

Summary
Membrane contact sites are recognized as critical means of intercompartmental communication. Here, we describe a protocol for engineering and validating a synthetic bridge between the inner and outer mitochondrial membranes to support functioning of the endogenous mitochondrial contact site and cristae organizing system (MICOS). A chimeric protein, MitoT, is stably expressed in cultured mammalian cells to bridge the mitochondrial membranes. This approach can be a valuable tool to study the function of the MICOS complex and associated proteins.
For complete details on the use and execution of this protocol, please refer to Viana et al. (2021).
Subject areas: Biotechnology and bioengineering, Cell Biology, Cell culture, Cell isolation, Cell Membrane, Flow Cytometry/Mass Cytometry, Metabolism, Microscopy, Molecular Biology
Graphical abstract
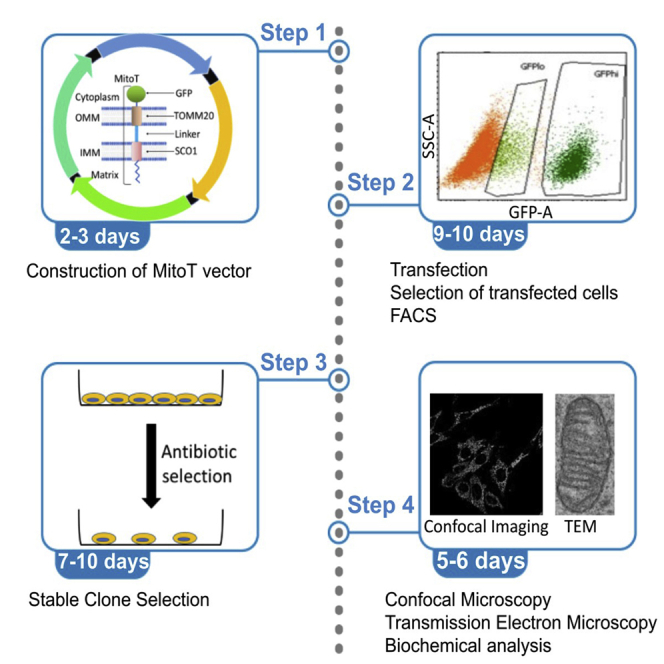
Highlights
-
•
Protocol for generating the molecular tool, MiotT
-
•
Synthetic protein for bridging membranes in mammalian mitochondria
-
•
Assessment of membrane contacts by confocal imaging and transmission electron microscopy
-
•
A combination of optimized experiments to use MitoT in mammalian cells
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Membrane contact sites are recognized as critical means of intercompartmental communication. Here, we describe a protocol for engineering and validating a synthetic bridge between the inner and outer mitochondrial membranes to support functioning of the endogenous mitochondrial contact site and cristae organizing system (MICOS). A chimeric protein, MitoT, is stably expressed in cultured mammalian cells to bridge the mitochondrial membranes. This approach can be a valuable tool to study the function of the MICOS complex and associated proteins.
Before you begin
This paper describes a step-by-step protocol for designing and expressing a synthetic chimeric protein to support the stability of mitochondrial intermembrane contacts. The MICOS complex is a multi-subunit protein complex that connects the inner mitochondrial membrane to the outer membrane and helps in maintaining mitochondrial architecture and function (Harner et al., 2011; Hoppins et al., 2011; Pfanner et al., 2014; Rabl et al., 2009; Van der Laan et al., 2016; Wollweber et al., 2017). The absence or reduced functionality of the MICOS complex or related proteins disrupts mitochondrial architecture, leading to mitochondrial dysfunction (Alkhaja et al., 2012; Harner et al., 2011; Head et al., 2011; Hoppins et al., 2011; John et al., 2005; Rabl et al., 2009). To mitigate such undesirable scenarios, we designed a mammalian chimeric protein designated MitoT to support the MICOS complex and mitochondrial intermembrane contacts.
MitoT is a first-in-class 435 amino acid-long synthetic protein that consists of four defined domains. The N-terminal portion of the protein contains a 161 amino acid-long N-terminal region of the inner mitochondrial membrane-anchored SCO1 protein, which includes the protein’s mitochondrial targeting sequence. This segment is followed by an Escherichia coli LacI protein-derived 12 amino acid-long unstructured linker region connected to a C-terminal moiety harboring 24 amino acid residues of the outer mitochondrial transmembrane segment of the mitochondrial import receptor protein TOMM20 and a cytosol-exposed eGFP tag.
The MitoT fusion protein not only augments the function of the MICOS complex but – owing to the presence of the eGFP tag – provides a handle to visualize the effect on mitochondrial architecture. The MitoT fusion protein is inspired by a previous study in the Saccharomyces cerevisiae yeast genetic model to compensate for MICOS depletion (Aaltonen et al., 2016). The Viana et al. study (Viana et al., 2021) used this strategy in mammalian cells to unravel the functional interaction between OMA1 metallopeptidase and the MICOS complex. Although the exact mechanism of how MitoT works to stabilize MICOS remains to be investigated, our results suggest a model whereby MitoT forms stable bridges between the outer (OM) and the inner boundary (IBM) membranes, thus stabilizing labile MICOS complex-mediated intermembrane OM-IBM contacts. The MitoT synthetic protein can be further modified as per intended for experimental purpose and scientific questions. The strengths of the present protocol include a novel combination of experiments designed and optimized to study first-in-class synthetic chimeric protein that is able to support the stability of mitochondrial intermembrane contacts in mammalian mitochondria.
Prepare buffers, media
Timing: 0.5–2 h
Prepare standard cell culture media, if needed.
Note: 500 mL of media is sufficient for the entire experiment.
Obtain or thaw cells
Obtain, prepare, or thaw cells as needed.
Cell culture
-
1.Cell revival.
-
a.Take out frozen MEF cells from liquid nitrogen storage container and revive as per standard revival protocol.Note: This protocol is described for mouse embryonic fibroblasts (MEFs) isolated as in Quiros et al., 2012 (Quirós et al., 2012). The protocol can be applied to other mammalian cell lines.Note: Ensure there are sufficient cell numbers to stain, seed, and backfill at appropriate time points.Note: A minimum of 200,000 MEF cells (passage 2–4) per well in a 6-well plate is recommended. Cell numbers must be adjusted if using a different size well, cell culture plate, or cell line.Note: A coated culture dish may be required for certain cell lines. Use coating material according to literature for specific cells of interest.
-
b.Transfer cell suspension into a T25 cell culture flask containing 5 mL of prewarmed FBS-containing DMEM media and mix well to ensure uniform distribution across the culture flask.
