

Abstract
The targeted delivery of therapeutics to diseased tissue enhances their efficacy and decreases their side effects. Here, we report enucleated mesenchymal stromal cells (MSC) as vehicles for the targeted delivery of therapeutic compounds. The cell nuclei are removed by density gradient centrifugation following the genetic integration of disease-targeting proteins that can be directed to improve the tissue-specific homing properties of the cells. Enucleated MSCs do not proliferate or permanently engraft in the host, yet retain organelles for energy and protein production, undergo integrin-regulated adhesion to inflamed endothelial cells, and actively home to chemokine gradients released by diseased tissues. In preclinical mouse models of acute inflammation and of pancreatitis, the systemic administration of enucleated MSCs, expressing two chemokine receptors and an adhesion molecule, improved the delivery of the cytokine interleukin-10 to target tissue, attenuating inflammation and ameliorating pancreatitis, compared to control exosomes and non-engineered MSCs. Enucleated MSCs are therapeutic delivery vehicles that bridge the functional properties of cells with the cargo-loading characteristics of extracellular vesicles and could be applied to treat a wide range of diseases.
Systemic administration of conventional therapeutic agents is often accompanied by unwanted side effects and toxicity due to accumulation in off-target sites, the lack of drug selectivity, and the inability to completely access diseased tissues1, 2. For example, cytokines such as IL-10 and IL-12 have potent immunomodulatory functions and potential to treat various diseases including acute pancreatitis, inflammatory bowel diseases, and cancer3, 4. However, their short half-life in the circulation, low bioavailability in target tissue, and adverse effects related to systemic administration hinder clinical successes3, 4. Several targeted-delivery platforms have been developed over the years to address these limitations and improve efficacy including nanotechnology-based drug delivery systems, antibody-drug conjugates, and bioinspired natural delivery methods2. Bioinspired natural delivery technologies are especially attractive because they closely mimic innate chemical and biological characteristics of biological surfaces in chemical composition, membrane fluidity, 3D protein presentation, and in some cases, possess innate disease targeting abilities2, 5–7. Naturally-derived drug delivery vehicles have been shown to display improved biodistribution, biodegradation, immune compatibility, and reduced toxicity compared to synthetically-derived nanotechnology-based drug delivery systems2. Several different bioinspired platforms are currently under investigation for drug delivery that utilize bacteria, viruses, erythrocytes, immune cells, stem cells, exosomes, membrane vesicles, and isolated membrane ghosts2, 8. Among these platforms, MSCs have gained appreciable attention as therapeutic delivery vehicles largely due to their innate disease targeting and paracrine secretion capacities8, 9.
Eukaryotic cells have evolved elegant innate communication systems to guide migrating stem and immune cells to injured or diseased tissues based on the unique expression profiles of chemoattractants, chemokines, selectins, integrins or other cell-surface adhesion molecules10–12. The physiological process by which cells leave the circulation and home to diseased/injured tissues in response chemical signals is referred to as chemotactic homing12. The initial stage of cell homing is mediated in a highly precise and regulated manner by specific selectins expressed by endothelial cells (ECs), which induce transient cell tethering and rolling on the endothelium at sites of disease12. This initial adhesion step also facilitates cell exposure to specific chemokines displayed on the surface of ECs, which bind to their cognate chemokine receptors to activate a cascade of intracellular signaling responses and lead to high affinity integrin activation12. High affinity integrin receptors promote firm cell arrest on the endothelium and cell sequestration from the circulation12. Upon firm adhesion, sequestered cells undergo transendothelial migration (TEM), penetrate through the basement membrane, and then move into the perivascular space12. Cells continue to move through the extravascular tissue to sites of disease or injury in response to chemoattractant gradients12. After cells arrive at diseased foci, they continue to position themselves near injured cells and immune cells using chemotactic mechanisms, so that they can communicate through membrane connections and secreted factors needed for proper immune cell communication and healing9, 10, 12. Significant works have shown that chemotactic migration uses complex gradient sensing mechanisms, signal transduction cascades, substantial amounts of energy for actin-mediated cell shape changes, and actin/myosin-mediated force production, to actively drive cell propulsion through complex tissues to sites of disease9, 10, 12.
The specific ability of cells to decipher chemical guidance cues, produce energy, and generate propulsive forces to extravasate through the vasculature uniquely sets them apart from other nanoparticle and bioinspired drug delivery systems which lack these innate machineries8. For example, cell-free delivery vehicles (CFDV) have been developed in the past decade, including extracellular vesicles (EVs)5, erythrocyte-based vehicles6, and cell membrane-derived/coated vehicles (nanoparticles or nanoghosts)7. However, CFDVs have been denuded of critical organelles and crucial cellular machineries needed to produce energy, activate high affinity integrin-mediated endothelial adhesion, decipher chemical gradients, and generate propulsive forces. Therefore, current CFDVs cannot actively chemotax through the vascular barrier nor can they physically propel themselves deeply into complex tissues in response to chemical signals, which can reduce therapeutic efficacy8.
Ideally, cell-based delivery vehicles could be engineered with enhanced chemotactic abilities for improved targeted drug delivery12, but such delivery systems face several limitations before translation to the clinic8. For example, while cells such as MSCs have natural disease trophic properties, the inconsistent and insufficient homing of MSCs to target tissues following systemic infusion is considered a major contributor to the inadequate efficacy of MSC-based therapies12. Notably, MSCs are largely limited by initial entrapment in pulmonary capillaries after intravenous (i.v.) injection, inconsistent expression of chemokine homing receptors, and lack of expression of endothelial adhesion molecules12–14. This is further compounded by the inability to control cell fate in vivo, such as the secretion of undesired therapeutic factors, unpredictable engraftment into the body, or differentiation into unwanted cell types in vivo, all of which significantly limit predictable and reproducible therapeutic delivery15. While cell and genetic engineering approaches can overcome many of these hurdles, obtaining regulatory approval for cells with multiple genomic modifications remains difficult16. Here, we use recent bioengineering techniques to endow MSCs with multiple chemoattractant receptors and endothelial cell adhesion molecules that mediate homing to inflamed and diseased tissues. To render bioengineered MSCs safe and controllable, we use large-scale physical enucleation to generate a new type of disease homing CFDV, named “Cargocytes”. Cargocytes retain important intrinsic cellular functions such as protein translation and secretion, active chemotactic migration towards defined chemokine signals, and regulated high affinity integrin activation and endothelial cell adhesion in vitro. In two independent animal disease models, we demonstrate that bioengineered Cargocytes robustly home and deliver therapeutic cytokine cargos to diseased tissues, and ameliorated disease pathology. To our knowledge, this is the first bioengineered CFDV that utilizes active chemotaxis and endothelial adhesion machineries to deliver therapeutic cargos efficiently and specifically to targeted tissues.
Results
Cargocytes retain important organelles and cellular functions
The principle for physically enucleating mammalian cells using Cytochalasin B and Ficoll density gradient ultracentrifugation was established over 40 years ago17. However, research of the resulting enucleated cells has been primarily limited to in vitro studies18–20. To our knowledge, the potential of these artificially enucleated cells to be bioengineered for in vivo therapeutic delivery has not yet been explored (Fig. 1a). While various cell types can be enucleated (Supplementary Table 1)18–20, we selected hTERT-immortalized adipose-derived MSCs (hT-MSC) for generating prototype Cargocytes because MSCs have proven therapeutic potential in clinical studies and the immortalized phenotype provides a homogenous cell population with consistent characteristics to facilitate further cell and genetic engineering15, 21. We optimized protocols to routinely achieve >95% enucleation efficiency of hT-MSCs with an 80–90% recovery rate (Supplementary Table 1). The hT-MSC-derived Cargocytes (“Cargocyte”) are significantly smaller than hT-MSCs in suspension (Fig. 1b and c) and readily attach to tissue culture plates with well-organized cytoskeletal structure (Fig. 1d). Cargocytes are viable for up to 72 hours post-enucleation, as shown by the trypan blue dye exclusion assay (Fig. 1e). They contain crucial subcellular organelles, including Golgi, ER, mitochondria, lysosomes, and endosomes (Supplementary Fig. 1) and retain MSC surface marker protein expression for at least 48 hours (Supplementary Fig. 2a). Interestingly, EVs isolated from conditioned media (CM) of Cargocytes and hT-MSCs had similar characteristic cup-shaped morphology by electron microscopy (Fig. 1f and Supplementary Fig. 2b), similar size distribution under Nanosight analysis (Fig. 1g), and similar amount produced (0.59μg/1E6 Cargocytes versus 1.07 μg/1E6 hT-MSCs) as measured by BCA assay, suggesting that Cargocytes may actively produce and release EVs. Moreover, Cargocytes have similar zeta potential as parental hT-MSCs (Fig. 1h) and similar Annexin V staining for at least 48 hours post enucleation (Supplementary Fig. 2c), suggesting the integrity of the bi-layered plasma membrane. Importantly, following transfection with in vitro synthesized GFP mRNA, epi-fluorescent images and flow cytometry analyses showed that Cargocytes express cytoplasmic GFP protein comparable to hT-MSCs (Fig. 1i and j, and Supplementary Fig. 2d). Notably, Cargocytes transfected with exogenous Gaussia Luciferase (Gluc) mRNA secreted active Gluc in CM at levels similar to parental cells (Fig. 1k). To our knowledge, this is the first evidence that enucleated cells can translate exogenous mRNAs and secrete functional proteins, demonstrating their functional mRNA translation and protein secretory machineries. Together, these results indicate that Cargocytes retain critical cell structures and functions, and therefore have potential for in vivo homing and delivery of genetically encoded therapeutics.
Fig. 1 |. Enucleated cells retain important cellular functions.
