

Abstract
Stroke and ischemic heart diseases are among the most common causes of death and disability throughout the world. Even more worrisome is the suggestion that stroke rates may further increase in certain developing nations. The purpose of this article is to review the particular subtype of stroke known as cardioembolic stroke. A cardioembolic stroke occurs when the heart pumps unwanted materials into the brain circulation, resulting in the occlusion of a brain blood vessel and damage to the brain tissue. The etiology, clinical manifestations, diagnosis and management of cardioembolic stroke are reviewed.
Keywords: Cardioembolic, diagnosis, etiology, ischemic, management, stroke
Introduction
Stroke and ischemic heart disease are among the most common causes of death and disability in the world.[1,2] Even more worrisome is the suggestion that stroke rates may actually be increasing in certain developing nations. For example, when the stroke rates vary according to the study, there has been a suggested escalation in the prevalence and incidence of stroke in India over the last 30 years.[3]
The frequency of the specific causes of ischemic stroke differs around the world, as well.[4] The purpose of this review is to explore the subtype of stroke known as cardioembolic stroke. A cardioembolic stroke occurs when the heart pumps unwanted materials into the brain circulation, resulting in the occlusion of a brain blood vessel and damage to the brain tissue. The etiology, clinical manifestations, diagnosis and management of cardioembolic stroke will be discussed.
Etiology
Diagnostic criteria for cardioembolic stroke were previously very strict. In the past, cardiogenic cerebral embolism was diagnosed only when sudden focal neurologic signs, maximal at onset, developed in patients with peripheral systemic embolism and recent myocardial infarction or rheumatic mitral stenosis.[5] With these criteria, cardioembolic infarcts were diagnosed in 3-8% of stroke patients. [6,7,8,9] However, in various current stroke registries, approximately 10-20% of patients diagnosed with cardioembolic strokes did not have maximal symptoms at the onset of their stroke.[9,10,11] In addition, certain cardiac arrhythmias, presently the well-accepted sources of embolic stroke, were not included in the old diagnostic criteria. Finally, another problem with the prior criteria is that only approximately 2% of patients with cardiogenic brain embolism have clinically recognized peripheral emboli. While necropsy studies of patients with brain embolism note that infarcts are often found in the spleen, kidneys and other organs, the symptoms of peripheral embolism are typically very minor and nonspecific (e.g., transient abdominal discomfort, and leg cramp) that they are rarely diagnosed correctly.[12]
As a result, the criteria for the diagnosis of cardiac embolism remain controversial even today. Cardiogenic cerebral embolism is now thought to be responsible for an estimated 20% of ischemic strokes with potentially even higher rates in developing countries.[4,11,12,13,14,15,16,17] As more advanced diagnostic techniques have been developed, additional causative cardiac abnormalities (and their association with stroke) have been recognized.[12]
Causes of cardioembolic strokes can be divided into three basic groups:[5,12] (1)cardiac wall and chamber abnormalities -cardiomyopathies, hypokinetic and akinetic ventricular regions after myocardial infarction, atrial septal aneurysms, ventricular aneurysms, atrial myxomas, papillary fibroelastomas and other tumors, septal defects and patent foramen ovale; (2)valve disorders -rheumatic mitral and aortic valve disease, prosthetic valves, bacterial endocarditis, fibrous and fibrinous endocardial lesions, mitral valve prolapse and mitral annulus calcification; and (3)arrhythmias,particularly atrial fibrillation and "sick-sinus" syndrome.
Some cardiac sources of stroke have a considerably higher risk of initial and recurrent embolism than other cardiac causes. Accordingly, the Stroke Data Bank[18] divided the potential cardiac causes of stroke into strong sources (prosthetic valves, atrial fibrillation, sick-sinus syndrome, ventricular aneurysm, akinetic segments, mural thrombi, cardiomyopathy and diffuse ventricular hypokinesia) andweak sources(myocardial infarct in earlier months, aortic and mitral stenosis, aortic and mitral regurgitation, congestive heart failure, mitral valve prolapse, mitral annulus calcification and hypokinetic ventricular segments).
The risk of brain embolism also varies even within individual cardiac abnormalities according to a various other factors. For example, in patients with atrial fibrillation, associated heart disease, age, duration of arrhythmia, chronicvsintermittent fibrillation and atrial size all influence embolic risk. It is also important to remember that the presence of a possible cardiac source of embolism does not necessarily mean that the stroke was caused by an embolus from the heart. Coexisting atherosclerotic cerebrovascular disease is a common cause of stroke in these patients as well.[12,16]
Atrial fibrillation (persistent and paroxysmal) is a potent predictor of first and recurrent stroke. In more developed nations, patients with cardioembolic stroke have a history of nonvalvular atrial fibrillation in roughly half of the cases, a history of left ventricular thrombus in almost one-third of the cases and a history of valvular heart disease in one-fourth of the cases.[11,12,19]
Intracavitary thrombusdue toacute myocardial infarction(MI) occurs in approximately one-third of patients within the first two weeks after anterior MI and in an even a larger proportion of those with large left ventricular apex infarcts.[13,19] Patients with chronic ventricular dysfunction due to coronary disease, hypertension and dilated cardiomyopathy can also develop ventricular thrombi. Stroke is less common among uncomplicated MI patients, but may occur in up to 12% of patients with acute MI complicated by a left ventricular thrombus [Figure 1]. The rate of stroke is higher in those patients with anterior rather than inferior infarcts and may reach up to 20% in those with large anteroseptal MI. Whether the increased stroke risk in these anterior wall MI patients is due to increased aneurysm formation, left ventricular dysfunction or other cause remains unclear. Regardless, the incidence of embolism appears to be highest during the period of active thrombus formation in the first 1-3 months, with substantial risk remaining even beyond the acute phase in patients who have persistent myocardial dysfunction, congestive heart failure or atrial fibrillation.[12,19,20]
Figure 1.
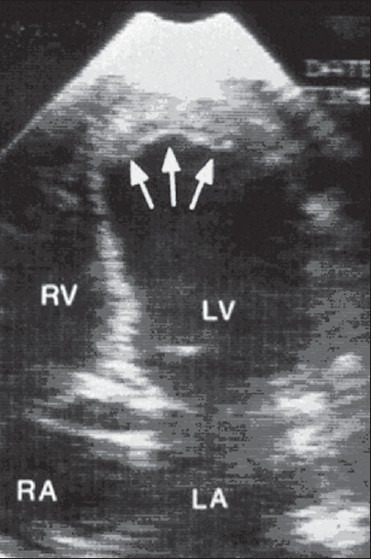
Transthoracic echocardiography with left ventricular thrombus (arrows)
Congestive heart failureaf fects greater than four million Americans and increases stroke risk by a factor of 2-3, accounting for roughly 10% of ischemic strokes in the United States and other industrialized nations.[14,19] In patients with nonischemic dilated cardiomyopathy, the rate of stroke is similar to that of cardiomyopathy due to ischemic heart disease. An estimated 72,000 initial strokes annually have been associated with left ventricular systolic dysfunction and the 5 year recurrent stroke rate in patients with cardiac failure has been reported as high as 45%.[12,19,21]
Valvular heart disease
Rheumatic mitral valve disease. Although the incidence of rheumatic fever and rheumatic heart disease has dramatically declined, rheumatic heart disease is still a very important cause of brain embolism, particularly in developing countries such as Mexico, India and Iran.[4,17,22,23] The mitral valve is most often involved. Second in frequency is the dual involvement of the mitral and aortic valves, while isolated rheumatic aortic valve disease is unusual. Pulmonic or tricuspid valve involvement occurs rarely in rheumatic heart disease.[9]
Recurrent embolism occurs in 30-60% of patients with rheumatic mitral valve disease and a history of a previous embolic event.[19,24,25,26,27] Sixty to sixty-five percent of these recurrences develop during the first year and many occur within the first 6 months.[19,24,25] Rheumatic mitral stenosis is a more frequent cause of brain embolism than is mitral regurgitation: among individuals with embolism in one series, 93% had mitral stenosis, while only 7% had mitral insufficiency.[9,28] Although embolism does occur in patients with mitral stenosis who have normal sinus rhythm, the development of atrial fibrillation greatly increases the risk of embolism.[9] Similarly, the incidence of silent brain infarction - asymptomatic cerebral infarcts detected with neuroimaging - was found to be 24.5% in patients with rheumatic mitral stenosis. The presence of left atrial enlargement and atrial fibrillation also increased the incidence of silent stroke in patients with rheumatic mitral stenosis.[29] Mitral valvuloplasty does not appear to significantly eliminate the risk of embolism.[19,30,31]
Mitral valve prolapse (MVP) is the most common form of valve disease in adults and is generally benign.[32,33] MVP as a source of embolic stroke is still controversial.[5] That being said, several small clinical series have reported cerebral embolism in MVP patients who lacked other possible embolic sources. [34,35,36,37] Occasional patients with MVP have thrombi attached to the valve leaflets of their myxomatous valves. It is important to remember, however, that patients with MVP also may have other conditions such as atrial fibrillation and migraine that in turn can potentially elevate stroke risk. The rate of recurrent stroke in patients with MVP as the lone cause is very low.[36,37] Given the very high incidence of MVP, the frequency of solely MVP-related stroke is exceptionally low.[12,37]
Mitral annulus calcification (MAC) is an important, often under-recognized, cause of embolism. Several series show a convincing relationship between MAC and brain emboli and stroke.[5,38,39,40] Bacterial endocarditis can develop on the MAC, increasing the cardioembolic stroke risk. While antibiotics may help reduce risk of future embolism in the case of endocarditis, anticoagulation does not prevent calcific emboli. The decision to use antiplatelet agentsvsanticoagulants in patients with MAC should include the consideration of other potential comorbid factors such as: atrial fibrillation (that can occur 12 times more often in patients with MAC in comparison to those without MAC) or endocarditis.[12,33,41]
Isolated aortic valve disease is not typically associated with systemic embolism. While there are rare case reports of patients with strokes from spontaneous aortic valve calcific emboli, only a few studies have analyzed series of patients with stroke and aortic valve disease.[19,42] One prospective analysis of 815 patients with calcification of the aortic valve (with or without stenosis) showed no association between either of the two aortic valvular lesions and stroke.[43] As a result, current treatment recommendations in these cases tend to be based on larger antiplatelet trials of stroke and Transient Ischemic Attack (TIA) patients.[12,19]
Paradoxical embolism
Thus far, the most common potential intracardiac shunt is a residual patent foramen ovale (PFO). The high frequency of PFOs in the normal adult population has made it difficult for physicians to be certain in an individual stroke patient with a PFO whether (1) a paradoxical embolism through the PFO was the cause of their stroke or (2) the PFO itself was merely an incidental finding observed during stroke work-up. Autopsy series have shown that up to 30% of adults have a probe patent foramen ovale at necropsy.[44] Interestingly, despite this observation, echocardiographic studies have shown that PFOs are more common in patients with an undetermined cause of stroke than in those in whom another etiology has been determined. [45,46,47,48]
Neuroimaging studies are not conclusive with regard to the link between patent foramen ovale and embolic stroke. However, in 1998, Steiner et al. reported on a series of 95 patients with first stroke who had PFOs.[12,49] Those with large PFOs had more features of embolic strokes with brain imaging in comparison to the patients with small PFOs.