-
c.Allow cells to grow overnight (∼12–16 h) in a CO2 incubator at 5% CO2 and 37°C.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH5α competent cells | ATCC | Cat# 68233 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin | AMRESCO | Cat# 0339 |
| G-418 | US Biological | Cat# 108321-42-2 |
| QIAquick gel extraction kit | QIAGEN | Cat# 28706 |
| QIAquick spin miniprep kit | QIAGEN | Cat# 27106 |
| Plasmid midi kit | QIAGEN | Cat# 12143 |
| Luria Bertani Agar | Millipore | Cat# MP113002032 |
| Luria Bertani Broth Miller | US Biological | Cat# L1520 |
| Gel/PCR Purification kit | QIAGEN | Cat# 28004 |
| BamHI-HF restriction enzyme | NEB | Cat# R3101S |
| EcoRI-HF restriction enzyme | NEB | Cat# R3136S |
| T4 DNA Ligase enzyme | Thermo Fisher Scientific | Cat# EL0011 |
| 10× T4 DNA Ligase buffer | Thermo Fisher Scientific | Cat# B69 |
| Lipofectamine 3000 reagent | Thermo Fisher Scientific | Cat# L3000015 |
| Hoechst 33342 | Invitrogen | Cat# H3570 |
| MitoTrackerTM DeepRed FM | Thermo Fisher Scientific | Cat# M22426 |
| FluroBrite DMEM medium | Thermo Fisher Scientific | Cat# A1896702 |
| RNaseZap RNase decontamination solution | Thermo Fisher Scientific | Cat# AM9782 |
| DMEM-High Glucose | HyClone | Cat# SH30243.01 |
| FBS | HyClone | Cat# SH30396.03 |
| Pen/Strep Mix | HyClone | Cat# 15070063 |
| PBS (calcium, magnesium free) | HyClone | Cat# SH30256.FS |
| Trypsin-EDTA | HyClone | Cat# SH30236.02 |
| Opti-MEM I medium | Gibco | Cat# 31985070 |
| Phusion Hot Start II High-Fidelity PCR Master Mix | Thermo Fisher Scientific | Cat# F565L |
| Sodium cacodylate trihydrate | Electron Microscopy Sciences | Cat# 12300 |
| Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) | Sigma-Aldrich | Cat# C2759 |
| Glutaraldehyde (50%) | Electron Microscopy Sciences | Cat# 16320 |
| Agarose, low melting temperature | Bio-Rad | Cat# 162-0017 |
| Osmium tetroxide (4%) | Electron Microscopy Sciences | Cat# 19170 |
| Lead citrate trihydrate | Electron Microscopy Sciences | Cat# 17810 |
| Uranyl acetate | Electron Microscopy Sciences | Cat# 22400 |
| Spurr, Low Viscosity Embedding kit | Electron Microscopy Sciences | Cat# 14300 |
| EMS grid 200 mesh, Nickle | Electron Microscopy Sciences | Cat# EMS-200-Ni |
| Double-end molds | Ted Pella | Cat# 10590 |
| Nuclease-free water | Thermo Fisher Scientific | Cat# AM9937 |
| SuperscriptTM IV first-strand synthesis system | Thermo Fisher Scientific | Cat# 18091050 |
| HEPES | Sigma-Aldrich | Cat# 54457 |
| D-Sorbitol | Sigma-Aldrich | Cat# S1876 |
| Sucrose | Sigma-Aldrich | Cat# S0389 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | Cat# E9884 |
| Experimental models: Cell lines | ||
| Mouse Embryonic Fibroblasts, Wild Type | Gift from C. Lopez-Otin (U. Oviedo), Quirós et al. (2012) | N/A |
| Mouse Embryonic Fibroblasts, OMA1_KO | Gift from C. Lopez-Otin (U. Oviedo), Quirós et al. (2012) | N/A |
| Oligonucleotides | ||
| Fwd BamHI-SCO1 primer 5′AAAGGATCCATGGCGATGCT GGTCCTAGTACCC-3′ |
Eurofins (Viana et al., 2021) | N/A |
| Rev SCO1-LacI primer 5′GACATCGTATAACGTTACT GGTTTCTTGACGTGCTTCATTCCAGC-3′ |
Eurofins (Viana et al., 2021) | N/A |
| Fwd LacI TOMM20 primer 5′AGTAACGTTATACGATGTCGCAG AGATGGTGGGTCGGAACAGCGC-3′ |
Eurofins (Viana et al., 2021) | N/A |
| Rev TOMM20-His-EcoRI primer 5′CAAGAATTCGTGATGGTGATGGTG ATGGAAGTAGATGCAGTACCCA-3′ |
Eurofins (Viana et al., 2021) | N/A |
| Recombinant DNA | ||
| pcDNA3-eGFP | Addgene | Cat# 13031; RRID: Addgene_13031 |
| Software and algorithms | ||
| ImageJ Software | NIH (Schindelin et al., 2012) | https://imagej.net/software/fiji/downloads version 1.53h |
| Microsoft Excel 365 | Microsoft Inc. | https://www.microsoft.com/en-us/microsoft-365 |
| FlowJo 10 | FlowJo Inc. | https://www.flowjo.com/solutions/flowjo/downloads |
| Other | ||
| 35 mm No. 1.0 glass bottom dishes | MatTek Corp. | Cat# P35G-1.0-14-C |
| 0.2 μm Filter | Fisherbrand | Cat# 09-720-511 |
| FACSCalibur II | BD Biosciences | N/A |
| Cytek DxP10 flow cytometer | Cytek Biosciences | N/A |
| Nikon A1R-Ti2 confocal microscope system | Nikon Inc. | N/A |
| Countess automated cell counter | Thermo Fisher Scientific | Cat# C10281 |
| Leica UC7 ultramicrotome | Leica microsystems | N/A |
| Hitachi H7500 transmission electron microscope | Hitachi | N/A |
| Vacuum Oven | Thermo Fisher Scientific | Cat# 3618-1CE |
| T100 Thermal cycler | Bio-Rad | N/A |
| Beckman DU-640 UV-Vis spectrophotometer | Beckman | N/A |
Materials and equipment
LB agar plates containing ampicillin antibiotic
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Premixed LB Agar powder | 1× | 37 g |
| Ultrapure water | N/A | Adjust to 1 L |
| Ampicillin | 100 μg/mL | 1 mL |
| Total | 1 L |
Antibiotic added LB agar plates can be stored at 4°C for 2–3 weeks.
Note: Autoclave LB agar media and cool down to 45°C–50°C before adding ampicillin.
LB broth containing ampicillin antibiotic
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Premixed LB broth powder | 1× | 25 g |
| Ultrapure water | N/A | Adjust to 1 L |
| Ampicillin | 100 μg/mL | 1 mL |
| Total | 1 L |
Note: Autoclaved LB broth can be stored at room temperature (20°C–22°C) for 1–2 months. Add ampicillin just before use.
Note: Autoclave LB broth and cool down to room temperature (20°C–22°C) before adding ampicillin.
FBS-containing DMEM-high glucose medium
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| DMEM-high glucose media | 1× | 445 mL |
| FBS | 10% | 50 mL |
| P/S antibiotic solution | 1× | 5 mL |
| Total | 500 mL |
Note: FBS and antibiotic-containing DMEM can be stored at 4°C for 1 month.