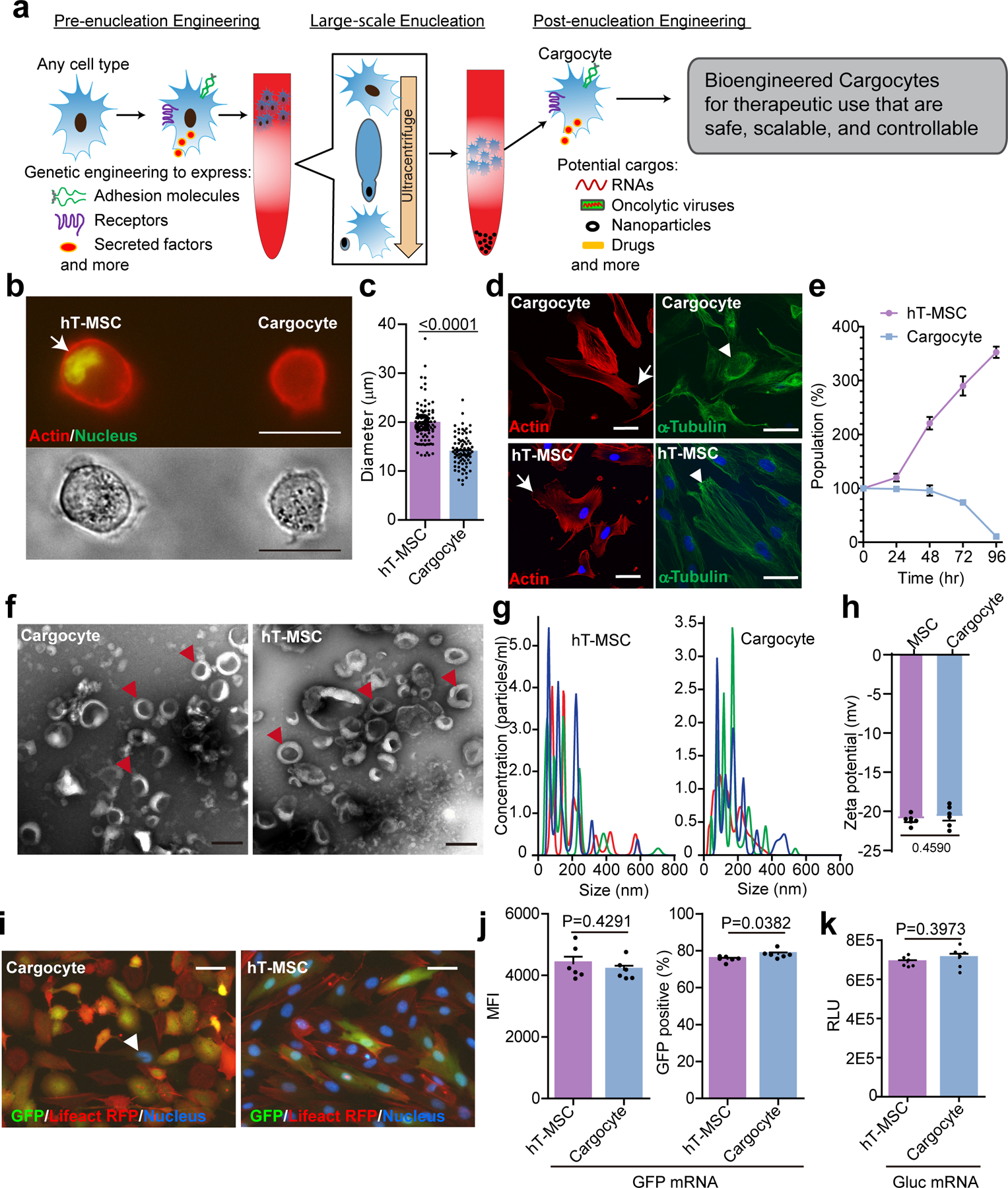
a, Schematic of workflow for therapeutic uses of bioengineered enucleated cells (Cargocytes). b, Fluorescent image (upper) and phase image (down) show hT-MSCs and Cargocytes in suspension. Representative images out of 30 images obtained are shown. Cortical actin was labeled with LifeAct-RFP. Arrow points to the cell nucleus stained with Vybrant Dyecycle Green. Scale bar, 20μm. c, Bar graph shows the average diameter of hT-MSCs or Cargocytes in suspension. Mean ± SEM; n=80 individual cells/Cargocytes. P value, two-tailed unpaired t-test. d, Fluorescent confocal images of hT-MSCs/Cargocytes stained with rhodamine phalloidin for F-actin cytoskeleton (left), or anti-α-Tubulin antibody for microtubule network (right), and Hoechst 33342 for nucleus. Representative images out of 30 images obtained are shown. Arrows point to the F-actin cytoskeleton; arrowheads point to the microtubule network. Scale bar, 50μm. e, Graphs show the percentage of viable hT-MSCs/Cargocytes versus initial population over time. Mean ± SEM; n=6 biological replicates. f, Electron microscopy images of extracellular vesicles (EVs) from hT-MSCs and Cargocytes. Representative images out of 10 images obtained are shown. Arrowheads point to typical EVs. Scale bar, 200 nm. See also Supplementary Fig.2b for higher resolution images. g, Histograms showing the size distribution of EVs from conditioned media of MSCs or Cargocytes analyzed by NanoSight. The red, green, and blue colored lines show technical triplicates from each sample. h, Bar graph shows the zeta potential of MSCs or Cargocytes measured by dynamic light scattering (DLS) technique using a Zetasizer Nano ZS analyzer and analyzed by Dispersion Technology Software. i, Epi-Fluorescent images show LifeAct RFP (red) hT-MSCs/Cargocytes stained with Hoechst 33342 (blue) 24hr (hour) post transfection of GFP (green) mRNA. Representative images out of 30 images obtained are shown. Arrowhead points to a single nucleus in the Cargocyte panel. Scale bar, 50μm. j, Bar charts show the mean fluorescent intensity (MFI) of GFP (left) or GFP positive ratio (right) of cells treated as in (i) and analyzed by flow cytometry. k, Bar graph shows Gaussia luciferase (Gluc) activity of conditioned medium from cells 48hr post-transfection with Gluc mRNA. RLU=Relative luminescence units. For (h), (j), and (k), Mean ± SEM; n=6 biological replicates; P value, two-tailed unpaired t-test. All data are representative of at least 2 independent experiments.
Cargocytes chemotax towards chemoattractant gradients
Chemotaxis is a critical step controlling the active cell homing in vivo14, so we first investigated the chemotactic ability of Cargocytes. Consistent with previous studies22, Cargocytes sensed and migrated towards various chemoattractants in an in vitro Boyden chamber assay (Fig. 2a to c), and invaded through 3D Matrigel-coated membranes towards FBS gradients in a basement membrane invasion assay (Supplementary Fig. 3b and c). Additionally, we used a microfluidic device in which LifeAct-RFP-labeled hT-MSCs and Cargocytes migrate along a FBS gradient through confined 3D-constrictions that mimic interstitial pores23. Cell chemotaxis was imaged by time-lapse confocal microscopy to record the time required for cells to migrate through individual constrictions. Notably, Cargocytes efficiently passed through small constrictions (≤2×5μm in cross section), whereas hT-MSCs were often trapped in confined constrictions (Fig. 2d and e, Supplementary Movie1 and 2). This result suggests that Cargocytes may have better deformability compared to parental cells, which is consistent with previous studies showing that the nucleus is 2 to 10 times stiffer than the surrounding cytoplasm and physically decoupling the nucleus from the cytoskeleton is sufficient to increase cytoskeletal deformability24–27. Together these findings demonstrate that Cargocytes retain intrinsic chemotaxis activity, basement membrane invasion, and have better deformability than parental MSCs, which may better facilitate passing through small capillaries, endothelial cell layers, or interstitial spaces in vivo.
Fig. 2 |. Cargocytes have chemotaxis activities and migrate better through confined spaces.
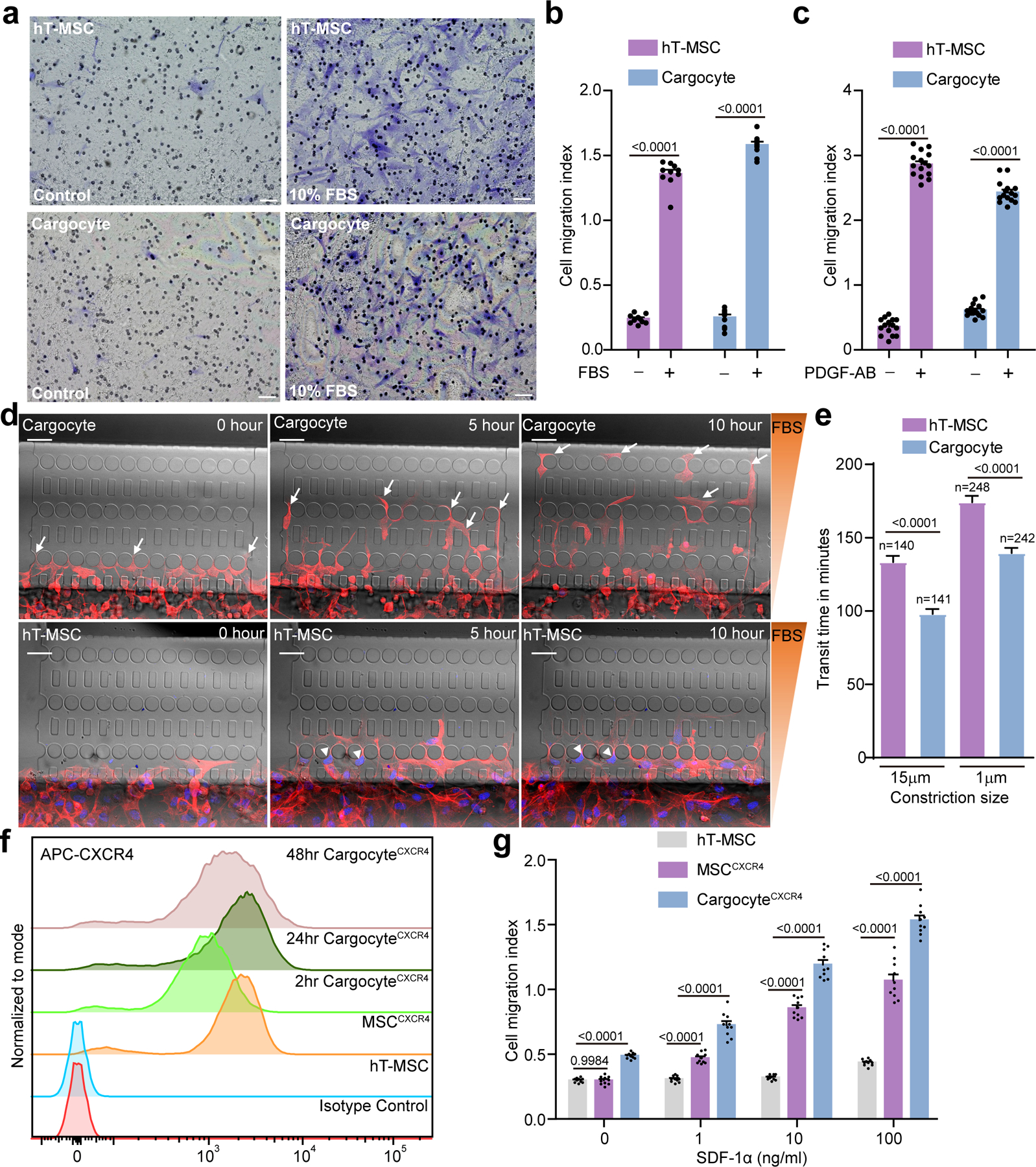
a, MSCs/Cargocytes migrated in Boyden chambers towards FBS (Fetal bovine serum) gradients for 2hr. Representative brightfield images of MSCs or Cargocytes that migrated to the underside of 8.0μm porous filters were stained with Crystal Violet. Scale bar, 50μm. b, Bar graph represents the cell migration index (migrated MSCs/Cargocytes versus loading control). Mean ± SEM; n=10 independent fields from 3 biological replicates. See also Supplementary Fig.3a for attachment control. c, Bar graph represents cell migration index of MSCs/Cargocytes towards PDGF-AB gradients. Mean ± SEM; n=15 independent fields from 5 biological replicates. d, Time-lapse image sequence of hT-MSCs and Cargocytes moving through constrictions (≤2×5μm in cross section) along an FBS gradient in a microfluidic device. F-actin cytoskeleton (red) was labeled with LifeAct-RFP and cell nucleus (blue) was stained with Hoechst 33342. Arrows point to migrating Cargocytes and arrowheads point to hT-MSCs trapped in the confined constrictions. Scale bar, 50μm. See also Supplementary Movies 1 and 2. e, MSCs and Cargocytes were treated as in (d). Bar graph shows the average time required for cells to migrate through an individual microfluidic constriction. Data for both confined (≤2μm×5μm) and unconfined (15μm×5μm) constrictions are shown. Mean ± SEM. Data were pooled from 3 independent experiments. Exact number in each group shown above the bar. P value, one-way ANOVA with Bonferroni’s multiple comparison tests. f, Graph shows cell surface expression of CXCR4 by flow cytometry. Data analyzed in Flowjo and normalized to mode. MSCCXCR4, CXCR4 lentivirus-engineered hT-MSC; 2hr/24hr/48hr Cargocytes, MSCCXCR4-derived Cargocytes analyzed at indicated time points post-enucleation; Parental hT-MSC, non-engineered hT-MSC; Isotype control, MSCCXCR4 stained with isotype-matching IgG. g, MSCs/Cargocytes migrated in Boyden chambers towards the indicated concentrations of SDF-1α for 2hr. Bar graph represents the cell migration index (migrated MSCs/Cargocytes versus loading control). Mean ± SEM; n=10 independent fields from 3 biological replicates. P value, two-way ANOVA with Bonferroni’s correction for multiple testing. All data are representative of at least 2 independent experiments.
