. Genetics. 1997;145:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Kurjan J. Sexual agglutination in budding yeasts: structure, function, and regulation of adhesion glycoproteins. Microbiol Rev. 1992;56:180–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AMet al. . Population genomics of domestic and wild yeasts. Nature. 2009;458:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz K, Cohen BA. Causal variation in yeast sporulation tends to reside in a pathway bottleneck. PLoS Genet. 2014;10:e1004634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini L, Dudin O, Martin SG. Mate and fuse: how yeast cells do it. Open Biol. 2013;3:130008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomycescerevisiae is essential for mating. Mol Cell Biol. 1988;8:1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara TR, Holmer SM, Selvig Ket al. . Cryptococcus neoformans Rim101 is associated with cell wall remodeling and evasion of the host immune responses. mBio. 2013;4:e00522–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orejas M, Espeso EA, Tilburn Jet al. . Activation of the Aspergillus PacC transcription factor in response to alkaline ambient pH requires proteolysis of the carboxy-terminal moiety. Genes Dev. 1995;9:1622–32. [DOI] [PubMed] [Google Scholar]
- Ost KS, O'Meara TR, Huda Net al. . The Cryptococcusneoformans alkaline response pathway: identification of a novel Rim pathway activator. PLoS Genet. 2015;11:e1005159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penalva MA, Arst HN. Recent advances in the characterization of ambient pH regulation of gene expression in filamentous fungi and yeasts. Annu Rev Microbiol. 2004;58:425–51. [DOI] [PubMed] [Google Scholar]
- Read T, Richmond PA, Dowell RD. A trans-acting variant within the transcription factor RIM101 interacts with genetic background to determine its regulatory capacity. PLoS Genet. 2016;12:e1005746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena EP, Radin DN, Fogel S. Synchronous mating in yeast. Proc Natl Acad Sci USA. 1973;70:1373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. [DOI] [PubMed] [Google Scholar]
- Sprague GFJ. Assay of yeast mating reaction. Methods Enzymol. 1991;194:77–93. [DOI] [PubMed] [Google Scholar]
- Su SS, Mitchell AP. Identification of functionally related genes that stimulate early meiotic gene expression in yeast. Genetics. 1993;133:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder Set al. . STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treco DA, Lundblad V. Preparation of yeast media. Curr Protoc Mol Biol. 1993;23:13.11.11–7. [DOI] [PubMed] [Google Scholar]
- Zhao F, Li J, Lin Ket al. . Genome-wide screening of Saccharomycescerevisiae deletion mutants reveals cellular processes required for tolerance to the cell wall antagonist calcofluor white. Biochem Biophys Res Commun. 2019;518:1–6. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shen ZM, Kahn PCet al. . Interaction of alpha-agglutinin and a-agglutinin, Saccharomycescerevisiae sexual cell adhesion molecules. J Bacteriol. 2001;183:2874–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Soufan O, Ewald Jet al. . NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.