MTE buffer
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| 1 M HEPES, pH 7.6 | 20 mM | 2 mL |
| 1 M Sorbitol | 220 mM | 22 mL |
| 1 M Sucrose | 70 mM | 7 mL |
| 0.5 M EDTA | 1 mM | 200 μL |
| Total | 100 mL |
Note: Always prepare fresh before use.
Alkaline sodium bicarbonate solution
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Sodium bicarbonate (Na2CO3) | 0.1 M | 106 mg |
| Ultrapure water | N/A | Adjust to 100 mL |
| Total | 100 mL |
Adjust to pH 7.5 and 11.5 with 1N NaOH.
Note: Always prepare fresh before use.
TEM wash buffer
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Sodium cacodylate | 100 mM | 21.4 g |
| Ultrapure water | N/A | Adjust to 1 L |
| Total | 1 L |
Adjust to pH 7.4 with 1N HCl.
Note: Always prepare fresh before use.
2.5% Glutaraldehyde fixing solution
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Glutaraldehyde (50%) | 2.5% | 0.5 mL |
| TEM wash buffer | 100 mM | 9.5 mL |
| Total | 10 mL |
Note: Always prepare fresh before use.
1% Osmium tetroxide solution
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Osmium tetroxide (4%) | 1% | 2.5 mL |
| Ultrapure water | N/A | 7.5 mL |
| Total | 10 mL |
Note: Prepare 1 day before use in a brown glass bottle and store at 4°C.
Spurr’s epoxy solution
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Vinyl cyclohexene dioxide (ERL-4221) | 22.3% | 9 mL |
| Diglycidyl ether of polypropylene glycol (D.E.R. 736) | 17.3% | 7 mL |
| Nonenyl succinic anhydride (NSA) | 59.5% | 24 mL |
| N,N-dimethylethanolamine (DMAE) | 0.74% | 0.3 mL |
| Total | 40.3 mL |
Note: Combine all the reagents into a flask and mix by shaking and swirling.
Note: Always prepare fresh before use.
1% Uranyl acetate solution
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Uranyl acetate | 1% | 0.5 g |
| Ultrapure water | N/A | 49.5 mL |
| Total | 50 mL |
Note: Prepare 1 day before use and store at 4°C.
1% Lead citrate solution
| Reagent | Final concentration | Amount/volume |
|---|---|---|
| Lead citrate | 1% | 0.1 g |
| Ultrapure water | N/A | Up to 10 mL |
| Add 3 drops of 10 N NaOH. | ||
| Total | 10 mL | |
Note: Prepare 1 day before use and store at 4°C.
Step-by-step method details
Prepare vector
This section describes steps to construct the MitoT expression vector. The vector is universal and should be readily transferable between mammalian cell lines.
Day 1: Cell harvesting, RNA isolation, and cDNA preparation
-
1.Cell harvesting and RNA isolation.
-
a.Harvest 5 million cells/sample as per standard cell harvesting protocol.
 CRITICAL: Prior to starting RNA isolation, clean workbench, pipettes, and vial stands with RNaseZap reagent. Use RNase-free polypropylene tips and vials.
CRITICAL: Prior to starting RNA isolation, clean workbench, pipettes, and vial stands with RNaseZap reagent. Use RNase-free polypropylene tips and vials. -
b.Add TRIzol reagent to the cell pellet (1 mL per 5 × 106 cells).Note: The volume of TRIzol reagent depends on the cell number. Refer to product manual to determine appropriate TRIzol volume according to cell number.
-
c.Homogenize cells by vigorous pipetting 10 times.Note: RNA is stable in TRIzol reagent. Therefore, samples can be stored at this step at −20°C for further use.
-
d.Add 0.2 mL chloroform to the homogenized cell suspension.Note: Handle chloroform under fume hood and use polypropylene tips or glass pipette for aspiration.
-
e.Shake tube vigorously or vortex for 15 s and incubate at room temperature (20°C–22°C) for 2–3 min.
-
f.Centrifuge at 12,000 g for 15 min at 4°C.
-
g.Aspirate the upper, colorless aqueous phase into a separate vial.CRITICAL: Be very careful when removing the upper layer; do not aspirate or disturb the middle white ring-like layer. Approximately 400 μL of aqueous phase can be easily aspirated out. Use a 200-μL pipette instead of a 1-mL pipette to minimize the chances of middle layer aspiration.
-
h.Add 280 μL (70% volume of the aspirated aqueous phase) of isopropanol, and mix by gently inverting the tube. Incubate for 10 min at room temperature (20°C–22°C).
-
i.Centrifuge at 12,000 g for 30 min at 4°C.
-
j.Carefully remove the supernatant and wash the pellet twice with 1 mL of 75% ethanol (room temperature- 20°C–22°C) by centrifuging at 7,500 g for 5 min at 4°C.Note: Pellet is usually not readily visible and loosely attached to tube’s wall. About 20–30 μL of supernatant can be left to prevent the loss of RNA pellet during aspiration.
-
k.After the final ethanol washing step, remove supernatant completely.
-
l.Air dry the pellet for 5 min at room temperature (20°C–22°C).Note: Repeatedly monitor the evaporation of ethanol. As soon as the pellet is dry, proceed to the next step.CRITICAL: Over-drying of RNA pellet will reduce RNA solubility and thus the overall yield.
-
m.Dissolve RNA in 30 μL of RNase-free water by gentle pipetting and incubate for 5 min at 55°C.Alternatives: Diethyl pyrocarbonate (DEPC)-treated water can be used if commercial RNase-free water is not available.
-
n.Determine RNA concentration and quality using nanodrop or UV-Vis spectrophotometer at 260 and 280 nm wavelengths.Note: A good quality RNA prep has a λ260/280 ratio of ∼2.0.Alternatives: RNA quality and integrity can be checked by separating the RNA of interest in a denaturing agarose gel.CRITICAL: Following resuspension in water, RNA becomes highly prone to degradation by RNase. From this step onwards, keep RNA on ice and avoid multiple freeze-thawing cycles. For long term storage, store the samples at −80°C.Note: If possible, try to prepare cDNA on the same day as RNA isolation to minimize freezing and thawing of RNA samples.
-
a.
-
2.
cDNA preparation.
Use SuperscriptTM IV (SSIV) first-strand cDNA synthesis system for cDNA synthesis.Note: Preset PCR machine at 65°C before starting the experiment.Note: Keep all the PCR tubes and reagents on ice.-
a.Prepare RNA-primer mix by adding 100 ng-2 μg RNA, 1 μL of Oligo-DT primer and 1 μL of dNTPs mix.
-
b.Adjust the final volume to 11 μL by adding RNase-free water.
-
c.Mix and briefly centrifuge the mixture.