We next determined if Cargocytes can be genetically engineered de novo to express exogenous functional chemoattractant receptors. We chose CXCR4 since it is not expressed by hT-MSCs and the CXCR4/SDF-1α chemotactic migration axis is well understood and clinically relevant31. We engineered hT-MSCs with CXCR4 expression (MSCCXCR4) via lentivirus infection and drug selection. After enucleation, Cargocytes derived from MSCCXCR4 (CargocytesCXCR4) showed stable surface expression of CXCR4 by flow cytometry for up to 48 hours (Fig. 2f). Importantly, CargocytesCXCR4 migrated towards chemokine gradients of the cognate ligand SDF-1α in a dose-dependent manner (Fig. 2g). Similarly, Cargocytes engineered with the chemokine receptor CCR2 (CargocyteCCR2) retained stable surface expression of CCR2 for at least 48 hours post-enucleation (Supplementary Fig. 3d) and showed significant induced migration towards the cognate ligand Ccl2 (Supplementary Fig. 3e). These findings demonstrate that Cargocytes can be engineered de novo with chemoattractant receptors that promote chemotaxis and homing. The cell-like chemotaxis activity of Cargocytes is a unique feature compared to other CFDVs like exosomes, nanoghosts and erythrocytes that lack the ability for directional migration.
Bioengineered Cargocytes interact with adhesion molecules
Besides chemoattractant receptors, bioengineering Cargocytes with endothelial adhesion molecules like PSGL-1 could be a powerful mechanism to target i.v. administered Cargocytes specifically to the vasculature of diseased and injured tissues, since they commonly express the PSGL-1 ligands E- and P-selectin12. Therefore, we engineered hT-MSCs to express PSGL-1 with fucosyltransferase 7 (FUT-7, for functional modification of PSGL-1) (MSCPSGL-1) via lentivirus infection. Flow cytometry showed that CargocytePSGL-1 retained stable surface expression of PSGL-1 for at least 48 hours post-enucleation and showed dramatically increased binding to P-/E-selectin (Fig. 3a), indicating the engineered PSGL-1 on the surface of Cargocytes is functional.
Fig. 3 |. Cargocytes interact with adhesion molecules.
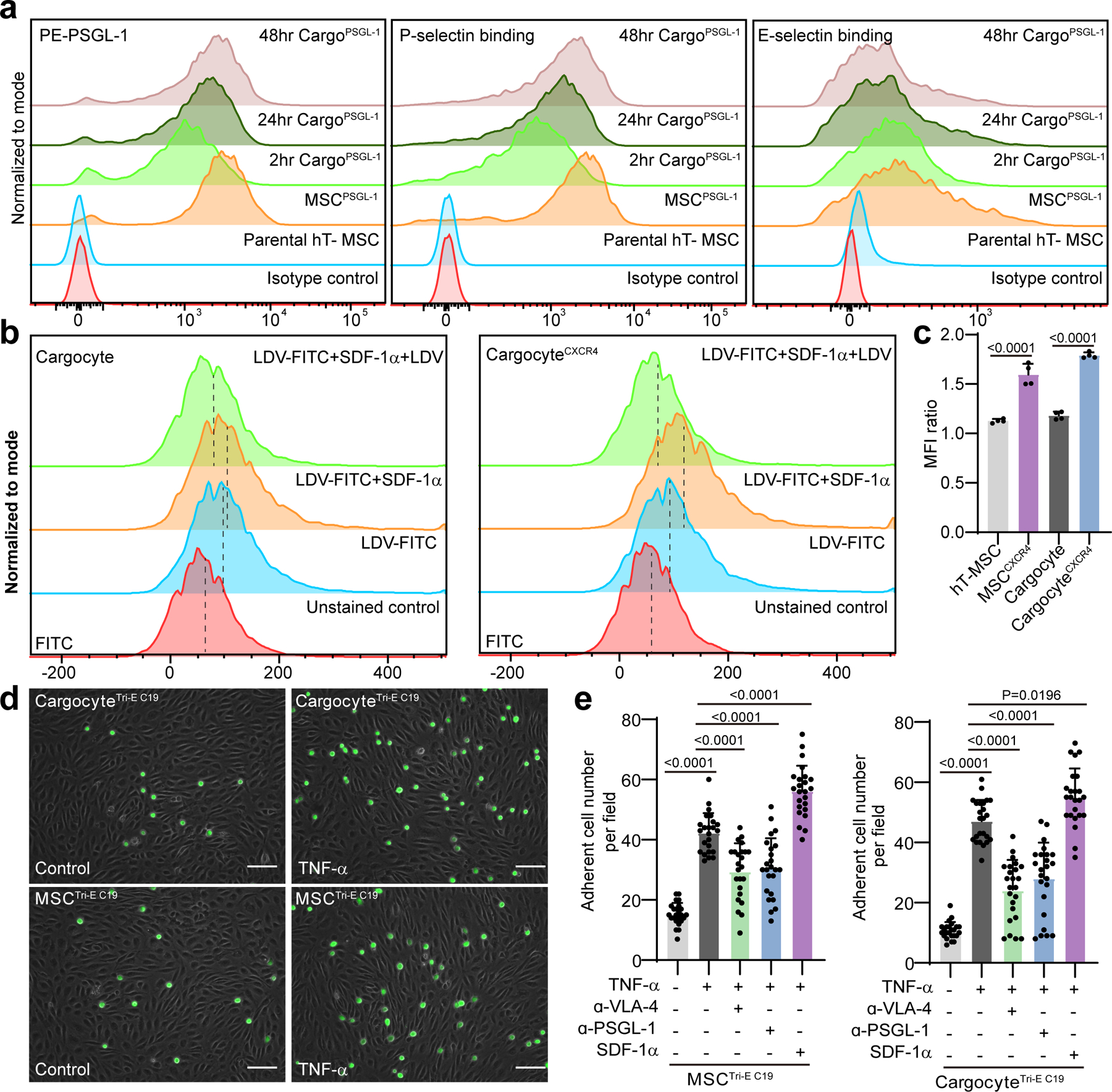
a, Graphs show surface expression of PSGL-1 (left), P-selectin binding (middle), and E-selectin binding (right) of hT-MSCs or Cargocytes analyzed by flow cytometry. Data were analyzed in Flowjo and normalized to mode. MSCPSGL-1, PSGL-1/Fut7 lentivirus-engineered MSC; 2hr/24hr/48hr Cargo PSGL-1, MSC PSGL-1-derived Cargocytes analyzed at indicated time point after enucleation; Parental MSC, Non-engineered MSC; Isotype control, MSC PSGL-1 stained with isotype matching IgG. b, Graphs show LDV-FITC binding on un-engineered Cargocytes or CargocytesCXCR4 analyzed by flow cytometry. Data were analyzed in Flowjo and normalized to mode. Unstained control, no LDV-FITC control; LDV-FITC, 4nM LDV-FITC; LDV-FITC+SDF-1α, 4nM LDV-FITC+500ng/ml SDF-1α; LDV-FITC+SDF-1α+LDV, 4nM LDV-FITC+500ng/ml SDF-1α+1μM LDV (unconjugated competitor). Dashed line labels the position of median fluorescence intensity. c, Bar graphs represents the MFI change of LDV-FITC binding intensity before and after SDF-1α treatment. MFI ratio = (MFILDV-FITC+ SDF-1α - MFIunstained control)/ (MFILDV-FITC - MFIunstained control). n=4 biological replicates. P value, one-way ANOVA with Tukey’s multiple comparisons test. d, Representative images show the adherent MSCs or Cargocytes on control HUVECs or inflamed HUVECs (TNF-α treated). Representative images out of 24 images obtained are shown. Scale bar, 100μm. e, Bar graphs represent the adherent cell numbers per field (100X magnification). TNF-α, HUVECs pre-treated with 10ng/ml TNF-α for 6hr. SDF-1α, 500ng/ml SDF-1α; a-PSGL-1, 10μg/ml anti-PSGL-1 antibody pre-treatment; a-VLA-4, 10μg/ml anti-VLA-4 antibody pre-treatment; n= 24 random fields of 4 biological replicates. P value, one-way ANOVA with Tukey’s multiple comparisons test. All data are representative of at least 2 independent experiments.