The review of a series of patients with paradoxical embolism[50,51,52] through a PFO as well as the experience of the author allows the derivation of five criteria that when four or more are met, establish the presence of paradoxical embolism-related cerebral infarct with a high degree of certainty.[5] The findings are (1) a situation that promotes thrombosis of leg or pelvic veins (e.g., sitting for long in one position such as a prolonged car trip, airplane flight, being bedridden or recent surgery); (2) increased coagulability (e.g., dehydration, the use of oral contraceptives and presence of V Leiden factor); (3) activity that includes a valsalva maneuver or that promotes right-to-left shunting of blood (the sudden onset of stroke during sexual intercourse, coughing, straining at stool or weight lifting) (4) pulmonary embolism within a short time before or after the neurologic ischemic event and (5) the absence of other clear causes of stroke after thorough evaluation.[12]
Fibrous and fibrinous lesions of the heart valves and endocardium are associated with particular medical conditions.[5] These valvular lesions traditionally occur in patients with systemic lupus erythematosus (Libman-Sacks endocarditis[53] ), antiphospholipid antibody syndrome[54] and other debilitating diseases such as cancer (nonbacterial thrombotic endocarditis). Mobile fibrous strands are also often found during echocardiography.[5,12,55,56,57] Fibrin-platelet aggregates may attach to these fibrous and fibrinous lesions.
Infective Endocarditis commonly causes embolic complications in patients .[5,58] Mycotic aneurysms can result in fatal subarachnoid bleeding. Bleeding can also result due to vascular necrosis as a result of an infected embolus.[58] Embolization usually stops once the infection is controlled.[9,55] Warfarin does not prevent embolization in these cases and is probably contraindicated unless there are other important lesions such as prosthetic valves or life-threatening pulmonary embolism. In children and young adults with congenital heart defects, particularly those with right-to-left shunts and polycythemia, brain abscess is an important complication.[12]
Cardiogenic embolism in underdiagnosed. Clinical features and brain investigations such as computed tomography (CT), magnetic resonance imaging (MRI) and angiography (CT, MR and digital subtraction angiography) may suggest emboli, but often a clear source is not identified. These cases that are termed infarcts of unknown causes (IUC) in the Stroke Data Bank include as many as 40% of patients.[10,12,59,60]
Clinical Findings and Course
Strokes can have a wide range of different clinical manifestations. Warning signs of stroke may include sudden hemiparesis, hemisensory loss, confusion, trouble speaking, difficulty understanding, visual loss, diplopia, ataxia, vertigo or even sudden severe headache with no known cause. Most embolic events occur during typical activities of daily living but some embolic strokes start during rest or sleep. Sudden coughing, sneezing or rising at night to urinate are also activities that are known to precipitate embolism.[5,9] Although the deficit from a cardioembolic stroke is typically maximal at outset, 11% of these patients in the Harvard Stroke Registry had a stuttering or stepwise course, whereas 10% had fluctuations or progressive deficits. Later progression, if it occurs, often develops within the first 48 h. When progression occurs, it is usually due to distal passage of emboli.[12]
In recent years, a high incidence of acute MCA blockage in patients with sudden-onset hemispheric strokes has been shown via transcranial Doppler (TCD) sonography. Eventually, the recanalization of the MCA and normalization of the intracranial blood velocities do occur.[5,61] With regard to any large infarct, cerebral edema may develop during the 24 to 72 h after stroke, manifested typically by headache, diminished alertness and a worsening of neurologic symptoms. The edema from an ischemic stroke is traditionally cytotoxic (with swelling occurring inside brain cells), and thus, it unfortunately does not tend to respond to corticosteroid therapy.[12]
Diagnostic Testing
Emboli mainly occlude distal arterial branches within the brain, resulting in surface infarcts that appear triangular in shape with the base of the triangle at the brain's surface and the apex pointing inward. The presence of embolism is suggested on CT or MRI by the location and shape of the lesion,[62] the presence of superficial wedge-shaped infarcts in multiple different vascular territories, hemorrhagic infarction and visualization of thrombi within arteries [Figure 2]. Among 60 patients with cardiogenic sources of embolism studied by CT in whom occlusive atherosclerotic cerebrovascular disease had been excluded, 56 had the above mentioned, superficial, large or small cortical or subcortical infarcts and only 4 had deep infarcts.[62] Thus, although embolic infarcts are typically triangular, it is possible for emboli on occasion to block the MCA and cause a lone deep infarct if the superficial territory has good collateral flow. Tiny emboli may cause small deep or superficial infarcts.[5,9,12,62,63]
Figure 2.
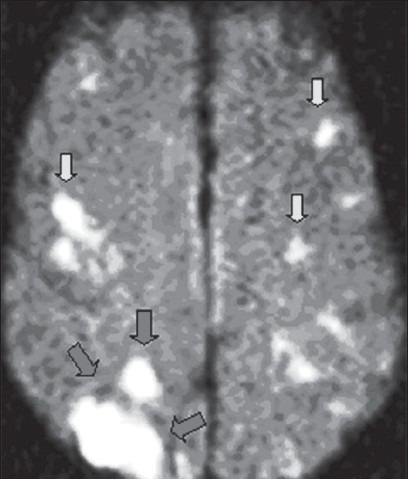
Positive diffusion-weighted imaging MRI scan of the brain that shows a cardiogenic shower of emboli. Typical wedge-shaped infarct (area outlined by large arrows) as well as other bilateral hemispheric smaller infarcts (small arrows)
Magnetic resonance imaging is more sensitive for the detection of acute brain infarcts in comparison to CT, particularly with the use of MR diffusion-weighted and MR gradient-recalled echo (GRE) imaging and it is also superior in detecting hemorrhagic infarction by imaging hemosiderin. For a long time, hemorrhagic infarction has been considered as the characteristic of embolism, particularly when the artery leading to the infarct is patent.[64] Hemorrhagic infarction results due to the reperfusion of ischemic zones, which can occur with either spontaneous passage of the embolus, iatrogenic opening of an occluded artery (e.g., endarterectomy, fibrinolytic treatment) or with restoration of the circulation after a period of systemic hypoperfusion. Typically, hemorrhage occurs into proximal reperfused regions of brain infarcts.[5,9,65] CT can image subacute infarcts well and at times, it is also possible to image the acute embolus itself on CT.[5,12,66,67]
Transthoracic echocardiography (TTE) has been variably useful in detecting sources of embolism in stroke patients.[5,68,69,70] This technique is certainly useful in patients with known heart diseases to clarify the potential embolic sources and cardiac function[9] in young patients without known stroke risk factors and in stroke patients who do not have lacunar infarction or ultrasound evidence of intrinsic atherostenosis of a major extracranial and intracranial artery. Transesophageal echocardiography (TEE) provides much better visualization of the atria, cardiac valves, septal regions and aorta. This technique reports suggest that its diagnostic yield is 2-10 times that of TTE.[71,72,73,74] Further, TEE clearly shows the aortic plaques, atrial septal aneurysms and atrial septal defects [Figure 3]. Utilizing an echo-enhancing agent (such as agitated saline) while performing a TTE or TEE can also help reveal an intracardiac shunt.[12]
Figure 3.