-
d.Incubate for 5 min at 65°C and keep on ice for 1 min immediately after.Note: Before proceeding to the next step, vortex and briefly centrifuge 5× SSIV reverse transcriptase buffer and heat PCR machine to 50°C. Also heat a water bath or heat block to 80°C.
-
e.Prepare reverse transcriptase mix by adding 4 μL of 5× SSIV buffer, 1 μL 100 mM DTT, 1 μL of ribonuclease inhibitor, and 1 μL of SSIV reverse transcriptase enzyme.CRITICAL: DTT is a toxic chemical and needs to be handled with caution. It gets oxidized easily in the presence of air and is unstable at room temperature (20°C–22°C). It should be stored at −20°C in small aliquots.
-
f.Add freshly prepared reverse transcriptase mix to RNA-primer mix.
-
g.Incubate combined reaction mixture for 10 min at 50°C.
-
h.Heat-inactive the reverse transcriptase by incubating for 10 min at 80°C.
-
i.Add 2 units of RNase H and incubate for 20 min at 37°C.Note: Produced cDNA is stable and can be stored at −20°C for future use.
-
a.
Day 2–3: Amplification and cloning of MitoT fragment into pcDNA3-eGFP vector
-
3.
PCR amplification and digestion of MitoT fragment.
Take 5 μM of each primer pair, prepare PCR master-mix, and perform overlap extension PCR as follows (Figure 1A).-
a.Reaction 1:
-
i.Use 10 ng of cDNA and the primers Fwd BamHI-SCO1 and Rev SCO1-LacI.
-
ii.Follow the protocol provided by Phusion Hot Start II High-Fidelity PCR Master Mix with 68°C setting for annealing temperature.
-
iii.Following PCR amplification, separate the samples on a 1% agarose gel.
-
iv.Observe ethidium bromide-stained relevant DNA bands under a transilluminator and cut out the 370 bp-fragment from the gel.Note: Always use a fresh scalpel/blade for gel cutting.CRITICAL: Long UV exposure is harmful for both the DNA in the agarose gel and the researcher. Use protective wear to protect eyes and body. Quickly make cuts around the amplified band and switch off the transilluminator.
-
v.Purify the amplicon by following the gel extraction protocol as per kit manual.
-
i.
-
b.Reaction 2:
-
i.Use 10 ng of cDNA and the primers Fwd LacI-TOMM20 and Rev TOMM20-His-EcoRI.
-
ii.Follow the manufacturer’s protocol on using Phusion Hot Start II High-Fidelity PCR Master Mix with 68°C for the annealing temperature.
-
iii.Following PCR amplification, separate the samples on a 1.5% agarose gel.
-
iv.Observe relevant DNA bands under a transilluminator and excise the 115-bp fragment from the gel.
-
v.Purify the amplicon by following the gel extraction protocol as per kit manual.
-
i.
-
c.Reaction 3:
-
i.Use an equal amount of the PCR products from the Reactions 1 and 2 and the primers Fwd BamH1-SCO1 and Rev TOMM20-His-EcoRI.Note: We suggest using 1 ng of each product for this reaction.
-
ii.Follow the manufacturer’s protocol on using Phusion Hot Start II High-Fidelity PCR Master Mix with 68°C for the annealing temperature.
-
iii.Following PCR amplification, separate the samples on a 1% agarose gel.
-
iv.Observe relevant DNA bands under a transilluminator and cut the 468-bp fragment from the gel. This final amplicon constitutes the complete MitoT DNA fragment.
-
v.Purify the amplicon by following the gel extraction protocol as per kit manual.
-
vi.Digest the PCR product from Reaction 3 using BamHI and EcoRI restriction enzymes.
-
vii.Purify the digested fragment using the QIAquick gel extraction kit per manufacturer’s protocol.
-
viii.Determine the purity and concentration of the purified MitoT DNA fragment using a UV-Vis spectrophotometer.
-
i.
-
a.
-
4.Amplification and digestion of pcDNA3-eGFP vector (Figure 1B).
-
a.Transform E. coli DH5α competent cells with the pcDNA3-eGFP plasmid as per standard protocol.
-
b.Inoculate a single colony in 5 mL of LB media containing ampicillin and incubate for 8 h at 37°C.
-
c.Take 100 μL of grown starter culture and inoculate in 100 mL of LB media containing ampicillin.
-
d.Pellet the cells by centrifuging at 5,000 g for 15 min at room temperature (20°C–22°C).
-
e.Isolate the plasmid using a plasmid midi prep endotoxin-free kit per manufacturer’s instructions.
-
f.Quantitate plasmid concentration using a UV-Vis spectrophotometer.
-
g.Digest the plasmid using BamHI and EcoRI restriction enzymes using standard NEB digestion protocol.
-
h.Separate the digested plasmid on a 1% agarose gel, cut out the desired band, and purify it using a gel purification kit.
-
i.Determine the purity and concentration of the purified digested plasmid using a UV-Vis spectrophotometer.
-
a.
-
5.Ligation of the MitoT DNA fragment and digested pcDNA3-eGFP vector.
-
a.Set up ligation reaction (Figure 1B).
Vector only control reaction Ligation reaction Digested pcDNA3-eGFP vector 100 ng 100 ng Digested MitoT insert – 20 ng 10× T4 DNA Ligase Buffer 2 μL 2 μL T4 DNA ligase 1 μL 1 μL Nuclease Free Water Adjust volume to 20 μL with ddH2O Adjust volume to 20 μL with ddH2O -
b.Incubate for 1 h in a thermal cycler at 16°C.Alternatives: The reaction can also be done by incubating the reaction mix overnight (∼12–16 h) at 4°C.Alternatives: The construct can be synthesized using other molecular biology approaches such as the Gibson assembly approach.
-
c.Transform 5 μL of ligation mix into E. coli DH5α competent cells (Figure 1C).
-
d.Select single colonies and inoculate in 5 mL LB containing ampicillin in a glass tube.
-
e.Following 16 h incubation at 37°C, pellet the cells by centrifuging at 5,000 g for 15 min at room temperature (20°C–22°C) and isolate plasmid for analysis.
-
f.Digest isolated plasmid with EcoRI and BamHI restriction enzymes and electrophoretically separate the digest on a 1% agarose gel to confirm the presence of the MitoT insert (468-bp band).
- g.
-
h.Inoculate 5 mL of LB-Ampicillin medium with an E. coli clone harboring confirmed MitoT plasmid.
-
i.After 8 h incubation at 37°C, measure the OD600 of the initial inoculum, and inoculate 100 μL in 100 mL LB medium containing ampicillin antibiotic.
-
j.Pellet the cells by centrifuging at 5,000 g for 15 min at room temperature (20°C–22°C).
-
k.Isolate plasmid using a midiprep endotoxin-free plasmid purification kit per manufacturer’s instructions.
-
l.Determine the purity and concentration of isolated plasmid using a UV-Vis spectrophotometer.
-
a.
Figure 1.