While PSGL-1 and E-/P-selectin binding can initiate endothelial cell adhesion, localized cytokine-induced high affinity activation of integrins like VLA-4 are required for complete cell arrest on the vascular endothelium and subsequent chemotactic events leading to extravasation12. The activation of integrin VLA-4 by CXCR4-SDF-1α occurs via “inside-out signaling” and is a rapid process (typically minutes) that occurs independent of gene transcription32. To determine if endogenous VLA-4 on CargocytesCXCR4 can be activated by SDF-1α, we used a small fluorescence-labeled peptide LDV-FITC, whose binding can be used to monitor VLA-4 affinity changes33. Compared to non-engineered Cargocytes, the binding of LDV-FITC on CargocyteCXCR4 was greatly enhanced by rapid SDF-1α stimulation (2 minutes) (Fig. 3b and c), even though they have similar VLA-4 surface expression (Supplementary Fig. 4a). While the baseline binding of LDV-FITC on Cargocytes is lower than parental MSCs due to Cargocytes’ smaller size (Fig. 3b and Supplementary Fig. 4b), the increase in mean fluorescence intensity (MFI) ratio of CargocyteCXCR4 upon SDF-1α stimulation was similar with MSCCXCR4 treated with SDF-1α (Fig. 3c). These findings demonstrate that Cargocytes effectively activate the high affinity state of VLA-4 integrins in response to external chemoattractant signals, suggesting that Cargocytes have innate capacity for integrin-mediated firm adhesion. This capability is a unique feature among CFDV platforms which lack fundamental mechanisms to mediate integrin activation.
Cargocytes can be engineered with multiple homing molecules
Cell homing to disease foci involves a multi-step process requiring precise coordination of integrins, adhesion molecules, and chemokine receptors12, 14. This prompted us to determine if Cargocytes can be engineered to simultaneously express multiple functional chemokine receptors and adhesion molecules important for cell homing. In proof-of-principle studies, we bioengineered hT-MSCs to express CCR2, CXCR4, and PSGL-1/FUT-7, and cells with high expression of all three cell surface markers (named MSCTri-E) were enriched by FACS. These homing receptors commonly mediate leukocyte cell homing to most inflammatory tissues10, 12. We established 19 single cell clones from MSCTri-E with high expression of all three markers (Supplementary Table 2), and selected Clone 19 (MSCTri-E C19, i.e., Triple-engineered Clone 19) for subsequent experiments based on surface expression, growth rate, and cell size. MSCTri-E C19-derived CargocyteTri-E C19 showed robust migration towards Ccl2 and SDF-1α gradients compared to single engineered Cargocytes (Supplementary Fig. 4c) and robust interaction with P-Selectin and E-Selectin (Supplementary Fig. 4d). CargocyteTri-E C19 also showed improved adhesion to TNF-α -treated endothelial cells in vitro (Fig. 3d and e), which mimics the inflamed endothelium in vivo with upregulated expression of VCAM-1 and E-Selectin, but not P-Selectin (Supplementary Fig. 4e). CargocyteTri-E C19 adhesion to inflamed ECs was largely abrogated by pre-treatment of anti-PSGL-1 or anti-VLA-4 antibodies (Fig. 3e), suggesting the interaction was partially dependent on the expression of PSGL-1 and VLA-4 on CargocyteTri-E C19. Interestingly, the increased adhesion between CargocyteTri-E C19 and inflamed-EC was further enhanced by SDF-1α stimulation (Fig. 3e), suggesting SDF-1α-CXCR4 signaling may increase the VLA-4 affinity on CargocyteTri-E C19, which is consistent with the LDV-FITC binding assay findings (Fig. 3b and c). Together our findings indicate Cargocytes are a genetically tractable platform that can be armed with functional endothelial adhesion molecules and multiple chemoattractant receptors with potential to improve in vivo homing and therapeutic delivery.
Cargocytes bioengineered with homing molecules specifically home to inflamed tissue in vivo
We next wanted to determine if bioengineered Cargocytes administered i.v. can home to sites of inflammation in vivo. However, one major obstacle for therapeutic cells administered i.v. is the initial trapping in the lung capillaries, which not only reduces homing efficiency13, but may also induce severe complications such as pulmonary embolism34, 35. Since Cargocytes are smaller (Fig. 1c), more deformable, and can transit through small constrictions more effectively than nucleated parental hT-MSCs (Fig. 2d and e), we hypothesized that Cargocytes may also pass through microvasculature (i.e. pulmonary capillaries) more efficiently than parental cells, which may facilitate better in vivo homing to the target tissue. To investigate this possibility, hT-MSCs or Cargocytes were double-labeled with LifeAct-RFP and vital dye Vybrant-DiD prior to i.v. injection in mice. Twenty-four hours post-injection, flow cytometry analysis of lung tissue detected significantly fewer Cargocytes compared to parental cells (> 2-fold decrease, Fig. 4a). To further decrease lung entrapment, we cultured MSCs in hanging drops to generate 3D-cultured MSCs that were smaller than traditional 2D-cultured MSCs and showed decreased lung trapping (Fig. 4a and Supplementary Fig. 5b) as previously reported36. When 3D-cultured MSCs were enucleated, the resulting 3D-Cargocytes were the smallest and had the least lung trapping (Fig. 1c, 4a and Supplementary Fig. 5b). Subsequent in vivo homing assays used 3D-hT-MSCs and 3D-Cargocytes unless noted otherwise.
Fig. 4 |. Bioengineered Cargocytes actively and specifically home to the inflamed ear.
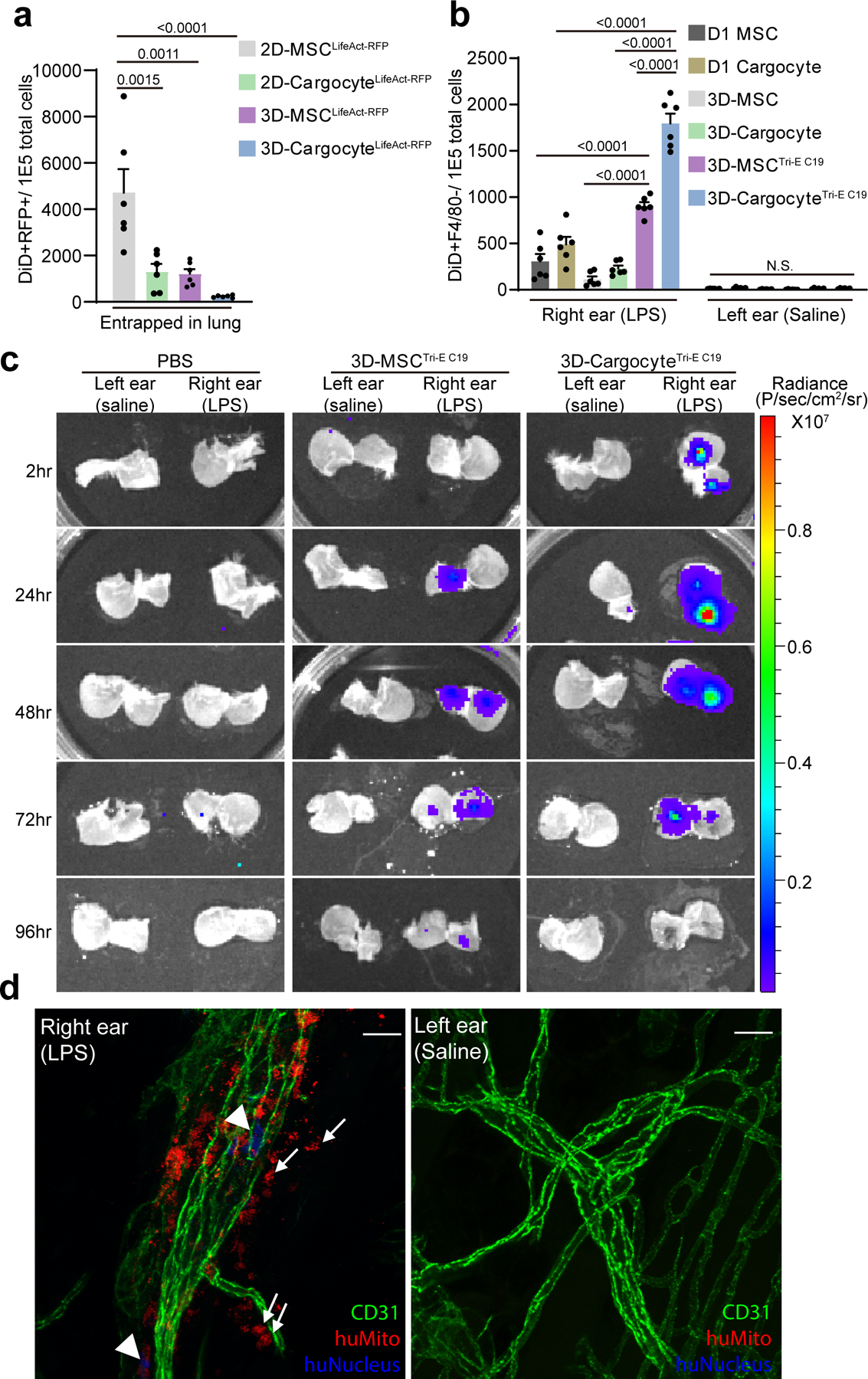
a, Bar graphs show the number of DiD+RFP+ double-positive cells out of 1E5 total cells harvested from mouse lung 24hr post-intravenous (i.v.) injection and detected by flow cytometry. Mean ± SEM, n=6 mice. b, Mice were intradermally (i.d.) injected with LPS in the right ear and saline in the left, followed by i.v. injection of DiD-labeled MSCs or Cargocytes 6hr later. Bar graphs show the number of DiD+F4/80− cells out of 1E5 total cells harvested from mouse ears 24hr post-injection and detected by flow cytometry. Mean ± SEM, n=6 mice. c, Mice were treated as in (b) and then i.v. injected with 1E6 3D-MSCTri-E C19 or 3D-CargocyteTri-E C19 transfected with firefly luciferase mRNA. Mice were euthanized at indicated time points after i.v. injection. The dorsal skin of the mouse ears was peeled from the underlying cartilage and the subcutaneous surfaces of the tissue were directly soaked in VivoGlo™-luciferin substrate and immediately subjected to bioluminescence imaging with IVIS Lumina Series III. See also Supplementary Fig.7a for in vitro control of luciferase activity. d, Mice treated as in (b) were i.v.-injected with 1E6 3D-CargocyteTri-E C19. After 24hr, mouse ears were harvested and whole-mount stained with anti-mouse CD31 (green), anti-human Mitochondrial (red), and anti-human nucleus antigen (blue). Confocal images from Olympus FV1000 were analyzed with Fiji ImageJ. Maximum projection of Z-stacks of images of ears from the same mouse were shown. Arrows point to human Cargocytes, and arrowheads point to human nuclei. Representative images out of 10 images obtained are shown. Scale bar, 20μm. For (a) and (b), adjusted P values shown above the bars, one-way ANOVA with Tukey’s multiple comparisons test. All data are representative of at least 2 independent experiments.