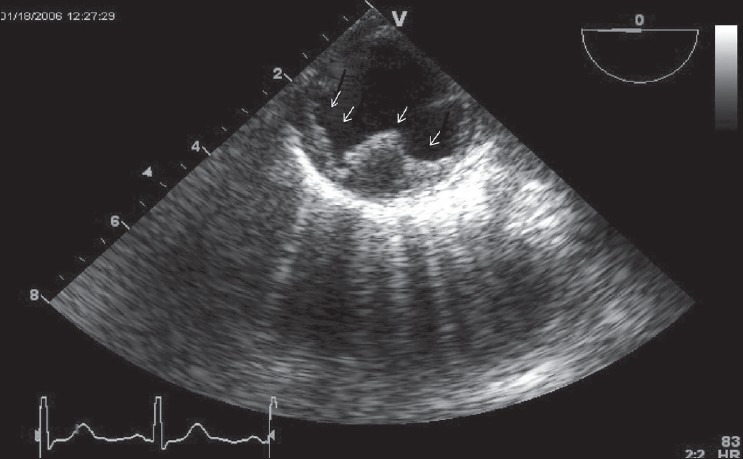
Transesophageal long-axis (90 degree) view of thoracic aorta with complex plaque (white arrow)
There are definite limitations to echocardiography. Particles with the size of 2 mm can block major brain arteries, but they are beyond the imaging resolution of current echocardiographic technology.[75] In addition, thromboembolism is a dynamic process: when a clot forms in the heart and embolizes, there may be no residual evidence until another intracardiac clot reforms.[5,76] Sequential echocardiograms image cardiac thrombi differently;[5,77] it has been shown that even large thrombi seen on one echocardiogram can disappear on in the later studies.[12,77]
Transcranial Doppler (TCD) monitoring can detect cerebral embolic signals.[5,78,79] Embolic particles passing under TCD probes produce transient, short-duration, high-intensity signals referred to as HITS (high-intensity transient signals). Examples of HITS are shown in Figure 4 and 5. A relatively high frequency of embolic signals has been detected with TCD in patients with atrial fibrillation,[80] cardiac surgery,[81] prosthetic valves, left ventricular assist devices,[82] carotid artery disease and carotid endarterectomy. The monitoring of emboli with TCD may guide treatment decisions (e.g., performing TCD pre- and postinitiation of anticoagulation to assess whether HITS cease).[12]
Figure 4.
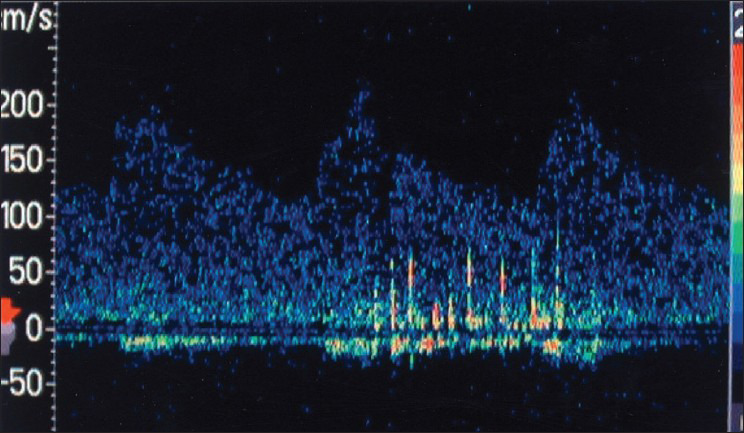
Transcranial Doppler during carotid angioplasty shows a ß urry of microembolic signals (HITS) in the middle cerebral artery
Figure 5.
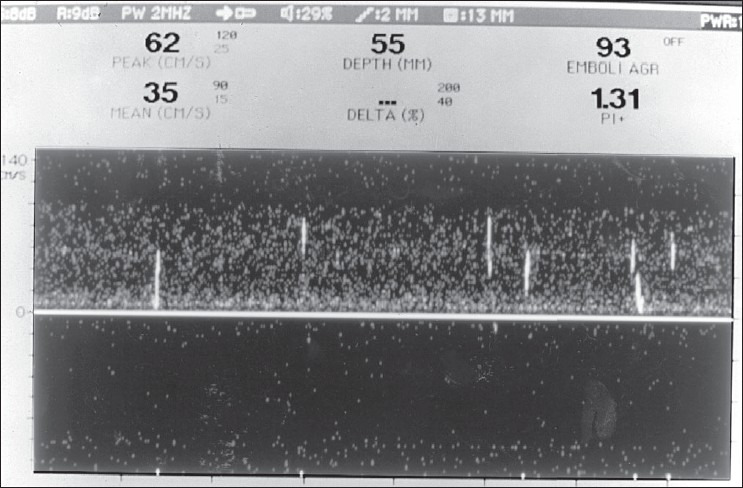
Transcranial Doppler recording from the middle cerebral artery during cardiac bypass surgery at a time when the aorta was manipulated. The white streaks (HITS) represent microemboli
Prevention and Treatment
Atrial fibrillation (rheumatic and non rheumatic)
Studies in patients with both rheumatic mitral stenosis and atrial fibrillation have shown that warfarin is effective in preventing brain embolism.[9,28,83,84] The use of aspirin for primary cardioembolic stroke prevention in the setting of rheumatic mitral stenosis and atrial fibrillation is not supported.[83] In early studies, the level of anticoagulation was higher than that currently used and brain hemorrhages and other bleeding complications were commonplace. Trials have now shown that low-dose warfarin [international normalized ratio (INR): 2.0-3.0] is also effective in preventing brain emboli in these patients as well as in patients with nonrheumatic AF. Mitral valve repair and mitral valve replacement are also considered in the prevention of cerebral embolism for patients with hemodynamically significant rheumatic mitral stenosis or insufficiency.[9,12]
In the Copenhagen Atrial Fibrillation Aspirin Anticoagulation (AFASAK) study, 1007 patients (median age: 74.2 years) with chronic, nonrheumatic AF were assigned to warfarin (INR: 2.8-4.2), aspirin (75 mg/day) or placebo.[85] This study was halted prematurely when the analysis of effectiveness reached a predetermined level of significance in favor of warfarin treatment. The principal outcome measured was the composite of ischemic or hemorrhagic stroke, transient ischemic attack (TIA) and systemic embolism. The observed reduction for warfarin in comparison to placebo was 64%, an absolute risk reduction of 3.5% per year. An analysis by intention to treat, excluding TIA and minor stroke, indicated a risk reduction of an estimated 50% ( P< 0.05) and an absolute reduction of approximately 1.5% annually.[12]
Investigators for the Stroke Prevention in Atrial Fibrillation (SPAF) study evaluated warfarin and aspirin in nonrheumatic AF patients.[86,87] In this study, two groups of patients were evaluated on the basis of their eligibility for warfarin. In the first group, 627 patients determined to be eligible for warfarin were randomized to open label warfarin (INR: 2.8-4.5; prothrombin time: 1.3-1.8 times control) or in a double-blinded fashion to either aspirin (325 mg daily, enteric-coated) or a matching placebo. In the second group, 703 patients ineligible for warfarin were randomized (double-blind) to aspirin (325 mg daily, enteric-coated) or placebo. During the mean follow-up of 1.3 years, the principal outcome - a composite of ischemic stroke and systemic embolism - was significantly decreased by warfarin and aspirin. Warfarin reduced the outcome of disabling ischemic stroke or vascular death by 54% (P=.11): an absolute reduction of 2.6% per year. Aspirin reduced the outcome of disabling stroke or death by 22% (P=.33): an absolute reduction of approximately 1% per year. The SPAF investigators later compared low-intensity fixed-dose warfarin (INR 1.2 to 1.5) plus aspirin (325 mg/day) with adjusted dose warfarin (INR 2.0 to 3.0) in elderly patients with one or more risk factors for embolism.[88] Ischemic stroke and systemic embolism were present in 7.9% of patients on fixed dose warfarin plus aspirinvsonly 1.9% on adjusted-dose warfarin. SPAF investigators later studied the effectiveness of 325 mg aspirin in patients with low risk and found that the rate of ischemic stroke was low in this particular group (2% per year).[12,89]
Three risk factors for thromboembolism were identified by the SPAF study-recent congestive heart failure, history of hypertension and previous thromboembolism.[90,91] The results of this study suggested that anticoagulation with warfarin was not indicated in patients without any of the three risk factors (i.e., those who were at low risk for thromboembolism). In such patients, the danger of anticoagulant therapy was thought to possibly outweigh its benefits. In conclusion, aspirin (325 mg daily) is probably a reasonable and safe therapy for patients with lone, nonrheumatic AF who are under 60 years of age and have none of the three identified risk factors.[90,91,92] In all other patients with AF, long-term oral warfarin therapy (INR: 2.0-3.0) should be used unless it is contraindicated.[12,89,92,93]
In the Boston Area Anticoagulation Trial for Atrial Fibrillation (BAATAF), 420 patients with nonrheumatic AF (mean age 68 years) were randomized (unblinded)to warfarin (target prothrombin time ratio, 1.2 to 1.5 x control; INR: 1.5 to 2.7) or to a control group who were allowed to take aspirin.[94] Ischemic stroke or systemic embolism was the principal outcome measured and the mean follow-up was 2.2 years. The incidence of stroke was reduced by 86% in the warfarin group compared to control (P= 0.002) This was equivalent to an absolute risk reduction of 2.6% per year. There was no demonstrable benefit noted with aspirin, but this specific study was also not designed to test aspirin efficacy.[12]
In the Canadian Atrial Fibrillation Anticoagulation (CAFA) study, 191 patients were randomized to placebo and 187 to warfarin (INR target range 2.0 to 3.0).[95] The composite of nonlacunar stroke, non-CNS embolism and fatal or intracranial hemorrhage was the principal outcomes. The relative risk reduction for warfarin was 37% (P= 0.17). This study ended prematurely when the results of both the Copenhagen AFASAK and SPAF studies became known.[12]
The question of the optimal level of anticoagulation was addressed by the European Atrial Fibrillation Trial (EAFT) Study Group.[96] No treatment effect was noted with anticoagulation responses below INRs of 2.0. The rate of thromboembolic events was lowest at INRs from 2 to 3.9 and most major hemorrhages occurred at INRs greater than 5.0. Thus, a target INR of 3.0 with a range from 2 to 5.0 was recommended by the EAFT group.[96] Similar to the SPAF findings, fixed-dose warfarin with a target of 1.3 to 1.5 was not as effective as standard adjusted-dose warfarin at an average INR of 2.4, even when aspirin 325 mg/day was added to the low fixed-dose warfarin in another study.[12]