Design and cloning of MitoT construct
(A) Relevant indicated fragments of the IM-resident SCO1 and OM-residing TOMM20 proteins were amplified individually by overlap extension PCR strategy. In the second round of PCR amplifications, the respective amplicons were used to generate a single recombinant DNA fragment (MitoT) containing indicated portions of SCO1, TOMM20, a hexa-histidine tag, an unstructured linker region (E. coli LacI sequence), and BamH1 and EcoRI restriction sites at the 5′- and 3′-flanking ends, respectively.
(B) The MitoT DNA fragment generated by overlap extension PCR and its host pCDNA3-eGFP vector were digested with BamHI and EcoRI restriction enzymes. The digested fragments were purified and ligated to yield pCDNA3-MitoT-eGFP vector.
(C) The E. coli DH5α competent cells were transformed with the resulting pCDNA3-MitoT-eGFP vector and transformed clones identified by plasmid isolation and restriction digestion. The verified positive clone was further propagated for endotoxin free plasmid isolation and sequence validation.
Cell culture, transfection, and FACS-based selection
The following steps provide details about transfection of the pcDNA-MitoT-eGFP vector in MEF cells followed by antibiotic selection and FACS-based selection of GFP-expressing cells (Figure 2).
-
6.Transfection and selection.
-
a.Twenty-four hours prior to transfection, seed 5 × 105 cells per well into 6-well plate or 35 mm dish.
-
b.Transfect the cells with 1 μg of control vector pcDNA3-eGFP and pcDNA3-MitoT-eGFP plasmid using Lipofectamine 3000 transfection reagent.
-
c.Change the media after 16 h of transfection.
-
d.At 48 h post-transfection, start the selection of G418-resistant clones using 2 mg/mL of G418.Note: Before treatment, an individual effective concentration of G418 needs to be determined for every cell line used. It is essential to make a titration curve to identify the lowest concentration of antibiotic that kills 90% of wildtype cells within 3–5 days.
-
e.Keep the cells under antibiotic pressure for at least 7 days, replacing the medium every 2 days.
-
f.Confirm expression and establishment of a MitoT-expressing stable cell line by fluorescence microscopy or by western blotting using anti-GFP antibody.
-
a.
-
7.FACS sorting.
-
a.Harvest the cells and make a cell suspension with approximately 5 × 106 cells per mL.Note: Use Ca2+- and Mg2+-free PBS to prevent the formation of cell aggregates.CRITICAL: To decrease the aggregate formation and maintain cell viability, supplement PBS with 1% FBS, 5 mM EDTA, and 25 mM HEPES, pH 7.4.
-
b.Sort the MitoT-GFP-positive cells using a 488-nm laser with a 530/30 bandpass filter.
-
c.Use 100 μm nozzle size, 20 psi for fluid sheath pressure, and 6,000 events/sec maximum rate.
-
d.Use non-transfected cells to determine proper threshold gate for eGFP-negative cells.
-
e.Sort the cells in 10% FBS-containing DMEM medium containing 20 mM HEPES, pH 7.4.CRITICAL: In our experiments using OMA1 knockout MEF cells transfected with pcDNA3-MitoT-eGFP, we observed two populations of GFP-expressing cells (Figure 2). The population of brighter cells with eGFP intensity greater than 104 on the histogram (referred to as GFPhi) results from protein localization error. In these cells, most of the eGFP signal was in the cytoplasm. The population of cells with GFP intensity close to 103 (referred to as GFPlo) displayed eGFP co-localization with mitochondria. These values can vary among different cell types and FACS instruments. It is important to carefully analyze isolated cells under a fluorescence microscope to confirm proper subcellular localization of the eGFP signal.
-
f.After sorting and validating the cells, centrifuge cells at 300 g for 10 min at room temperature (20°C–22°C), remove the medium, and transfer the cells to the 10% FBS-containing DMEM medium and dish containing 2 mg/mL of G418.CRITICAL: The cells must always be kept in G418 to ensure the retention of the MitoT-eGFP construct.
-
a.
Figure 2.

Generation and isolation of MitoT-expressing cells
Mouse embryonic fibroblasts were transfected with pCDNA3-MitoT-eGFP vector, cultured and harvested as explained in the text. Antibiotic-resistant cells were then profiled and processed by FACS. Importantly, FACS profiling revealed two distinct cell populations with low (GFPlo) and high (GFPhi) eGFP expression. Cells from the GFPlo group were confirmed to be the desired cell population and collected for further analyses.
Clone selection
This section describes the process of stable clone generation of FACS-sorted GFP positive cells by antibiotic selection. This is a final step of clone selection by antibiotic treatment after FACS-sorting of the GFPlo MEF cell population (Figure 3). It is expected that under antibiotic selection pressure the surviving cells should harbor stably integrated MitoT-eGFP expression construct.
-
8.Culturing sorted cells.
-
a.Re-seed the FACS-sorted cells in a 6-well plate.
-
b.Add 10% FBS-containing DMEM medium containing 2 mg/mL of active G418 antibiotic.
-
c.Change the medium every other day with antibiotic selection.
-
d.After 7 days culturing under the selective conditions, reconfirm plasmid retention by checking for the presence of eGFP signal under a fluorescence microscope.
-
e.Trypsinize and re-seed surviving cells into a fresh 6-well plate.
-
f.Allow cells to grow to 90% confluency.
-
g.Cryofreeze some of the cells for future use.
-
h.Seed the cells into glass-bottom 35-mm dishes.
-
a.
Figure 3.

Selection of stable MitoT-expressing clones
The selected cells were cultured under antibiotic selective conditions to produce stable clones of the MitoT-eGFP-expressing cells. The surviving clones were preserved and used in subsequent imaging studies.
Live cell imaging by confocal fluorescence microscopy
This section describes steps to prepare samples for fluorescence imaging. Set up a confocal fluorescence microscope according to the manufacturer’s instructions prior to sample preparation for imaging.
-
9.Live cell imaging (Figure 4A).
-
a.Prepare a fresh 1 mM stock solution of MitoTracker DeepRed dye in DMSO.Note: For storage, keep 1 mM MitoTracker DeepRed dye solution at −20°C.
-
b.Dilute the dye at 1:1000 dilution (final working concentration 1 μM) in FluoroBrite DMEM medium.
-
c.Remove the culture medium and wash the cells once with 1 mL of 1× PBS.
-
d.Add 1 mL of MitoTracker dye-containing medium to the 35-mm dish.
-
e.Incubate for 30 min at 37°C.
-
f.Remove medium and wash cells twice with 1 mL FluoroBrite DMEM medium only.
-
g.Add 1 mL of FluoroBright DMEM medium containing 1:2000 diluted Hoechst 33342 (final working concentration 5 μg/mL) nuclear staining dye.
-
h.Incubate for 10 min (protected from light!) and then wash twice with FluoroBright DMEM medium.
-
i.Add 1 mL of FluoroBrite DMEM medium supplemented with 1% FBS.Note: Using 1% FBS prevents cell starvation without increasing background fluorescence on images.