We first tested the in vivo homing ability of bioengineered MSCs and Cargocytes in an established mouse model of acute ear inflammation (Supplementary Fig.5a). Bacterial-derived lipopolysaccharide (LPS) was intradermally (i.d.) injected into the pinna to induce acute, local inflammation, while saline was i.d. injected into the contralateral ear as a control37. This model allows examination of therapeutic cell homing quantitatively between an inflamed and non-inflamed contralateral tissue within the same animal37. Previous studies suggest chemokines and endothelial adhesion molecules play important roles in cell homing to inflamed tissues10, 12. Consistent with these studies, SDF-1α, Ccl2, Vcam1, Icam1, and P-Selectin, but not E-Selectin, were upregulated in inflamed ears compared to controls, starting 6 hours post-LPS injection (Supplementary Fig. 5c). 3D-MSCs were labeled with Vybrant-DiD and i.v. injected into mice 6 hours after LPS injection. Mouse tissues were harvested 24 hours later and analyzed by flow cytometry for DiD+F4/80− cells. The pan-macrophage marker F4/80− was used to exclude the possibility of non-specific Vybrant-DiD incorporation into mouse macrophages38. Independent expression of either CCR2, CXCR4, or PSGL-1 improved MSC specifically homing to the inflamed ear compared to non-engineered hT-MSCs, indicating that these proteins are functional in vivo and contribute to homing (Supplementary Fig. 5d). Notably, MSCTri-E simultaneously expressing all 3 surface proteins showed the greatest homing (Supplementary Fig. 5d), suggesting that bioengineering cells with multiple adhesion and chemoattractant receptors greatly improves disease targeting potential.
Based on these findings, we next tested the homing ability of CargocyteTri-E C19 in this model. 3D-CargocyteTri-E C19 robustly homed to the inflamed ear, but not to the uninflamed control ear from the same mouse, suggesting the homing is specific (Fig. 4b). While non-engineered 3D-Cargocytes and 3D-CargocyteTri-E C19 had similar low levels of lung trapping (Supplementary Fig. 5e), 3D-CargocyteTri-E C19 had a >6-fold increased homing to the inflamed ear (Fig. 4b), suggesting that bioengineering with CCR2, CXCR4, and PSGL-1 enabled Cargocytes to specifically and efficiently home to the target tissue in vivo. 3D-CargocyteTri-E C19 also showed significantly better homing compared to mouse D1 MSCs or D1 Cargocytes (Fig. 4b), which is a syngeneic MSC line from BALB/c mice that possesses endogenous homing abilities39. Importantly, 3D-CargocyteTri-E C19 had an approximate 2-fold increase in homing to inflamed tissue compared to parental 3D-MSCTri-E C19, which is likely due to reduced lung trapping by Cargocytes (Supplementary Fig. 5e and 6).
The homing result was independently confirmed using a bioluminescence assay with firefly-luciferase (Fluc). It is notable that firefly-luciferase is an intracellular enzyme that requires ATP for functional bioactivity40. Therefore, the bioluminescence assay only detects viable cells/Cargocytes expressing Fluc in different tissues at the time of imaging. MSCTri-E C19 and CargocyteTri-E C19 transfected with Fluc mRNA showed comparable bioluminescence in vitro (Supplementary Fig. 7a). Consistent with our results using Vybrant-DiD labeling, the bioluminescence assay showed 3D-CargocyteTri-E C19 had decreased lung trapping but dramatically increased homing to the inflamed ear compared to 3D-MSCTri-E C19 as early as 2 hours post i.v. injection (Fig. 4c and Supplementary Fig. 7b). There was minimal accumulation in other organs such as liver and kidney (Supplementary Fig. 8 and 9).
We also investigated 3D-CargocyteTri-E C19 homing to inflamed ears and their ability to extravasate out of the vasculature using whole-mount immunostaining with specific antibodies against human mitochondria and the vascular endothelial marker CD31 (Supplementary Fig. 10a). Confocal imaging revealed 3D-CargocyteTri-E C19 were only in the inflamed ear outside vessel lumina and in the interstitial connective tissue, indicating that 3D-CargocyteTri-E C19 were not passively trapped in the ear vasculature but extravasated into surrounding tissues (Fig. 4d and Supplementary Fig. 10b). Taken together, these findings demonstrate that Cargocytes bioengineered with cell adhesion molecules and chemoattractant receptors can specifically and efficiently home to inflamed tissue, with minimum lung trapping and accumulation in non-inflamed organs.
Cargocytes deliver bioactive cytokine to attenuate tissue inflammation and damage
Next, we investigated the ability of bioengineered Cargocytes administered i.v. to home and deliver anti-inflammatory biologics to inflamed tissues using the same ear inflammation model. As proof-of-principle, we chose human IL-10 (hIL-10) since it is a potent and clinically important anti-inflammatory cytokine, for which clinical applications require more efficient and specific delivery methods4. Cargocytes transfected with in vitro synthesized human IL-10 mRNA (Supplementary Fig. 11a) (Cargocyte-IL-10) produced IL-10 for up to 72 hours in vitro, which was similar to parental MSCs transfected with IL-10 (MSC-IL-10) (Fig. 5a). Non-transfected hT-MSCs did not secrete detectable IL-10 in vitro (Fig. 5a) and CM collected from MSC-IL-10 or Cargocyte-IL-10 activated Stat3 phosphorylation in mouse RAW macrophages in vitro, confirming that secreted hIL-10 was biologically active on mouse cells (Fig. 5b).
Fig. 5 |. Bioengineered Cargocytes efficiently deliver bioactive IL-10 to inflamed ears and ameliorate local inflammation.
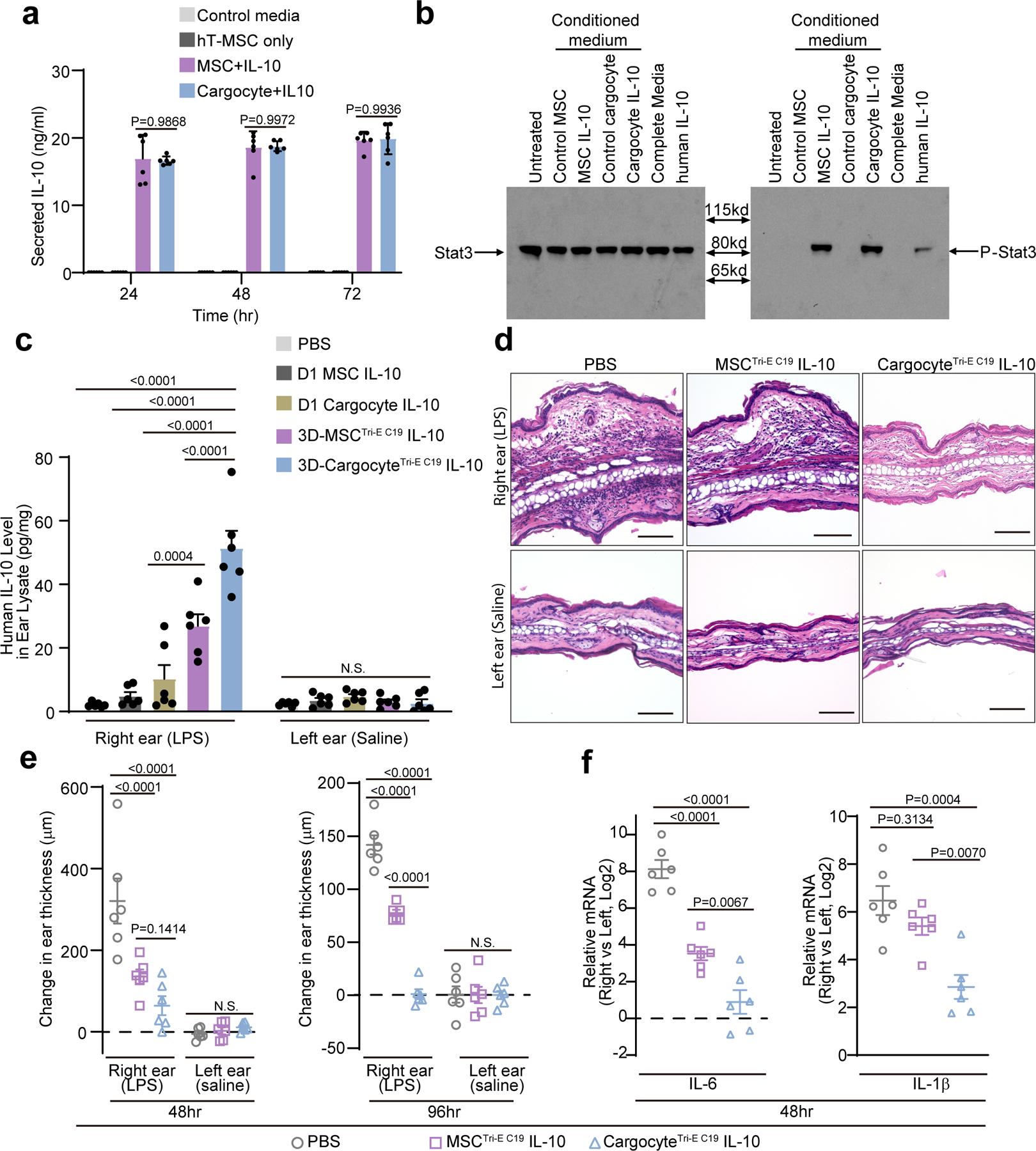
a, Graph shows the secreted IL-10 concentration measured by ELISA in conditioned media of IL-10 mRNA transfected MSCs (MSC-IL-10) and Cargocytes (Cargocyte-IL-10), non-transfected cells (hT-MSC only) or control media. Mean ± SEM; n=6 biological replicates from two independent experiments. b, Mouse RAW macrophage cells were treated with indicated conditioned media or recombinant IL-10 protein (rIL-10, 1ng/ml) for 30 mins. The phosphorylation of Stat3 was determined by western blot. c, Mice treated as in Fig.4b were i.v.-injected with indicated MSCs or Cargocytes transfected with human IL-10 mRNA. Bar graph shows the level of human IL-10 protein detected by ELISA from indicated mouse ears at 24hr post-injection. d, Light microscopy images of ears from mice treated as in (c) and harvested at 48hr post-injection and processed for hematoxylin and eosin staining. Scale bar, 100μm. e, Mice treated as in (c) received indicated i.v. injections. Graph shows change in ear thickness as measured by digital micrometer prior to LPS/Saline injections, 48hr post-injection and 96hr post-injection. f, Mice treated as in (e) had ears harvested and analyzed by real-time RT-PCR 48hr after LPS injection. Graphs show the fold change (Log2) of the indicated mRNA markers between LPS-treated (right) and saline-treated (left) ears. For (c, e, and f), Mean ± SEM, n=6 mice; each point represents 1 mouse. All statistics are one-way (f) or two-way ANOVA (a, c, and e) with Tukey’s multiple comparisons test. Adjusted P values shown above the bars. All data are representative of at least 2 independent experiments.