Warfarin is approximately 50% more effective than aspirin in preventing stroke in patients with atrial fibrillation who do not have valvular disease. Data suggests that the optimal intensity of oral anticoagulation for stroke prevention in patients with atrial fibrillation appears to be a target INR of 2.0-3.0. Unfortunately, warfarin is not without any problems. The narrow therapeutic margin of warfarin and its known associated food and drug interactions require the INR values to be followed closely. Dosage adjustments are frequently needed. These liabilities obviously contribute to the underutilization of warfarin and alternative therapies are sorely needed.[12] An additional study, Atrial Fibrillation Clopidogrel Trial with Irbesartan for Prevention of Vascular Events (ACTIVE), evaluated the safety and efficacy of the combination of aspirin plus clopidogrel in atrial fibrillation patients. Data suggests coumadin was superior to the aspirin/clopidogrel combination in this setting.[97]
Studies have also assessed ximelagatran's efficacy in stroke prevention.[19]
Ximelagatran is a direct thrombin inhibitor that is orally administered, has stable pharmacokinetics independent of the hepatic P 450enzyme system and has a low potential for food and drug interactions. Two large studies, Stroke Prevention Using the Oral Direct Thrombin Inhibitor Ximelagatran in Patients with Atrial Fibrillation SPORTIF-III and SPORTIF-V compared fixed-dose ximelagatran (36 mg BID) with dose-adjusted warfarin (INR: 2.0-3.0) in high-risk patients with AF. This study was performed on 7329 patients. In both the trials, ximelagatran was not inferior to warfarin and had fewer major and minor bleeding complications. However, serum alanine-aminotransferase levels rose transiently to >3 times normal in 6% of patients with ximelagatran (usually within the first 6 months).[12,19,98]
Patent foramen ovale
Treatment options at this time for future stroke prevention in patients with PFO and a prior cryptogenic ischemic stroke include medical therapy, open or minimally invasive cardiac surgical closure and transcatheter closure. With regard to medical therapies, antiplatelet therapy is a reasonable choice for future stroke prevention in stroke patients with an isolated PFO and first ischemic stroke/TIA. Warfarin is considered to be an appropriate treatment option in the subgroup of PFO/ischemic stroke patients who have been diagnosed with a concomitant hypercoagulable state, venous thrombosis or atrial-septal aneurysm. There is no clear evidence at present that open surgical closure is superior to medical therapy for secondary stroke prevention. Transcatheter closure may ultimately show a benefit over medical therapy for future stroke prevention: a recent review of 10 nonrandomized, unblended transcatheter closure studies for secondary stroke prevention reported a 1 year rate of recurrent, neurological events of 0-4.9% in transcatheter closure patientsvs3.8-12.0% in medically treated patients. The incidence of minor and major procedural complications was 7.9% and 1.45%, respectively. Additional randomized trials evaluating the efficacy of transcatheter closure devices are in progress.[12,19,99] At the time of this publication, trancatheter closure is not FDA approved for use in the United States after the first ischemic stroke or TIA in PFO patients; however, in PFO patients who fail medical therapy and have a second ischemic cerebral event, it is a treatment option that may be considered.
Other cardiac conditions
The effectiveness of anticoagulation on embolic stroke prevention from other cardiac conditions has not been well studied. In the United States, the use of aspirin plus warfarin in MI patients with left ventricular thrombus is based on ACC/AHA guidelines for patients with ST-segment elevation MI.[19,100] As for other conditions, the rate of recurrent stroke in patients with MVP is so low that warfarin is not recommended for stroke prophylaxis except when a thrombus is noted on echocardiography. Warfarin is also not thought to be effective in preventing calcific, myxomatous, bacterial and fibrin-platelet emboli. Finally, warfarin has even been suspected to worsen cholesterol crystal embolization.[12,101]
General stroke prevention considerations
When anticoagulation was initiated at the authors' institution, the protocols shown in Tables 1 and 2 were used. On the occasions that warfarin is used, the time to initiate anticoagulation post-stroke remains controversial. Embolic brain infarcts have the potential to become hemorrhagic, and serious cerebral hemorrhage has occurred after anticoagulation. [102,103,104,105,106] Large infarcts, hypertension, large bolus doses of heparin and excessive anticoagulation are factors that have been associated with the potential to develop brain hemorrhage. Because most hemorrhagic transformations occur within 48 h of stroke onset, the recommendations of the Cerebral Embolism Task Force are used to avoid early anticoagulation in patients with large infarcts or in those patients with hemorrhagic transformation noted on repeat CT.[107,108]
Table 1.
Heparin treatment protocol

Table 2.
Warfarin treatment protocol

In the vast majority of studies, patients with cerebral and cerebellar hemorrhagic infarction are noted to have an embolic cause. Despite the aforementioned cautions, their hemorrhagic infarctions have been noted to occur equally with and without anticoagulation with the development of hemorrhagic infarction rarely accompanied by clinical worsening.[109,110] Interestingly, the majority of patients with hemorrhagic transformation who were continued on anticoagulants did not worsen. Thus, the risk of re-embolism should be balanced against the small but definite risk of important bleeding. For example, if the patient has a large brain infarct, heparin should likely be delayed and when it is initiated, bolus heparin infusions should be avoided. If the risk for re-embolism is high, immediate heparinization is advisable (again, without using a bolus), whereas if the risk seems low, it is prudent to delay anticoagulants for at least 48 h if not longer. One study showed that in patients with atrial fibrillation with embolic strokes who were treated with well-controlled heparin anticoagulation soon after stroke onset fared better than the patients who were treated later.[12,111,112]
Acute stroke treatment: Chemical thrombolytics
For rational treatment to be given, the following basic facts should be known: (1) past and present medical history, including the time the patient was last known to be well; (2) the patient's medications; (3) vital signs; (4) laboratory data such as blood counts, glucose level and coagulability; (5) the location, nature and severity of the occlusive lesion; and (6) finally, the location, extent and reversibility of the brain lesion.[9,113] Treatment should not be guided solely by the temporal pattern of the symptoms, such as TIA, progressing stroke or so-called completed stroke.[9,113,114,115] These time courses do not predict the cause and mechanism of ischemia, do not suggest if an infarct is present and do not identify patients who will have further or recurrent ischemia.[12,115]
The first decision a physician should make is whether or not any thrombolytic therapy is indicated. Very severe neurologic deficits, serious intercurrent illnesses (dementia, cancer, etc.) and psychosocioeconomic considerations may make patients unsuitable for specific treatments. If treatment is a feasible option for the patient, the next questions to be considered are what brain tissue is at risk for further ischemia and what may be the benefit/risk ratio of specific treatments. To determine the tissue at risk, clinicians consider the cause and the deficit. For example, a man with a slight hemiplegia and numbness due to a small lacunar infarct in the anterior limb of the internal capsule may have infarcted the entire tissue supplied by an occluded small artery. In that case, the treatment of choice may consist of supportive care and ultimately controlling lacunar stroke risk factors (such as hypertension, diabetes or hypercholesterolemia). However, if that same patient has waxing and waning symptoms and a small cortical infarct with a clot in the middle cerebral artery trunk, the rest of the MCA territory is at risk for further ischemia. Aggressive thrombolytic treatment may be warranted in this case. Newer MRI techniques-diffusion-weighted and perfusion MRI- along with MRA, can show the brain that is already infarcted and brain tissue that is underperfused but not yet infarcted, even very soon after the symptoms begin.[9,12,116,117,118] Patients who have little tissue at risk are not optimal candidates for specific interventional therapy.