-
j.Observe cells using 60× oil objective.
-
k.Use ultraviolet 405 nm solid state laser for Hoechst 33342 excitation, 425–475 nm bandpass filter for emission and focus the cells according to optimum nuclear staining signal. The maximum emission wavelength for Hoechst 33342 dye is 461 nm.Note: For live cell imaging, it is beneficial to use stage top CO2 incubator to minimize cell stress and to preserve proper mitochondrial morphology.Note: Hoechst 33342 nuclear stain is used here only to focus cells hence it is optional yet highly recommended. Pre-focusing the cells using Hoechst signal minimizes the GFP bleaching due to long exposure of laser. Observe eGFP signal using 488 nm solid state excitation laser and 500–550 nm bandpass emission filter. The maximum emission wavelength for eGFP protein is 509 nm.
-
l.For MitoTracker DeepRed imaging, use 640 nm solid state excitation laser and 633–673 nm emission filter. The maximum emission wavelength for MitoTracker DeepRed is 665 nm.CRITICAL: If a stage-top incubator for CO2 control is not available, the medium must be supplemented with 25 mM HEPES buffer, pH 7.4.
-
a.
Figure 4.

Visualization of MitoT-GFP expression, its colocalization with mitochondria, and ultrastructure analysis
(A) Collected MitoT-eGFP-expressing cells (GFPhi and GFPlo) were treated with MitoTracker Deep Red dye (MTR) and live-imaged by confocal fluorescent microscopy to ensure proper mitochondrial localization of the MitoT-eGFP synthetic protein. Scale bar, 20 μm.
(B) Representative TEM images of OMA1 KO and OMA1 KO_MitoT cells that have been treated with an uncoupler CCCP or left untreated. Induced expression of MitoT in OMA1 KO cells mitigates abnormal mitochondrial cristae morphology that becomes visible upon uncoupling. Scale bar, 0.4 μm. Images adopted from Viana et al. (2021).
Transmission electron microscopy (TEM) imaging
This section describes steps of sample preparation and visualization of cristae stabilization by MitoT using transmission electron microscopy (TEM) (Figure 4B).
-
10.TEM Imaging.
-
a.Grow cell lines of interest in a 30-mm culture dish overnight (∼12–16 h).
-
b.Treat the cells with 2 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or without CCCP (mock control: DMSO) for 1 h at 37°C in a CO2 incubator.
-
c.Trypsinize the cells and wash three times with 500 μL PBS.
-
d.Count cells using an automated cell counter (Online Automated Cell Counting Protocol) and pellet 1 million cells for each cell line in question.Note: Prepare and process 4–5 replicates per cell line being analyzed.
-
e.Fix with 2.5% glutaraldehyde (prepared in TEM wash buffer) for 1 h at room temperature (20°C–22°C) and then keep overnight (∼12–16 h) at 4°C.CRITICAL: Prepare fresh fixative immediately before use.CRITICAL: Glutaraldehyde is a skin and respiratory tract irritant. It should be used in a fume hood with caution and protective wear.
-
f.Wash the cells with 500 μL TEM wash buffer 3× 5 min at 500 g.
-
g.Pellet the cells and embed in 1% low-melting agarose.
-
i.Dissolve 1% low-melting agarose at 65°C in TEM wash buffer.
-
ii.Add 250 μL lukewarm agarose to the cell pellets, vortex to resuspend and centrifuge at 9,500 g at room temperature (20°C–22°C) for 5 min.
-
iii.Allow agarose to solidify at 4°C for 3–4 h.
-
iv.Transfer agarose to a Petri dish using small spoon-scoop spatula and filter forceps. Cut out a 2-mm block of solidified agarose containing the cell pellet.
-
i.
-
h.Post-fix the cells with 1% osmium tetroxide in deionized water for 1 h.CRITICAL: Osmium tetroxide is a skin and respiratory tract irritant. It should be prepared and used in a fume hood with caution and protective wear. Prepare and store Osmium tetroxide in amber bottle at 4°C. Osmium tetroxide is a heavy metal and should not be drained off the sink. Follow institutional guideline for the proper disposal.
-
i.Wash the cells with 500 μL deionized water three times for 5 min each time.
-
j.Gradually dehydrate agarose block with embedded cells by washing it with 50%, 75%, 95% (5 min each wash) and 100% ethanol (twice, 10 min each wash).CRITICAL: Do not allow samples to air dry!
-
k.Follow below steps to embed the cells:
-
i.Keep the cell block in a double-end mold and incubate in 1:1 solution of 100% ethanol/Spurr’s solution for 2 h at room temperature (20°C–22°C).
-
ii.Incubate in pure Spurr’s solution for 2–3 h at room temperature (20°C–22°C).
-
iii.Keep for embedding in Spurr’s solution for 24–36 h at 60°C in a vacuum oven.
-
i.
-
l.Remove the dried block and trim the edges.
-
m.Cut 70–90 nm-thick sections using a Leica UC7 ultramicrotome.
-
n.Place the sections on EM grids and stain with 1% uranyl acetate for 10 min. Rinse thoroughly (up to 10 times) with double distilled water (ddH20).
-
o.Stain sections with 1% lead citrate for 5 min. Spot 100 μL of ddH20 at 8 places on a Petri dish and dip grid 10 times into each spot for washing.CRITICAL: Uranyl acetate and lead citrate are highly toxic heavy metals and should be used with utmost care. Do not drain off in sink; follow institutional guideline for proper disposal of the used solutions.
-
p.Image at 10,000× and 25,000× magnification with a Hitachi H7500 transmission electron microscope at 80 kV setting. Take 25–30 images for each section.
-
a.
Mitochondrial isolation and alkaline extraction of mitochondrial proteins
This part briefly describes steps of isolation of MitoT-expressing mitochondria and carbonate extraction of mitochondrial proteins to ensure membrane anchoring of MitoT (Figure 5).
-
11.Mitochondria Isolation.
-
a.Remove media, wash the cells with 5 mL 1× PBS and harvest 20 million MitoT-eGFP expressing MEF cells by scraping the cells with a rubber policeman.Note: Mitochondrial yield may vary depending on the cell type. Users may need to determine appropriate cell number for mitochondria isolation.
-
b.Pellet down cells at 800 g for 5 min at room temperature (20°C–22°C) and remove supernatant.
-
c.Resuspend cells in 2 mL pre-chilled MTE buffer per 10 million cells (add 10 μL of 0.2 M PMSF per mL of MTE buffer just before use).Note: Use 15 mL tubes containing no more than 6 mL of MTE buffer.CRITICAL: Avoid touching the sonicator probe with the walls of plastic tube.
-
d.Sonicate for 10 s at 20%–30% of amplitude.
-
e.Keep samples on ice for 20 s.
-
f.Repeat steps ‘d’ and ‘e’ two more times.
-
g.Centrifuge at 2,000 g for 10 min at 4°C.