Consistent with their improved homing potential to inflammatory tissues, 3D-CargocytesTri-E C19 resulted in the highest levels of hIL-10 delivery to the inflamed ear with lowest level of hIL-10 in the mouse plasma, compared to 3D-MSCTri-E C19 or non-engineered D1 MSC and D1 Cargocytes (Fig. 5c and Supplementary Fig. 11c). All contralateral (control) ears from treated animals had little detectable hIL-10 (Fig. 5c), indicating delivery of hIL-10 to inflamed ears was highly specific. We measured the hIL-10 levels in mouse ears and plasma at different time points post i.v. injection (Supplementary Fig. 11 d to g). The results suggest that the hIL-10 delivered by 3D-MSCTri-E C19 and 3D-CargocytesTri-E C19 reached a peak in inflamed ears at 24 hours post injection and then gradually decrease over time, while 3D-CargocytesTri-E C19 delivered more hIL-10 than 3D-MSCTri-E C19 (Supplementary Fig. 11 d and e). In contrast, recombinant hIL-10 injected i.v. was not detected in inflamed ears and was quickly eliminated in the circulation (Supplementary Fig. 11 f and g), which is consistent with its reported short plasma half-life41. Furthermore, while LPS inflamed ears from PBS-treated mice showed severe hemorrhage and edema with leukocyte infiltrates, mice treated with 3D-CargocytesTri-E IL-10 had minimal hemorrhage and edema and reduced leukocyte infiltration (Fig. 5d). It is notable that the increased fluid and cellular infiltration correspond to increased thickness of the pinna, which is a quantifiable marker of inflammation37. While the thickness of saline-injected control ears was unchanged and comparable across all groups, both engineered MSC and Cargocyte-treated animals had significantly thinner LPS-inflamed ears compared to control mice (Fig. 5e). Additionally, the expression of inflammatory markers IL-6, IL-1β and TNF-α were significantly downregulated in ears of mice treated with 3D-CargocytesTri-E C19 IL-10 compared to PBS-treated mice (Fig. 5f, and Supplementary Fig. 11h and 11i). Therefore, Cargocytes bioengineered with homing receptors and adhesion molecules specifically home to target inflamed tissues where they produce and deliver anti-inflammatory cytokines that effectively reduce local inflammation in vivo.
Bioengineered Cargocytes ameliorate acute pancreatitis
We further tested the therapeutic delivery of bioengineered Cargocytes in a disease model of acute pancreatitis (AP). AP is a severe disease with significant morbidity and mortality that currently lacks effective treatments42, 43. Caerulein is a decapeptide analog of hormone Cholecystokinin (CCK), which can stimulate exocrine pancreatic secretion and induce AP in pre-clinical mouse models44, 45. Previous studies suggested frequent systemic administration of high doses of anti-inflammatory cytokine IL-10 in pre-clinical AP models can greatly attenuate the inflammation and mitigate the disease46, 47. However, repeated high doses of IL-10 are not cost-effective in clinical applications and may also lead to unwanted severe complications such as anemia, suggesting a specific and efficient delivery vehicle may be necessary4. In early stage of caerulein-induced AP, chemokines such as Ccl2 and SDF-1α, and adhesion molecules such as E-/P-Selectins and Vcam1, are all significantly upregulated in the inflamed mouse pancreas (Supplementary Fig. 12a), suggesting bioengineered CargocytesTri-E C19 may be an ideal delivery vehicle to specifically deliver IL-10 to the inflamed pancreas. We assessed the homing of Cargocytes and parental MSCs in AP mice by Vybrant-DiD labeling and FACS analysis as in Fig. 4b. Compared to non-engineered 3D-Cargocytes, 3D-CargocytesTri-EC19 homed more efficiently (>11-fold) to the inflamed pancreas (Fig. 6a). 3D-CargocyteTri-E C19 also had a >2-fold increase in homing to the inflamed pancreas and decreased lung trapping compared to parental 3D-MSCTri-E C19 (Fig. 6a and Supplementary Fig. 12b). In the healthy non-treated pancreas, both 3D-CargocyteTri-E C19 and 3D-MSCTri-E C19 had minimum accumulation (Fig. 6a). This result was independently confirmed using the bioluminescence assay as in Fig. 4c (Supplementary Fig. 13). Importantly, compared to parental 3D-MSCTri-E C19, 3D-CargocyteTri-E C19 also delivered IL-10 protein more efficiently (> 2-fold) to the inflamed pancreas (Fig. 6b), which correlated with decreased expression of the inflammatory gene markers, Ccl2, TNF-α, IL-1β, and IL-6 (Fig. 6c and Supplementary Fig. 14d). Infusion of 3D-CargocyteTri-E C19 IL-10 also significantly reduced blood serum levels of lipase and amylase (Fig. 6d), which correlate with the severity of pancreas damage43. And histological analyses showed reduced acinar cell necrosis, lower interstitial edema, and less inflammatory cell infiltration in the damaged pancreas (Fig. 6e and f, Supplementary Fig. 14e and f). Notably, 3D-CargocyteTri-E C19 or 3D-MSCTri-E C19 without IL-10 did not significantly affect caerulein-induced pancreatitis (Fig. 6b to f). Together, our results demonstrate that bioengineered CargocyteTri-E C19 can efficiently deliver bioactive anti-inflammatory cytokine IL-10 to the inflamed pancreas, which greatly ameliorates the disease in an established clinically relevant AP model.
Fig. 6 |. Bioengineered Cargocytes ameliorate Caerulein-induced acute pancreatitis.