Thrombolytic drugs, especially recombinant-tissue-type plasminogen activator (rt-PA) and streptokinase, have been given intravenously and intraarterially in patients with acute brain ischemia. In a study in which the arterial lesions were not defined, intravenous therapy with rt-PA given within 90 min and 3 h of ischemia onset in the aggregate, provided a statistically significant benefit is obtained.[119] Unfortunately, in the present study and other studies, approximately 6-12% of patients treated with thrombolytic agents developed important intracranial bleeding. Uncontrolled studies show that patients with distal intracranial arterial embolic occlusions do well with intravenous thrombolytic therapy. [120,121,122,123,124,125] Patients with ICA occlusions in the neck and intracranially rarely reperfuse after intravenous thrombolytic therapy, particularly if collateral circulation is poor. Intraarterial therapy may be considered in these patients. Intraarterially administered prourokinase thrombolysis has also been shown to be very effective in opening blocked intracranial arteries within the anterior circulation.[126] The dose, timing, mode of delivery and target group for therapy remain unsettled. The authors believe that vascular imaging should precede the administration of thrombolytic agents. Brain and vascular imaging can guide physicians as to who should receive thrombolytics and by what route.[12,127]
Because all the stroke patients are at risk of developing more lesions, the control of risk factors is very important and should be begun in the hospital. Risk factors include smoking, hyperlipidemia, obesity, inactivity and hypertension. Blood pressure should not be excessively lowered during the acute ischemic period as this may decrease flow in collateral arteries. Blood pressure control can be instituted 3-4 weeks after the stroke. Rehabilitation must also begin early.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Goyal A, Yusuf S. The burden of cardiovascular disease in the Indian subcontinent. Indian J Med Res. 2006;124:235–44. [PubMed] [Google Scholar]
- 2.Lipska K, Sylaja PN, Sarma PS, Thankappan KR, Kutty VR, et al. Risk factors for acute ischaemic stroke in young adults in South India. J Neurol Neurosurg Psychiatry. 2007;78:959–63. doi: 10.1136/jnnp.2006.106831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee TK, Roy MK, Bhoi KK. Is stroke increasing in India-preventive measures that need to be implemented. J Indian Med Assoc. 2005;103:162. 164,166. [PubMed] [Google Scholar]
- 4.Ghandehari K, Izadi-Mood Z. Etiology of young adult onset brain infarction in Iran. Arch Iran Med. 1006;9:240–3. [PubMed] [Google Scholar]
- 5.Caplan LR. Brain embolism. In: Caplan LR, Hurst JW, Chimowitz MI, editors. Clinical Neurocardiology. New York: Marcel Dekker; 1999. pp. 35–185. [Google Scholar]
- 6.Aring C, Merritt H. Differential diagnosis between cerebral hemorrhage and cerebral thrombosis. Arch Intern Med. 1935;56:435–56. [Google Scholar]
- 7.Whisnant J, Fitzgibbons J, Kurland L, Sayre GP. Natural history of stroke in Rochester, Minnesota 1945-1954. Stroke. 1971;2:11–22. doi: 10.1161/01.str.2.1.11. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto N, Whisnant J, Kurland L, Okazaki H. Natural history of stroke in Rochester, Minnesota 1955-1969. Stroke. 1973;4:2–29. doi: 10.1161/01.str.4.1.20. [DOI] [PubMed] [Google Scholar]
- 9.Caplan LR. Stroke: A clinical approach. 3rd ed. Boston: Butterworth-Heinemann; 2000. [Google Scholar]
- 10.Mohr JP, Caplan LR, Melski JW, Goldstein RJ, Duncan GW, Kistler JP, et al. The Harvard Cooperative Stroke registry: A prospective study. Neurology. 1978;28:754–62. doi: 10.1212/wnl.28.8.754. [DOI] [PubMed] [Google Scholar]
- 11.Cardiogenic brain embolism: The second report of the Cerebral Embolism Task Force. Arch Neurol. 1989;46:727–43. [PubMed] [Google Scholar]
- 12.Leary MC, Caplan LR. Cerebrovascular disease and neurologic manifestations of heart disease. In: Fuster V, Alexander RW, O-Rourke RA, Roberts R, King 3rd, et al., editors. Hurst?s the Heart. 12th ed. New York: McGraw-Hill; 2007. [Google Scholar]
- 13.Fuster V, Halperin JL. Left ventricular thrombi and cerebral embolism. N Engl J Med. 1989;320:392–4. doi: 10.1056/NEJM198902093200610. [DOI] [PubMed] [Google Scholar]
- 14.Wolf PA, Abbot RD, Kannel WB. Atrial fibrillation: A major contributor to stroke in the elderly: The Framingham study. Arch Intern Med. 1987;147:1561–4. [PubMed] [Google Scholar]
- 15.Bogousslavsky J, Van Melle G, Regli F. The Lausanne Stroke Registry: Analysis of 1000 consecutive patients with first strokes. Stroke. 1988;19:1083–92. doi: 10.1161/01.str.19.9.1083. [DOI] [PubMed] [Google Scholar]
- 16.Bogousslavsky J, Cachin C, Regli F, Despland PA, Van Melle G, Kappenberger L. Cardiac sources of embolism and cerebral infarction-clinical consequences and vascular concomitants: The Lausanne Stroke Registry. Neurology. 1991;41:855–9. doi: 10.1212/wnl.41.6.855. [DOI] [PubMed] [Google Scholar]
- 17.Constante Sotelo JL, Mendez Dominguez A. Rheumatic heart disease: Cause of cerebrovascular disease at the National Intstitute of Cardiology "Ignacio Chavez". Arch Cardiol Mex. 2006;76:47–51. [PubMed] [Google Scholar]
- 18.Kittner SJ, Sharkness CM, Sloan MA, Price TR, Dambrosia JM, Tuhrim S, et al. Infarcts with a cardiac source of embolism in the NINDS Stroke Data Bank: Neurologic examination. Neurology. 1992;42:299–302. doi: 10.1212/wnl.42.2.299. [DOI] [PubMed] [Google Scholar]
- 19.Sacco RL, Adams R, Albers G, Alberts MJ, Benavente O, Furie K, et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: A statement for healthcare professionals from the American Heart Association/ American Stroke Association Council on Stroke: Co-sponsored by the Council on Cardiovascular Radiology and Intervention: The American Academy of Neurology affirms the value of this guideline. Stroke. 2006;37:577–617. doi: 10.1161/01.STR.0000199147.30016.74. [DOI] [PubMed] [Google Scholar]
- 20.Visser CA, Kan G, Meltzer RS, Lie KI, Durrer D. Long-term follow-up of left ventricular thrombus after acute myocardial infarction: A two-dimensional echocardiographic study in 96 patients. Chest. 1984;86:532–6. doi: 10.1378/chest.86.4.532. [DOI] [PubMed] [Google Scholar]
- 21.Sacco RL, Shi T, Zamanillo MC, Kargman DE. Predictors of mortality and recurrence after hospitalized cerebral infarction in an urban community: The Northern Manhattan Stroke Study. Neurology. 1994;44:626–34. doi: 10.1212/wnl.44.4.626. [DOI] [PubMed] [Google Scholar]
- 22.Chockalingam A, Gnanavelu G, Elangovan S, Chockalingam V. Clinical spectrum of chronic rheumatic heart disease in India. J Heart Valve Dis. 2003;12:577–81. [PubMed] [Google Scholar]
- 23.Srinivasan K. Ischemic cerebrovascular disease in the young: Two common causes in India. Stroke. 1984;15:733–5. doi: 10.1161/01.str.15.4.733. [DOI] [PubMed] [Google Scholar]
- 24.Carter AB. Prognosis of cerebral embolism. Lancet. 1965;2:514–9. doi: 10.1016/s0140-6736(65)91473-x. [DOI] [PubMed] [Google Scholar]
- 25.Wood P. Diseases of the heart and circulation. Philedelphia, PA: JB Lippincott; 1956. [Google Scholar]
- 26.Levine HJ. Which atrial fibrillation patients should be on chronic anticoagulation? J Cardiovasc Med. 1981;6:483–7. [Google Scholar]
- 27.Friedberg CK. Diseases of the Heart. Philedelphia, PA: WB Saunders; 1966. [Google Scholar]