-
h.Recover the supernatant, transfer recovered supernatant to a new tube and centrifuge at 12,000 g for 10 min at 4°C.
-
i.The pellet formed is the crude mitochondria fraction. Add 500 μL of MTE buffer containing 0.2 M PMSF, resuspend the pellet and aliquot in 50 μL fractions.
-
j.Determine total protein concentration of mitochondria-enriched fraction using Coomassie (Bradford) Protein Assay kit.Note: Flash-freeze crude mitochondrial fractions in liquid nitrogen and store aliquots at −80°C for later use.CRITICAL: Liquid nitrogen is a skin, eyes, and respiratory tract irritant. It should be used with caution and protective wear.
-
a.
-
12.Alkaline extraction of mitochondrial proteins.
-
a.Take 100 μg of mitochondria, pellet the organelles at 12,000 g for 10 min at 4°C, and resuspend pelleted mitochondria in 0.5 mL of 0.1 M alkaline sodium bicarbonate solution, pH 7.5 or 11.5. Use 0.5 mL of ultrapure water for mock-treatment sample.
-
b.Vortex and incubate samples on ice for 30 min.
-
c.Transfer samples into 5 mL polycarbonate tubes and adjust volume to 3 mL with 0.1 M sodium bicarbonate or ultrapure water. Ultracentrifuge at 175,000 g for 30 min at 4°C. Keep obtained pellets on ice for further steps.
-
d.Take supernatant fraction into a fresh 1.5 mL tubes and precipitate protein using 20% TCA and 0.07% (w/v) sodium deoxycholate. Incubate these samples on ice for 45 min.
-
e.Centrifuge at 16,000 g for 10 min at 4°C.
-
f.Wash the pellet with ice-cold acetone and centrifuge at 16,000 g for 2 min at 4°C. Repeat this step two times. Air dry the pellet after final wash.Note: The pellet obtained at step ‘c’ is the insoluble fraction and contains integral proteins, which do not solubilize in alkaline bicarbonate buffer. The soluble proteins are precipitated by using TCA buffer and obtained as a pellet after TCA precipitation and centrifugation.Note: The same soluble pellets containing TCA-precipitated proteins can be pooled together at this step.
-
g.Resuspend both pellets obtained at step ‘c’ and step ‘d’ in 10 μL of 0.1M NaOH and perform standard SDS-PAGE and immunoblotting with relevant antibodies.
-
a.
Figure 5.

Isolation of MitoT-GFP expressing mitochondria and alkaline extraction of mitochondrial proteins
(A) Workflow for isolation of MitoT-expressing mitochondria.
(B) Workflow for alkaline sodium carbonate extraction and fractionation of mitochondrial proteins.
(C) Representative immunoblots of MitoT-eGFP and control mitochondrial proteins at indicated pH or without Na2CO3 treatment (-). These experiments demonstrate retention of MitoT-eGFP in the extraction-resistant fractions (P), thereby indicating membrane anchoring of the protein. HSPD1 and NDUFA9 were used as controls for soluble and membrane associated mitochondrial proteins respectively. P = pellet fraction, S = Soluble fraction, T = Total protein (10% of fraction loaded).
Expected outcomes
The present protocol describes the design, synthesis, and application of a synthetic mitochondrial protein, MitoT. This construct bridges the outer and inner mitochondrial membranes and provides additional stability to the mitochondrial intermembrane contact sites. We used this protocol for MEF cells but recently it has been replicated successfully in U2OS cells (Li et al., 2022). This construct is useful in situations where mitochondria membrane stability or MICOS function gets compromised due to absence or mutation in mitochondrial quality control and structural proteins.
Figure 1 provides a schematic depiction of MitoT vector design and cloning procedures. The MitoT fusion protein comprises the N-terminal residues of the inner mitochondrial membrane protein SCO1, a 12 amino acid-long unstructured linker region, a transmembrane region of the outer mitochondrial membrane protein TOMM20, and an eGFP tag. This fusion construct was synthesized by an overlap extension PCR approach. The EcoRI and BamHI restriction sites were introduced in the 5′- and 3′- regions of the resulting DNA fragment, respectively. The amplified construct was gel-purified and examined by UV-Vis spectroscopy to assess its concentration and purity. Both the fusion construct and pcDNA3-eGFP vector were digested with EcoRI and BamHI restriction enzymes and purified using a PCR purification kit. The resulting DNA fragments were ligated and transformed into E. coli DH5α chemically competent cells. Transformant colonies were picked up individually and cultured for subsequent plasmid DNA isolation and insert validation by restriction digestion followed by re-isolation and DNA sequencing of positive plasmid constructs. The positive clones were then amplified and endotoxin-free purified for further applications.
Figure 2 shows the workflow of MitoT construct transfection in cultured MEF cells and FACS-based analysis and isolation of transfected cells. The MEF cells were revived and cultured in the indicated growth medium. The cells were transfected with the control vector (pcDNA3-eGFP) or MitoT-harboring plasmid. At 48 h post-transfection, cells were selected in G418 for 1 week. Post selection, cells were trypsinized and FACS-sorted based on eGFP expression intensity.
Figure 3 depicts the process of stable integration of MitoT vector through antibiotic selection. The eGFP-positive cells isolated via FACS procedure were re-cultured and treated with gentamycin antibiotic for 7 days. Cells that survived the antibiotic treatment were individually selected and cultured to obtain stable clones of MitoT-expressing MEFs.
Figure 4 shows microscopy images of MEF cells expressing eGFP vector or MitoT-eGFP. Mitochondria were stained with MitoTracker Deep Red dye and imaged using a live confocal fluorescence imaging system. The red (MitoTracker) and green (eGFP) signals were merged to demonstrate co-localization. The representative image shows that MitoT protein specifically localizes to mitochondria. TEM images of mock or CCCP-treated OMA1 KO MEF cells and OMA1 KO MEF cells expressing MitoT show the effect of CCCP treatment on cristae morphology. Specifically, the treatment of OMA1 KO cells with CCCP disrupts cristae morphology, whereas this effect is mitigated by MitoT overexpression.
Figure 5 shows the workflow of mitochondrial isolation from MitoT-eGFP expressing cells and subsequent alkaline extraction of mitochondrial proteins to ascertain membrane integration of MitoT. The representative figure shows immunoblot analyses of fractionated MitoT-eGFP and control mitochondrial proteins demonstrating the resistance of the former to alkaline carbonate extraction, thereby validating that MitoT is indeed an integral membrane protein.
Limitations
First, the MitoT has been only successfully tested in OMA1 knockout MEF cells and wild-type U2OS cells. The broader application of this system remains to be established. For instance, MitoT could be deployed to study mechanisms of mitochondrial membrane stabilization by many other proteins including the MICOS subunits. Second, the mechanism of MitoT-mediated cristae stabilization remains incompletely understood. Exactly how the expression of the inner and outer mitochondrial membrane-bridging polypeptide rescues the complex cristae architecture of mitochondria requires further investigation. Third, the extent to which MitoT is able to rescue mitochondrial functions also needs further testing under different stress conditions.