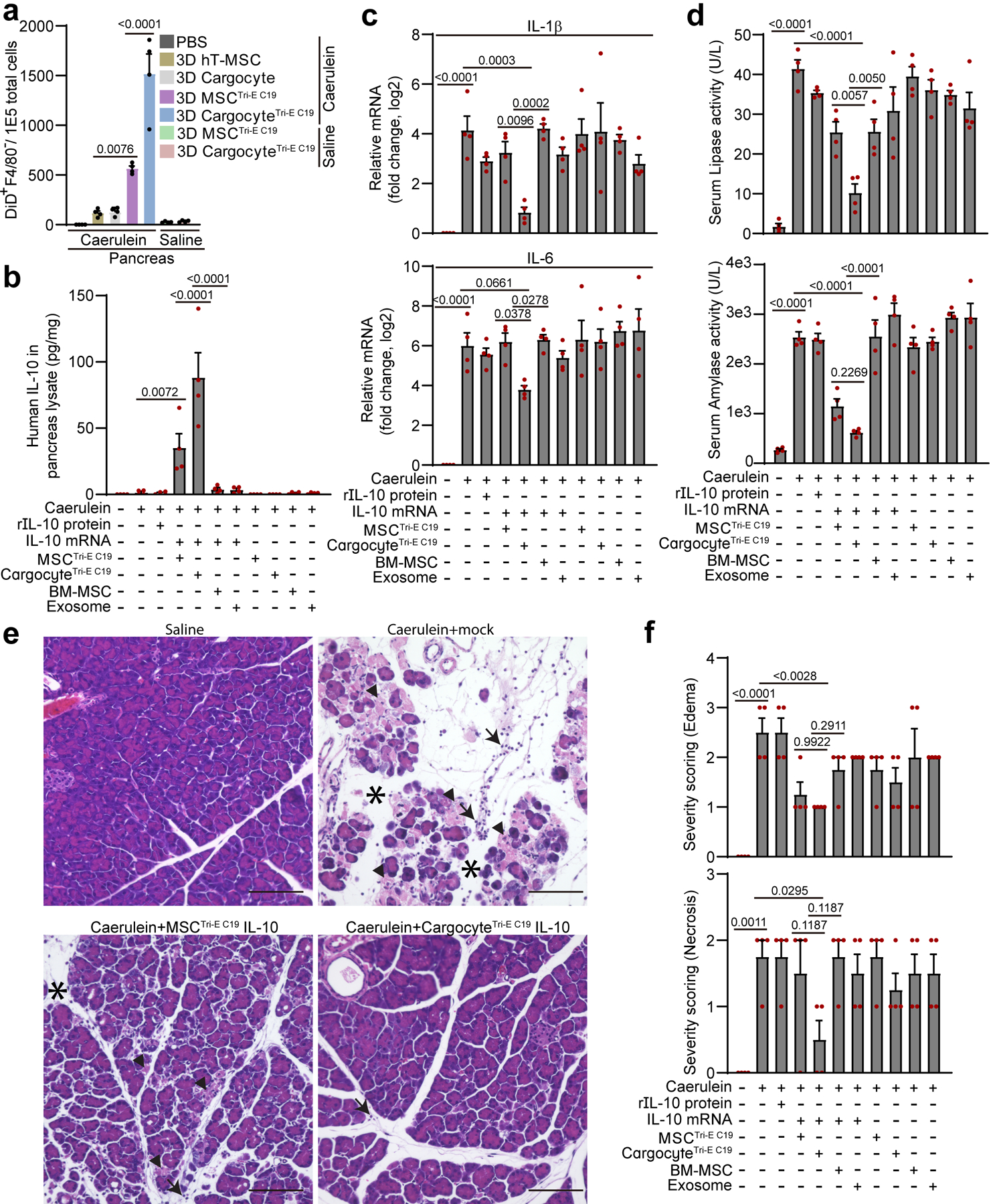
a, Acute pancreatitis was induced by intraperitoneal (i.p.) injection of Caerulein in BalB/c mice, followed by i.v. injection of DiD-labeled MSCs or Cargocytes. Bar graphs show the number of DiD+F4/80− cells out of 1E5 total cells harvested from mouse pancreas 16hr post-injection and detected by flow cytometry. Mean ± SEM, n=4 mice. From (b) to (e), Mice with Caerulein-induced AP were i.v.-injected with indicated treatments. Mouse tissues were harvested 16hr post-injection. b, Bar graph shows the level of human IL-10 protein detected by ELISA from mouse pancreas from indicated treatment. Mean ± SEM, n=4 mice. See also Supplementary Fig.14a and b for in vitro transfection controls. c, Bar graph shows the relative mRNA expression of IL-1β (upper) and IL-6 (lower) detected by real-time RT-PCR in the mouse pancreas from indicated treatment. Graphs show the fold change (Log2) of the indicated mRNA markers normalized to no Caerulein treatment group. Mean ± SEM, n=4 mice. d, Bar graph shows the lipase activity (upper) and amylase activity (lower) detected in the mouse serum from indicated treatment. Mean ± SEM, n=4 mice. e, Representative light microscopy images of pancreas from mice treated as in (c) and harvested at 16hr post-injection and processed for hematoxylin and eosin staining. Arrows point to infiltrated leukocytes, arrowheads point to necrosis, and asterisks label edema. Scale bar, 100μm. f, Histological analysis of pancreas treated as in (e). The severity of edema (upper) and necrosis (lower) were graded from 0 to 3 using established criteria. See also Supplementary Fig.14e and f for inflammatory cell infiltration. Mean ± SEM, n=4 mice. All statistics are one-way ANOVA with Dunnett’s correction for multiple testing, and P values for more post-hoc comparisons can be found in Supplementary Table 10. All data are representative of at least 2 independent experiments.
As expected, animals i.v. injected with a single dose (8μg/kg body weight) of recombinant hIL-10 protein had minimal effects in all inflammatory and AP markers, probably due to the short half-life of IL-10 protein in circulation (Fig. 6b to f)48. Also, as a comparison to recognized therapeutic delivery platforms, we tested commercially available bone marrow-derived primary MSCs (BM-MSC) and purified BM-MSC-derived exosomes (B-exosomes) loaded with IL-10 mRNAs in the AP model. While infusion of BM-MSCs+IL-10 significantly increased the level of IL-10 in the serum and in the lung (Supplementary Fig. 14c), the actual IL-10 levels detected in the inflamed pancreas were negligible (Fig. 6b), suggesting that BM-MSCs are inefficient at homing and delivering IL-10 to the inflamed pancreas. Similarly, animals injected with B-exosomes loaded with IL-10 mRNA showed no detectable IL-10 in the inflamed pancreas and only a slight increase in serum IL-10 levels (Fig. 6b and Supplementary Fig. 14c).
Of final note, administration of bioengineered, human MSC-derived Cargocytes in these mouse models did not produce any detectable negative health effects in over 300 mice, as determined by clinical observation and gross examination of tissues by a board-certified veterinary pathologist (C.N.A.). BALB/c mice i.v. injected with Cargocytes had no significant change in the plasma concentration of pro-inflammatory cytokines IL-6, IL-1β, TNF-α and IFN-γ (Supplementary Table 3), nor in complete blood counts (Supplementary Table 4) or in comprehensive blood chemistry analysis (Supplementary Table 5). Moreover, as a prototype for clinical use, we labeled cell nuclei with Histone 2B-GFP and generated Cargocytes of 99.999% purity through FACS, without loss of viability or migration ability (Supplementary Fig. 15a to c). Furthermore, Cargocytes recovered at slightly higher rates than hT-MSCs after long-time cryopreservation (>1 month) and maintained both viability and migration ability after thawing (Supplementary Fig. 15d to f), which facilitates the logistics of storage and delivery for clinical applications. While more comprehensive safety studies will be necessary before clinical use, we conclude that Cargocytes have the potential to be a safe and effective vehicle for targeted therapeutic delivery.
Discussion
Therapeutic agents administered systemically often result in unwanted side effects and toxicity due to accumulation in off-target sites, the lack of drug selectivity, and the inability to completely access diseased foci1, 2. Therefore, there is a substantial need for novel systems which can deliver therapeutics precisely to diseased tissues to decrease unwanted side effects and improve efficacy. The primary goal of this study was to develop a bioinspired delivery vehicle that specifically targets diseased tissues while maintaining a safe and controllable profile important for clinical applications. Our unique approach facilitates the introduction of multiple genetic elements into a cell without introducing major safety concerns, since the modified chromatin is removed during enucleation. Enucleated cells are distinct from other CFDVs in that they retain crucial organelles for energy and protein production, do not proliferate or permanently engraft in the host, undergo regulated integrin-mediated adhesion to inflamed endothelial cells, and actively home to specific chemokine gradients released by diseased tissues. Cargocytes are also distinct from CFDVs in that they actively extravasate out of the vasculature and synthesize/secrete therapeutics at the site of disease. In proof-of-principle, Cargocytes engineered with CXCR4, CCR2 and PSGL-1 showed significantly improved homing and delivery of anti-inflammatory cytokine IL-10 to target tissue compared to normal MSCs and exosome vehicles, which attenuated inflammation and ameliorated pancreatitis. These findings indicate that the innate cell-like functions of Cargocytes can be further enhanced through cell and genetic engineering to improve therapeutic delivery.
Cargocytes also possess several distinct features over intact cell delivery vehicles. First, it remains difficult to obtain regulatory approval for clinical use of extensively engineered cells (especially stem cells like MSCs) due to safety concerns16. Second, primary cells harvested from donors are usually heterogeneous and not amenable to creating proliferative clonal cell lines due to senescence after in vitro passaging, which limits bioengineering and therapeutic capacities49. Third, Cargocytes have a more defined and predictable cell fate in vivo because they cannot perform new gene transcription and therefore cannot be induced to differentiate or produce unpredictable paracrine factors, nor can they engraft and proliferate uncontrollably. This ensures that crucial therapeutic features established through bioengineering ex vivo are correctly conveyed in vivo, making Cargocyte therapeutic delivery more precise and reliable for clinical applications with reduced off-target effects. Fourth, compared to intact cells, Cargocytes are smaller and lack rigid nuclei (Fig. 1c, 2d, 2e and Supplementary Fig. 5b), which translates to decreased lung entrapment and improved biodistribution (Fig. 4a and Supplementary Fig. 5e, 6, 8, 9, and 12b), and improved homing to target tissues (Fig. 4b, 4c, 6a, and Supplementary Fig. 7b and 13). These unique attributes significantly improved therapeutic delivery to diseased tissues (Fig. 5c and 6b), compared to their larger nucleated parental MSCs. Importantly, because Cargocytes are more deformable and migrate through small vessel-like constrictions better than cells (Fig. 2d, 2e, and Supplementary Movies 1 and 2), patients may have a lower risk of severe adverse events such as pulmonary or cerebral emboli associated with systemic administration of cells with nuclei34, 35, 50. Therefore, Cargocytes are uniquely positioned between cells and other CFDV platforms (i.e., exosomes, erythrocytes, or nanoghosts) that are currently under development for clinical applications.
One of the greatest challenges of current CFDVs is the inability to actively migrate across the endothelium into target tissue interstitium to disease foci following systemic administration8. Moreover, other CFDVs cannot be genetically engineered to actively produce and secrete therapeutic proteins. Cargocytes on the other hand actively home to diseased tissues, exit the vasculature, synthesize and secrete defined therapeutic proteins within disease foci. Cargocytes can also be loaded with or engineered to produce various therapeutic agents alone or in combination such as small RNAs, small molecule drugs, peptides, antibodies, and cytoplasmic-replicating viruses (Supplementary Table 6)51. We envision that Cargocytes could be used as a stand-alone cell-like therapeutic or be combined with other therapeutics to treat specific diseases. For example, disease-homing Cargocytes could be used to transport drug-loaded nanoparticles across the vascular barrier deep into the tissue parenchyma or facilitate stem cell transplantation and regenerative medicine applications through priming the stem cell niche. Cargocytes can also be generated from many cell types with therapeutic potential (Supplementary Table 1) that could be exploited to safely and controllably modulate the immune system, produce cancer vaccines, or serve as diagnostic tools and imaging modalities for a wide range of clinical applications. In addition, Cargocytes are ideally suited for precision medicine applications due to their genetic tractability and payload versatility. For example, Cargocytes could be engineered with specific disease targeting moieties and therapeutic payloads tailored to each patient’s specific disease and unique biomarker profile.
Manufacturing and scalability of cells and CFDVs can be limiting for clinical applications52. Cargocytes can be generated from immortalized single cell clones cultured in scalable spinner flasks, which facilitate consistent large-scale production and banking of cryopreserved stocks. And because of the widely-accepted use of density gradient ultracentrifugation for clinical applications like virus and exosome purification53 and the commercially available large-scale continuous flow ultracentrifuge systems54, we envision that large-scale production of Cargocytes for clinical use is achievable. While Cargocytes have a defined lifespan, our results suggest most Cargocytes home to the target tissue in the first 24 hours after i.v. administration. Additionally, if necessary, the half-life of the Cargocyte payload can be increased to improve dosing regimens and clinical efficacy by recent bioengineering techniques (e.g. antibody/cytokine fusion to extracellular matrix binding domains, cytokines fused to albumin, or FC antibody fragments, etc.) 55. Finally, while our initial studies indicate that Cargocytes can be systemically administered in mice without detectable adverse health events, more comprehensive safety studies will be required prior to clinical applications. In summary, Cargocytes provide a new platform technology that combines cell enucleation and bioengineering techniques to generate a versatile therapeutic delivery vehicle with potential to treat a range of diseases in a controllable and effective manner with an improved safety profile.