- 28.Fleming HA, Bailey SM. Mitral valve disease, systemic embolism and anticoagulants. Postgrad Med J. 1971;47:599–604. doi: 10.1136/pgmj.47.551.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akdemir I, Dagdelen S, Yuce M, Davutoglu V, Akcay M, Akdemir N, et al. Silent brain infarction in patients with rheumatic mitral stenosis. Jpn Heart J. 2002;43:137–44. doi: 10.1536/jhj.43.137. [DOI] [PubMed] [Google Scholar]
- 30.Deverall PB, Olley PM, Smith DR, Watson DA, Whitaker W. Incidence of systemic embolism before and after mitral valvotomy. Thorax. 1968;23:530–6. doi: 10.1136/thx.23.5.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coulshed N, Epstein EJ, McKendrick CS, Galloway RW, Walker E. Systemic embolism in mitral valve disease. Br Heart J. 1970;32:26–34. doi: 10.1136/hrt.32.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeresaty RM. Mitral valve prolapse. New York, NY: Raven Press; 1979. [Google Scholar]
- 33.Salem DN, Hartnett Daudelin D, Levine HJ, Pauker SG, Eckman MH, Riff J. Antithrombotic therapy in valvular heart disease. Chest. 2001;119:207S–9S. doi: 10.1378/chest.119.1_suppl.207s. [DOI] [PubMed] [Google Scholar]
- 34.Barnett HJ, Jones MW, Boughner DR, Kostuk WJ. Cerebral ischemic events associated with prolapsing mitral valve. Arch Neurol. 1976;33:777–82. doi: 10.1001/archneur.1976.00500110045008. [DOI] [PubMed] [Google Scholar]
- 35.Barnett HJ, Boughner DR, Taylor DW, Cooper PE, Kostuk WJ, Nichol PM. Further evidence relating mitral valve prolapse to cerebral ischemic events. N Engl J Med. 1980;302:139–44. doi: 10.1056/NEJM198001173020303. [DOI] [PubMed] [Google Scholar]
- 36.Sandok BA, Giuliani ER. Cerebral ischemic events in patients with mitral valve prolapse. Stroke. 1982;13:448–50. doi: 10.1161/01.str.13.4.448. [DOI] [PubMed] [Google Scholar]
- 37.Lauzier S, Barnett HJ. Cerebral ischemia with mitral valve prolapse and mitral annulus calcification. In: Furlan AJ, editor. The Heart and Stroke. London: Springer-Verlag; 1987. pp. 63–100. [Google Scholar]
- 38.deBono DP, Warlow CP. Mitral-annulus calcification and cerebral or retinal ischemia. Lancet. 1979;2:383–5. doi: 10.1016/s0140-6736(79)90402-1. [DOI] [PubMed] [Google Scholar]
- 39.Korn D, DeSanctis RW, Sell S. Massive calcification of the mitral annulus. A clinicopathological study of fourteen cases. N Engl J Med. 1962;267:900–9. doi: 10.1056/NEJM196211012671802. [DOI] [PubMed] [Google Scholar]
- 40.Benjamin EJ, Plehn JF, D'Agostino RB, Belanger AJ, Comai K, Fuller DL, et al. Mitral annular calcification and the risk of stroke in an elderly cohort. N Engl J Med. 1992;327:374–9. doi: 10.1056/NEJM199208063270602. [DOI] [PubMed] [Google Scholar]
- 41.Savage DD, Garrison RJ, Castelli WP, McNamara PM, Anderson SJ, Kannel WB, et al. Prevalence of submitral (annular) calcium and its correlations in a general population-based sample (the Framingham study) Am J Cardiol. 1983;51:1375–8. doi: 10.1016/0002-9149(83)90315-6. [DOI] [PubMed] [Google Scholar]
- 42.Debruxelles S, Sibon I, Rouanet F, Orgogozo JM. Cerebral infarction by calcified embolism: A spontaneous complication of calcified aortic stenosis. Rev Neurol. 2004;160:582–4. doi: 10.1016/s0035-3787(04)70992-3. [DOI] [PubMed] [Google Scholar]
- 43.Boon A, Lodder J, Cheriex E, Kessels F. Risk of stroke in a cohort of 815 patients with calcification of the aortic valve with or without stenosis. Stroke. 1996;27:847–51. doi: 10.1161/01.str.27.5.847. [DOI] [PubMed] [Google Scholar]
- 44.Hagen PT, Scholz DG, Edwards WD. Incidence and size of patent foramen ovale during the first 10 decades of life: An autopsy study of 965 normal hearts. Mayo Clin Proc. 1984;59:17–20. doi: 10.1016/s0025-6196(12)60336-x. [DOI] [PubMed] [Google Scholar]
- 45.Lechat PH, Mas JL, Lascault G, Loron P, Theard M, Klimczac M, et al. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med. 1988;318:1148–52. doi: 10.1056/NEJM198805053181802. [DOI] [PubMed] [Google Scholar]
- 46.Di Tullio M, Sacco RL, Gopal A, Mohr JP, Homma S. Patent foramen ovale as a risk factor for cryptogenic stroke. Ann Intern Med. 1992;117:461–5. doi: 10.7326/0003-4819-117-6-461. [DOI] [PubMed] [Google Scholar]
- 47.Petty GW, Khanderia BK, Chu CP, Sicks JD, Whisnant JP. Patent foramen ovale in patients with cerebral infarction: A transesophageal echocardiographic study. Arch Neurol. 1997;54:819–22. doi: 10.1001/archneur.1997.00550190013008. [DOI] [PubMed] [Google Scholar]
- 48.Webster MW, Chancellor AM, Smith HJ, Swift DL, Shar pe DN, Bass NM, et al. Patent foramen ovale in young stroke patients. Lancet. 1988;2:11–2. doi: 10.1016/s0140-6736(88)92944-3. [DOI] [PubMed] [Google Scholar]
- 49.Steiner MM, Di Tullio MR, Rundek T, Gan R, Chen X, Liguori C, et al. Patent foramen ovale size and embolic brain imaging findings among patients with ischemic stroke. Stroke. 1998;29:944–8. doi: 10.1161/01.str.29.5.944. [DOI] [PubMed] [Google Scholar]
- 50.Jones HR, Jr, Caplan LR, Come PC, Swinton NW, Jr, Breslin DJ. Cerebral emboli of paradoxical origin. Ann Neurol. 1983;13:314–9. doi: 10.1002/ana.410130315. [DOI] [PubMed] [Google Scholar]
- 51.Biller J, Adams HP, Johnson MR, Kerber RE, Toffol GJ. Paradoxical cerebral embolism: Eight cases. Neurology. 1986;36:1356–60. doi: 10.1212/wnl.36.10.1356. [DOI] [PubMed] [Google Scholar]
- 52.Gautier JC, Durr A, Koussa S. Paradoxical cerebral embolism with a patent foramen ovale: A report of 29 patients. Cerebrovasc Disc. 1991;1:193–202. [Google Scholar]
- 53.Galve E, Candell-Riera J, Pigrau C, Permanyer-Miralda G, Garcia-Del-Castillo H, Soler-Soler J. Prevalence, morphology, types and evaluation of cardiac valvular disease in systemic lupus erythematosus. N Engl J Med. 1988;319:817–23. doi: 10.1056/NEJM198809293191302. [DOI] [PubMed] [Google Scholar]
- 54.Barbut D, Borer JS, Wallerson D, Ameisen O, Lockshin M. Anticardiolipin antibody and stroke: Possible relation of valvular heart disease and embolic events. Cardiology. 1991;79:99–109. doi: 10.1159/000174866. [DOI] [PubMed] [Google Scholar]
- 55.Nighoghossian N, Derex L, Loire R, Perinetti M, Honnorat J, Riche G, et al. Giant Lambl excrescences: An unusual source of cerebral embolism. Arch Neurol. 1997;54:41–4. doi: 10.1001/archneur.1997.00550130027011. [DOI] [PubMed] [Google Scholar]
- 56.Cohen A, Tzourio C, Chauvel C, Bertrand B, Crassard I, Bernard Y, et al. Mitral valve strands and the risk of ischemic stroke in elderly patients. Stroke. 1997;28:1574–8. doi: 10.1161/01.str.28.8.1574. [DOI] [PubMed] [Google Scholar]
- 57.Caplan LR. Mitral valve strands: What are they and what is their relation to stroke? Neurol Network Comment. 1998;2:11–4. [Google Scholar]
- 58.Kanter MC, Hart RG. Neurologic complications of infective endocarditis. Neurology. 1991;41:1015–20. doi: 10.1212/wnl.41.7.1015. [DOI] [PubMed] [Google Scholar]
- 59.Sacco RL, Ellenberg JH, Mohr JP, Tatemichi TK, Hier DB, Price TR, et al. Infarcts of undetermined cause: The NINCDS Stroke Data Bank. Ann Neurol. 1989;25:382–90. doi: 10.1002/ana.410250410. [DOI] [PubMed] [Google Scholar]
- 60.Mohr JP. Infarct of unclear cause. In: Furlan AJ, editor. The Heart and Stroke. London: Springer-Verlag; 1987. pp. 101–16. [Google Scholar]
- 61.Kushner MJ, Zanotte EM, Bastianiello S, Mancini G, Sacchetti ML, Carolei A, et al. Transcranial doppler in acute hemispheric brain infarction. Neurology. 1991;41:109–13. doi: 10.1212/wnl.41.1.109. [DOI] [PubMed] [Google Scholar]