Finally, we used an antibiotic-based selection approach to obtain stable clones of MitoT-expressing cells, which has a shortcoming of spontaneous reversal of cells to their original phenotype. Using lentiviral-based vectors may be advantageous for stable clone generation.
Troubleshooting
Problem 1
Very little or no GFPlo-cells are observed during the FACS procedure (step 7e).
Potential solution
The concentration of antibiotic used in the screening procedure is essential for obtaining a stable line of MitoT-expressing cells. Using >2 mg/mL of G418 may be beneficial in accelerating the attainment of stable cells; however, this may also lead to selecting cells with multiple copies of the plasmid, which would result in MitoT-eGFP overexpression, causing its unwanted accumulation in the cytosol. It is essential to make a titration curve to identify the lowest concentration of antibiotic that kills 90% of cells within 3–5 days.
Problem 2
Cell line is not responding to antibiotic treatment or shows a very low recovery of GFP-positive cells after FACS sorting irrespective of initial transfection efficiency (steps 8d and 7e).
Potential solution
Some immortalized cell lines may contain an endogenous (e.g., neomycin) antibiotic resistance gene. The presence of such a gene makes the selection of stable cells considerably more challenging. In this case, it is recommended to use vectors harboring different antibiotic resistance marker genes such as blasticidin or puromycin.
Problem 3
Loss of eGFP signal or plasmid by a presumably stable cell line after a few passages.
Potential solution
Stable cell line generation using this protocol is based on the efficiency of antibiotic selection and random integration of the gene of interest. Some cell cultures are not very prone to such random integration and consequently could lose the gene quickly, even under selective pressure conditions. To circumvent this issue and produce long-lasting stable cell lines, we recommend using vectors that carry a transposase such as piggyBac, Sleeping beauty, or even lentiviral particles.
Problem 4
Weak adherence of cells to glass-bottom dish (step 9c).
Potential solution
Glass bottom dishes are essentially required for live cell microscopy, but glass is a poor substrate for cell adherence and some cell lines may detach easily during washing or staining step. Coating glass surface with appropriate coating material can overcome this issue.
Problem 5
Low mitochondria yield (step 11j).
Potential solution
The mitochondrial yield depends on the cell type and culture media composition. To improve the mitochondrial yield, more cells can be cultured. Alternatively, culturing cells in 10 mM galactose-containing media for 48 h before harvesting also increases the cellular mitochondrial content and, ultimately, mitochondrial yield.
Resource availability
Lead contact
Requests for resources and materials should be directed to the corresponding author, Oleh Khalimonchuk (okhalimonchuk2@unl.edu).
Materials availability
All reagents are available from the lead contact upon request and with a completed materials transfer agreement.
Acknowledgments
We acknowledge help from the Flow Cytometry Service Center and the Morrison Microscopy Core Facility at the University of Nebraska-Lincoln. We thank Dr. Jennifer Fox for critically reading the manuscript. This work was supported, in whole or in part, by NIH grants GM108975 and GM131701-01 to O.K.
Author contributions
Conceptualization, M.P.V. and O.K.; investigation, M.P.V.; writing – original draft, G.P. and M.P.V.; writing – reviewing & editing, M.P.V., G.P., and O.K.; funding acquisition, O.K.; supervision, O.K.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101454.
Contributor Information
Gunjan Purohit, Email: gpurohit2@unl.edu.
Oleh Khalimonchuk, Email: okhalimonchuk2@unl.edu.
Supplemental information
Data and code availability
No datasets or codes were generated in this study.
References
- Aaltonen M.J., Friedman J.R., Osman C., Salin B., di Rago J.P., Nunnari J., Langer T., Tatsuta T. MICOS and phospholipid transfer by Ups2-Mdm35 organize membrane lipid synthesis in mitochondria. J. Cell Biol. 2016;213:525–534. doi: 10.1083/jcb.201602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhaja A.K., Jans D.C., Nikolov M., Vukotic M., Lytovchenko O., Ludewig F., Schliebs W., Riedel D., Urlaub H., Jakobs S., Deckers M. MINOS1 is a conserved component of mitofilin complexes and required for mitochondrial function and cristae organization. Mol. Biol. Cell. 2012;23:247–257. doi: 10.1091/mbc.E11-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harner M., Körner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U., Griffith J., Mann M., Reggiori F., Neupert W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011;30:4356–4370. doi: 10.1038/emboj.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B.P., Zulaika M., Ryazantsev S., Van Der Bliek A.M. A novel mitochondrial outer membrane protein, MOMA-1, that affects cristae morphology in Caenorhabditis elegans. Mol. Biol. Cell. 2011;22:831–841. doi: 10.1091/mbc.E10-07-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S., Collins S.R., Cassidy-Stone A., Hummel E., DeVay R.M., Lackner L.L., Westermann B., Schuldiner M., Weissman J.S., Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 2011;195:323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John G.B., Shang Y., Li L., Renken C., Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell. 2005;16:1543–1554. doi: 10.1091/mbc.E04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Straub J., Medeiros T.C., Mehra C., den Brave F., Peker E., Atanassov I., Stillger K., Michaelis J.B., Burbridge E., et al. Mitochondria shed their outer membrane in response to infection-induced stress. Science. 2022;375:eabi4343. doi: 10.1126/science.abi4343. [DOI] [PubMed] [Google Scholar]
- Pfanner N., van der Laan M., Amati P., Capaldi R.A., Caudy A.A., Chacinska A., Darshi M., Deckers M., Hoppins S., Icho T., et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014;204:1083–1086. doi: 10.1083/jcb.201401006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós P.M., Ramsay A.J., Sala D., Fernández-Vizarra E., Rodríguez F., Peinado J.R., Fernández-García M.S., Vega J.A., Enríquez J.A., Zorzano A., López-Otín C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl R., Soubannier V., Scholz R., Vogel F., Mendl N., Vasiljev-Neumeyer A., Körner C., Jagasia R., Keil T., Baumeister W., et al. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J. Cell Biol. 2009;185:1047–1063. doi: 10.1083/jcb.200811099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Laan M., Horvath S.E., Pfanner N. Mitochondrial contact site and cristae organizing system. Curr. Opin. Cell Biol. 2016;41:33–42. doi: 10.1016/j.ceb.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Viana M.P., Levytskyy R.M., Anand R., Reichert A.S., Khalimonchuk O. Protease OMA1 modulates mitochondrial bioenergetics and ultrastructure through dynamic association with MICOS complex. iScience. 2021;24:102119. doi: 10.1016/j.isci.2021.102119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollweber F., von der Malsburg K., van der Laan M. Mitochondrial contact site and cristae organizing system: a central player in membrane shaping and crosstalk. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:1481–1489. doi: 10.1016/j.bbamcr.2017.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets or codes were generated in this study.