- 62.Ringlestein EB, Koschorke S, Holling A, Thron A, Lambertz H, Minale C. Computed tomographic patterns of proven embolic brain infarcts. Ann Neurol. 1989;26:759–65. doi: 10.1002/ana.410260612. [DOI] [PubMed] [Google Scholar]
- 63.Bladin PF, Berkovic SF. Striatocapsular infarction. Neurology. 1984;34:1423–30. doi: 10.1212/wnl.34.11.1423. [DOI] [PubMed] [Google Scholar]
- 64.Fisher CM, Adams RD. Observations on brain embolism. J Neuropathol Exp Neurol. 1951;10:92–4. [PubMed] [Google Scholar]
- 65.Fisher CM, Adams RD. Observations on brain embolism with special reference to hemorrhagic infarction. In: Furlan AJ, editor. The Heart and Stroke. London: Springer-Verlag; 1987. pp. 17–36. [Google Scholar]
- 66.Gacs G, Fox AJ, Barnett HJ, Vinuela F. CT visualization of intracranial arterial thromboembolism. Stroke. 1983;14:756–63. doi: 10.1161/01.str.14.5.756. [DOI] [PubMed] [Google Scholar]
- 67.Tomsick T, Brott T, Barsan W. Thrombus localization with emergency cerebral computed tomography. Stroke. 1990;21:180. [PMC free article] [PubMed] [Google Scholar]
- 68.Bergeron GA, Shah PM. Echocardiography unwarranted in patients with cerebral ischemic events. N Engl J Med. 1981;304:489. doi: 10.1056/NEJM198102193040818. [DOI] [PubMed] [Google Scholar]
- 69.Greenland P, Knopman DS, Mikell FL, Asinger RW, Anderson DC, Good DC. Echocardiography in diagnostic assessment of stroke. Ann Intern Med. 1981;95:51–4. doi: 10.7326/0003-4819-95-1-51. [DOI] [PubMed] [Google Scholar]
- 70.Donaldson RM, Emmanuel RW, Earl CJ. The role of two-dimensional echocardiography in the detection of potentially embolic intracardiac masses in patients with cerebral ischemia. J Neurol Neurosurg Psychiatry. 1981;44:803–9. doi: 10.1136/jnnp.44.9.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tegeler CH, Downes TR. Cardiac imaging in stroke. Stroke. 1991;22:1206–11. doi: 10.1161/01.str.22.9.1206. [DOI] [PubMed] [Google Scholar]
- 72.Pop G, Sutherland GR, Koudstaal PJ, Sit TW, de Jong G, Roelandt JR. Transesophageal echocardiography in the detection of intracardiac embolic sources in patients with transient ischemic attacks. Stroke. 1990;21:560–5. doi: 10.1161/01.str.21.4.560. [DOI] [PubMed] [Google Scholar]
- 73.Zenker G, Ecbel R, Kramer G, Mohr-Kahaly S, Drexler M, Harnoncourt K, et al. Transesophageal echocardiography in young patients with cerebral ischemic events. Stroke. 1988;19:345–8. doi: 10.1161/01.str.19.3.345. [DOI] [PubMed] [Google Scholar]
- 74.Cohen A, Chauvel C. Transesophageal echocardiography in the management of transient ischemic attack and ischemic stroke. Cerebrovasc Dis. 1996;6:15–25. [Google Scholar]
- 75.Kase CS, White R, Vinson TL, Eichelberger RP. Shotgun pellet embolus to the middle cerebral artery. Neurology. 1981;31:458–61. doi: 10.1212/wnl.31.4.458. [DOI] [PubMed] [Google Scholar]
- 76.Caplan LR. Of birds and nests and brain emboli. Rev Neurol (Paris) 1991;147:265–73. [PubMed] [Google Scholar]
- 77.DeWitt LD, Pessin MS, Pandian NG, Paulker SG, Sonnenberg FA, Caplan LR. Benign disappearance of ventricular thrombus after embolic stroke: A case report. Stroke. 1988;19:393–6. doi: 10.1161/01.str.19.3.393. [DOI] [PubMed] [Google Scholar]
- 78.Markus HS, Droste DW, Brown MM. Detection of symptomatic cerebral embolic signals with Doppler ultrasound. Lancet. 1994;343:1011–2. doi: 10.1016/s0140-6736(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 79.Markus HS, Harrison MJ. Microembolic signal detection using ultrasound. Stroke. 1995;26:1517–9. doi: 10.1161/01.str.26.9.1517. [DOI] [PubMed] [Google Scholar]
- 80.Tong DC, Bolger A, Albers GW. Incidence of transcranial Doppler-detected cerebral microemboli in patients referred for echocardiography. Stroke. 1994;25:2138–41. doi: 10.1161/01.str.25.11.2138. [DOI] [PubMed] [Google Scholar]
- 81.Barbut D, Hinton RB, Szatrowski TP, Hartman GS, Bruefach M, Williams-Russo P, et al. Cerebral emboli detected during bypass surgery are associated with clamp removal. Stroke. 1994;25:2398–402. doi: 10.1161/01.str.25.12.2398. [DOI] [PubMed] [Google Scholar]
- 82.Nabavi DG, Georgiadis D, Mumme T, Schmid C, Mackay TG, Scheld HH, et al. Clinical relevance of intracranial microembolic signals in patients with left ventricular assist devices: A prospective study. Stroke. 1996;27:891–6. doi: 10.1161/01.str.27.5.891. [DOI] [PubMed] [Google Scholar]
- 83.Poungvarin N, Opartkiattikul N, Chaithiraphan S, Viriyavejakul A. A comparative study of coumadin and aspirin for primary cardioembolic stroke and thromboembolic preventions of chronic rheumatic mitral stenosis with atrial fibrillation. J Med Assoc Thai. 1994;77:1–6. [PubMed] [Google Scholar]
- 84.Al-Ahmad AM, Daudelin DH, Salem DN. Antithrombotic therapy for valve disease: Native and prosthetic valves. Curr Cardiol Rep. 2000;2:56–60. doi: 10.1007/s11886-000-0026-1. [DOI] [PubMed] [Google Scholar]
- 85.Petersen P, Boysen G, Godtfredsen J, Andersen ED, Andersen B. Placebo-controlled, randomized trial of warfarin and aspirin for prevention of thromboembolic complications in chronic atrial fibrillation: The Copenhagen AFASAK Study. Lancet. 1989;1:175–9. doi: 10.1016/s0140-6736(89)91200-2. [DOI] [PubMed] [Google Scholar]
- 86.Stroke Prevention in Atrial Fibrillation Study Group Investigators: Preliminary report of the Stroke Prevention in Atrial Fibrillation Study. N Engl J Med. 1990;322:863–8. doi: 10.1056/NEJM199003223221232. [DOI] [PubMed] [Google Scholar]
- 87.Stroke Prevention in Atrial Fibrillation Investigators: The stroke prevention in atrial fibrillation trial: Final results. Circulation. 1991;84:527–39. doi: 10.1161/01.cir.84.2.527. [DOI] [PubMed] [Google Scholar]
- 88.Stroke Prevention in Atrial Fibrillation Investigators. Adjusted-dose warfarin versus low-intensity fixed-dose warfarin plus aspirin for high-risk patients with atrial fibrillation. Stroke Prevention in Atrial Fibrillation III randomized clinical trial. Lancet. 1996;348:633–8. [PubMed] [Google Scholar]
- 89.Stroke Prevention in Atrial Fibrillation Investigators: Prospective identiþ cation of patients with nonvalvular atrial þ brillation at low risk during treatment with aspirin: Stroke prevention in Atrial Fibrillation III Study. Circulation. 1997;96:1–28. [Google Scholar]
- 90.The Stroke Prevention in Atrial Fibrillation Investigators. Predictors of thromboembolism in atrial fibrillation: I, Clinical features of patients at risk. Ann Intern Med. 1992;116:1–5. doi: 10.7326/0003-4819-116-1-1. [DOI] [PubMed] [Google Scholar]
- 91.The Stroke Prevention in Atrial Fibrillation Investigators. Predictors of thromboembolism in atrial fibrillation: II, Echocardiographic features of patients at risk. Ann Intern Med. 1992;116:6–12. doi: 10.7326/0003-4819-116-1-6. [DOI] [PubMed] [Google Scholar]
- 92.Pritchett EL. Management of atrial fibrillation. N Engl J Med. 1992;326:1264–71. doi: 10.1056/NEJM199205073261906. [DOI] [PubMed] [Google Scholar]
- 93.Singer DE. Randomized trials of warfarin for atrial fibrillation. N Engl J Med. 1992;327:1451–3. doi: 10.1056/NEJM199211123272009. [DOI] [PubMed] [Google Scholar]
- 94.The Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. The effect of low-dose warfarin on the risk of stroke in patients with nonrheumatic atrial fibrillation. N Engl J Med. 1990;323:1505–11. doi: 10.1056/NEJM199011293232201. [DOI] [PubMed] [Google Scholar]
- 95.Connolly SJ, Laupacis A, Gent M, Roberts RS, Cairns JA, Joyner C. Canadian Atrial Fibrillation Anticoagulation (CAFA) study. J Am Coll Cardiol. 1991;18:349–55. doi: 10.1016/0735-1097(91)90585-w. [DOI] [PubMed] [Google Scholar]
- 96.European Atrial Fibrillation Trial Study Group. Optimal oral anticoagulation therapy in patients with nonrheumatic atrial fibrillation and recent cerebral ischemia. N Engl J Med. 1995;333:5–10. doi: 10.1056/NEJM199507063330102. [DOI] [PubMed] [Google Scholar]
- 97.ACTIVE Writing Group of the ACTIVE Investigators. Connolly S, Pogue J, Hart R, Pfeffer M, Hohnloser S, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): A randomised controlled trial. Lancet. 2006;367:1903–12. doi: 10.1016/S0140-6736(06)68845-4. [DOI] [PubMed] [Google Scholar]
- 98.Halperin JL for the Executive Steering Committee, SPORTIF III and V Study Investigators. Ximelagatran compared with warfarin for prevention of thromboembolism in patients with nonvalvular atrial fibrillation:Rationale, objectives and design of a pair of clinical studies and baseline patient characteristics (SPORTIF II and V) Am Heart J. 2003;146:431–8. doi: 10.1016/S0002-8703(03)00325-9. [DOI] [PubMed] [Google Scholar]
- 99.Khairy P, O'Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: A systemic review. Ann Intern Med. 2003;139:753–60. doi: 10.7326/0003-4819-139-9-200311040-00010. [DOI] [PubMed] [Google Scholar]
- 100.Braunwald E, Antman EM, Beasley JW, Califf RM, Cheitlin MD, Hochman JS, et al. ACC/AHA 2002 guideline update for the management of patients with unstable angina and non-ST segment elevation myocardial infarction: Summary article: A report of the American College of Cardiology/American Heart Association Task Force on Practice guidelines (Committee on the Management of Patients With Unstable Angina) J Am Coll Cardiol. 2002;40:1366–74. doi: 10.1016/s0735-1097(02)02336-7. [DOI] [PubMed] [Google Scholar]
- 101.Moldveen-Geronimus M, Merriam JC., Jr Cholesterol embolization: From pathologic curiosity to clinical entity. Circulation. 1967;35:946–53. doi: 10.1161/01.cir.35.5.946. [DOI] [PubMed] [Google Scholar]
- 102.Shields RW, Jr, Laureno R, Lachman T, Victor M. Anticoagulant-related hemorrhage in acute cerebral embolism. Stroke. 1984;15:426–37. doi: 10.1161/01.str.15.3.426. [DOI] [PubMed] [Google Scholar]
- 103.Lieberman A, Hass WK, Pinto R, Isom WO, Kupersmith M, Bear G, et al. Intracranial hemorrhage and infarction in anticoagulated patients with prosthetic heart valves. Stroke. 1978;9:18–24. doi: 10.1161/01.str.9.1.18. [DOI] [PubMed] [Google Scholar]
- 104.Drake ME, Jr, Shin C. Conversion of ischemic to hemorrhagic infarction by anticoagulant administration: Report of two cases with evidence from serial computed tomographic brain scans. Arch Neurol. 1983;40:44–6. doi: 10.1001/archneur.1983.04050010064018. [DOI] [PubMed] [Google Scholar]
- 105.Cerebral Embolism Study Group. Immediate anticoagulation of embolic stroke: A randomized trial. Stroke. 1983;14:668–76. doi: 10.1161/01.str.14.5.668. [DOI] [PubMed] [Google Scholar]
- 106.Toni D, Fiorelli M, Bastianello S, Sacchetti ML, Sette G, Argentino C, et al. Hemorrhagic transformation of brain infarct. Neurology. 1996;46:341–5. doi: 10.1212/wnl.46.2.341. [DOI] [PubMed] [Google Scholar]
- 107.Cerebral Embolism Task Force. Cardiogenic brain embolism. Arch Neurol. 1986;43:71–84. [PubMed] [Google Scholar]
- 108.Cerebral Embolism Task Force. Cardiogenic brain embolism: The second report of the Cerebral Embolism Task Force. Arch Neurol. 1989;46:727–43. [PubMed] [Google Scholar]
- 109.Pessin MS, Estol CJ, Lafranchise F, Caplan LR. Safety of anticoagulation after hemorrhagic infarction. Neurology. 1993;43:1298–303. doi: 10.1212/wnl.43.7.1298. [DOI] [PubMed] [Google Scholar]
- 110.Chaves CJ, Pessin MS, Caplan LR, Chung CS, Amarenco P, Breen J, et al. Cerebellar hemorrhagic infarction. Neurology. 1996;46:346–9. doi: 10.1212/wnl.46.2.346. [DOI] [PubMed] [Google Scholar]
- 111.Chamorro A, Vila N, Ascaso C, Blanc R. Heparin in acute stroke with atrial fibrillation: Clinical relevance of very early treatment. Arch Neurol. 1999;56:1098–102. doi: 10.1001/archneur.56.9.1098. [DOI] [PubMed] [Google Scholar]
- 112.Caplan LR. When should heparin be given to patients with atrial fibrillation-related brain infarcts. Arch Neurol. 1999;56:1059–60. doi: 10.1001/archneur.56.9.1059. [DOI] [PubMed] [Google Scholar]
- 113.Caplan LR. Treatment of cerebral ischemia: Where are we headed? Stroke. 1984;15:571–4. doi: 10.1161/01.str.15.3.571. [DOI] [PubMed] [Google Scholar]
- 114.Caplan LR. TIAs-We need to return to the question, what is wrong with Mr.Jones? Neurology. 1988;38:791–3. doi: 10.1212/wnl.38.5.791. [DOI] [PubMed] [Google Scholar]
- 115.Caplan LR. Are terms such as completed stroke or RIND of continued usefulness? Stroke. 1983;14:431–3. doi: 10.1161/01.str.14.3.431. [DOI] [PubMed] [Google Scholar]
- 116.Caplan LR, DeWitt LD, Breen JC. Neuroimaging in patients with cerebrovascular disease. In: Greenberg J, editor. Neuroimaging, A companion to Adams and Victor's Principles of Neurology. New York: McGraw-Hill; 1999. pp. 493–520. [Google Scholar]
- 117.Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute human stroke studied by whole brain echo planar diffusion-weighted nagnetic resonance imaging. Ann Neurol. 1995;37:231–41. doi: 10.1002/ana.410370214. [DOI] [PubMed] [Google Scholar]
- 118.Sorensen AG, Buonanno FS, Gonzalez RG, Schwamm LH, Lev MH, Huang-Hellinger FR, et al. Hyperacute stroke: Evaluation with combined multisection diffusion-weighted and hemodynamically weighted echo-planar MR imaging. Radiology. 1996;199:391–401. doi: 10.1148/radiology.199.2.8668784. [DOI] [PubMed] [Google Scholar]
- 119.The National Institute of Neurological Disorders and Stroke rt-PA Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–7. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 120.del Zoppo GJ, Poeck K, Pessin MS, Wolpert SM, Furlan AJ, Ferbert A, et al. Recombinant tissue plasminogen activator in acute thrombotic and embolic stroke. Ann Neurol. 1992;32:78–86. doi: 10.1002/ana.410320113. [DOI] [PubMed] [Google Scholar]
- 121.Wolpert SM, Bruckmann H, Greenlee R, Wechsler L, Pessin MS, del Zoppo GJ. Neuroradiologic evaluation of patients with acute stroke treated with recombinant tissue plasminogen activator. AJNR Am J Neuroradiol. 1993;14:3–13. [PMC free article] [PubMed] [Google Scholar]
- 122.Pessin MS, del Zoppo GJ, Furlan AJ. Thrombolytic treatment in acute stroke: Review and update of selected topics. In: Moskowitz MA, Caplan LR, editors. Cerebrovascular Diseases: Nineteenth Princeton Stroke Conference. Boston: Butterworth-Heinemann; 1995. pp. 409–18. [Google Scholar]
- 123.Furlan A, Higashida R, Wechsler L, et al. Intra-arterial prourokinase for acute ischemic stroke The PROACT II study: A randomized controlled trial Prolyse in Acute Cerebral Thromboembolism. JAMA. 1999;282:2003–11. doi: 10.1001/jama.282.21.2003. [DOI] [PubMed] [Google Scholar]
- 124.Caplan LR, Mohr JP, Kistler JP, Koroshetz W. Should thrombolytic therapy be the first-line treatment for acute ischemic stroke? N Engl J Med. 1997;337:1309–13. doi: 10.1056/NEJM199710303371812. [DOI] [PubMed] [Google Scholar]
- 125.Caplan LR. Intracranial branch atheromatous disease. Neurology. 1989;39:1246–50. doi: 10.1212/wnl.39.9.1246. [DOI] [PubMed] [Google Scholar]
- 126.Caplan LR. Lacunar infarction: A neglected concept. Geriatrics. 1976;31:71–5. [PubMed] [Google Scholar]
- 127.Caplan LR. Binswanger's disease revisited. Neurology. 1995;45:626–33. doi: 10.1212/wnl.45.4.626. [DOI] [PubMed] [Google Scholar]