Methods
Animals
All animals were housed and treated according to the University of California San Diego animal welfare guidelines as described and approved by the UCSD Institutional Animal Care and Use Committee (protocol #S12005) in a specific pathogen-free vivarium. The light cycle was 6am to 6pm; the temperature was between 69 to 71 degrees F; the humidity was between 40–60%. BALB/cJ and C57BL/6J mice were purchased from Jackson Laboratory. The co-first author of the study, Dr. Christina N. Alarcón, is a board-certified veterinary pathologist (Diplomate, American College of Veterinary Pathologists, Veterinary Medical Board license #VET19413). She designed and supervised all experimental animal procedures including animal colony management, injections, tissue collection, tissue processing, organ histopathology, and daily animal health examinations. Mouse serum chemistry analyses and whole blood counts were performed in the ACP Diagnostic Services Laboratory at UCSD.
Cell culture
Human telomerase reverse transcriptase (hTERT)-immortalized, adipose-derived mesenchymal stromal cells (hereinafter referred to as “hT-MSC”) were purchased from ATCC (#SCRC-4000™). Primary human bone marrow-derived MSCs were obtained through Texas A&M College of Medicine’s Institute for Regenerative Medicine. Primary human umbilical cord-derived MSCs were a generous gift from Dr. Mana Parast (UCSD). MSCs were cultured in complete culture medium (CCM) including alpha MEM (Gibco #12561) supplemented with 16.5% Premium FBS (Atlanta Biologics S1150), 1% HEPES (Gibco #15630), 1% Glutamax 100X (Gibco #35050), and 1% Anti-Anti 100X (Gibco #15240). D1 MSC (ATCC® CRL-12424), NK-92 cell (ATCC® CRL-2407), RAW 264.7 cells (ATCC® TIB-71), HEK-293 cells (ATCC ® CRL-1573), HL-60 cells (ATCC® CCL-240), L929 cells (ATCC® CCL-1) and NIH3T3 cells (ATCC® CRL-1658) were purchased from ATCC and cultured according to ATCC instructions. Briefly, RAW 264.7, HEK-293, L929 and NIH3T3 cells were cultured in DMEM (Gibco, #11960) with 10% FBS; D1 MSCs were cultured in DMEM (Gibco, # 670116) with 10% FBS; NK-92 cells were cultured in RPMI with 10% FBS and 100 U IL-2/ml (Discovery Labware, #CB40043); and HL-60 cells were cultured in RPMI with 20% FBS. Human Umbilical Vein Endothelial Cells (HUVECs, C2517A) were purchased from Lonza and cultured in EGM-2 media (Lonza, # CC-3162) according to the manufacturer’s instructions. hT-MSCs were authenticated by IDEXX Laboratories, while other cells were authenticated by ATCC. All cell stocks tested mycoplasma negative by PCR.
Cell enucleation
Cell enucleation was modified from the previously published protocol53. Ficoll PM400 (GE Healthcare, #17–0300-500) was dissolved into a 50% (wt/wt) solution with ultrapure water (Invitrogen, #10977–015) by continual magnetic stirring for 24 hours at RT, followed by autoclaving. Refractive index was measured to ensure consistency (range 1.4230–1.4290) on a refractometer (Reichert, #13940000). The Ficoll stock was aliquoted and stored at −20 °C. To make discontinuous gradients, 50% stock Ficoll solution was diluted to 25% with 2X MEM (diluted from Gibco, #11430) and then further diluted with 1X MEM (diluted from 2X MEM) for the 17%, 16%, 15%, and 12.5% Ficoll solutions. Cytochalasin B (Sigma Aldrich, #C6762) was added to all Ficoll solutions at a final concentration of 10 μg/ml. 2ml 25%, 2ml 17%, 0.5ml 16%, 0.5ml 15%, and 2ml 12.5% Ficoll solutions were carefully layered into a 13.2ml ultra-clear tube (Beckman Coulter, #344059) and equilibrated at in a 37 °C tissue culture incubator overnight. Subconfluent cells were harvested with Accutase (Innovative Cell Technologies, # AT104–500) and re-suspended in 3.2 ml 12.5% Ficoll solution and then carefully loaded onto the prepared discontinuous gradients and topped off with 1X MEM. Tubes were balanced in swing buckets of the SW41 rotor, run in a pre-warmed Beckman Coulter L8–60M ultracentrifuge for 60 minutes at 26,000 rpm and 31 °C with minimal braking. Fractions were collected with low-binding pipette tips into low-binding tubes and washed with serum-free media 3 times. Cargocyte efficiency (purity, % enucleated out of input number of cells) was examined by epifluorescence microscope (Nikon Eclipse Ti) after staining with Vybrant Dyecycle Green (Invitrogen, #V35004) for 10 minutes, and the yield of viable Cargocytes were measured in Cell Countess (ThermoFisher). Diameters of Cargocytes and parental MSCs were determined by the NIS-Elements AR 3.0 software (Nikon).
Transfection of in vitro synthesized mRNA
GFP mRNA, Gaussia Luciferase mRNA, firefly luciferase mRNA, and human IL-10 were synthesized by TriLink Biotechnologies. The protein coding sequence was flanked by 5’- and 3’-untranslated regions (UTRs) from mouse α-globin. Full substitution of pseudouridine was used to synthesize transcripts. After adding 5’ cap structure (CleanCap® AG) and 3’ poly(A) tail (120A), the synthesized mRNAs were purified with silica membrane. The pre-made mRNAs from TriLink were directly used for mRNA transfection. Briefly, 1 μg synthesized mRNA was added to 49 μl warm opti-MEM and separately 4 μl Lipofectamine 3000 (ThermoFisher, # L3000008) was added to 46 μl Opti-MEM. The Lipofectamine and mRNA solutions were incubated for 5 minutes, then were mixed and incubated for another 15 minutes at room temperature (RT). MSCs or Cargocytes were resuspended in CCM without antibiotics at 1E6 cells/ml. 100 μl of mixed mRNA + lipofectamine-3000 solution was added to 1ml MSC or Cargocyte suspension, mixed thoroughly and incubated at 37 °C for 30 minutes. After transfection, MSCs or Cargocytes were washed twice with CCM, then either used immediately for in vivo experiments or plated on tissue culture plates. For GFP mRNA, transfected MSCs or Cargocytes were plated in 6-well-plate (1E5 per well), and GFP expression was analyzed with the epifluorescence microscope (Nikon Eclipse Ti) or by flow cytometry. For Gaussia Luciferase mRNA, transfected MSCs or Cargocytes were plated in 24-well-plate (25,000 per well, 1 ml CCM media). Conditioned medium was taken 48 hours after transfection, and luciferase activity was determined as relative luminescence units (RLU) using BioLux Gaussia Luciferase Assay Kit (NEB, E3300) on GloMax 96 microplate luminometer (Promega). For human IL-10 mRNAs, transfected MSCs or Cargocytes were either plated in a 24-well-plate (25,000 per well, 1 ml CCM media) for collecting conditioned media at indicated time points to analyze cytokine expression by ELISA, or plated in 6-well-plate (1E5 per well, 2ml CCM media), for collecting conditioned media at 24 hours to treat RAW macrophage cells. The phosphorylation of Stat3 was then analyzed by western blot. Exosome transfection with in vitro synthesized mRNA was performed using the kit (System Biosciences, # EXFT10A-1) according to manufacturer’s instructions. Briefly, 20 billion exosomes (Erivan Bio, #B-MSC Exosomes) from human bone marrow-derived MSC were transfected with 1 μg synthesized mRNA in the presence of Exo-fect solution and were incubated at 37°C in a shaker for 10 min. Transfection was stopped by ExoQuick-TC and exosomes were precipitated by centrifugation at 14,000 rpm for 3min. The exosome pellets were resuspended in 300 μl of PBS. To test the transfection efficiency, 100 μl of transfected exosomes was added to 293 cells (1E5 cells per well, 24 well plate). And conditioned media was collected 24 h later and IL-10 level was determined by ELISA.
3D cell culture by hanging drops
Generation of 3D MSC spheroids in hanging drops was modified from previously published protocols33, 54. Briefly, MSCs were plated as drops on an inverted plastic dish lid (Olympus Plastics, #32–106) in 35 μl of CCM at 30,000 cells/drop. The lid was then replaced to cover the plate bottom containing 15 ml Dulbecco’s phosphate-buffered saline (PBS, ThermoFisher, # 14190250) to prevent evaporation. Hanging drop cultures were grown at 37 °C for approximately 40 hours with 5% CO2. To obtain single cell suspension, spheroids were collected into tubes and incubated with Accutase (Innovative Cell Technologies, # AT104–500) at RT for 20 minutes with gentle pipetting every 5 minutes. The cell-Accutase suspension was diluted with CCM, and dissociated spheroids were further treated with 100 μg/ml DNase I (Sigma Aldrich, #10104159001) for 10 minutes at 37 °C. Cells were passed through a 70μm cell strainer (BioPioneer, # DGN258368) to obtain a single cell suspension.
3D cell culture by spinner flasks
Generation of MSCs spheroids via spinner flask was modified based on previously published protocols for less compaction and easier dissociation55, 56. Briefly, MSCs were plated at 0.8 million cells per plate (Olympus Plastics, #25–203) in 15 ml of culture medium around 4 days before input into the spinner flask. To prepare the spinner flask (Corning, #4500–500), the flask was coated with 2 ml of Sigmacote (Sigma, SL2–100ML) to prevent cells from sticking to the glass, then autoclaved for 30 minutes. On the day of collection, 50 million cells were collected using Accutase and incubated on ice for 1 hour before adding into the spinner flask. Cells were cultured at 37 °C for approximately 45 hours with 5% CO2 and the rotation speed was set to 60 rpm. To obtain a single cell suspension, cells were collected from the spinner flask after 45 hours and incubated with Accutase for 25 minutes in a 37°C water bath. Cells were pipetted every 5 minutes to promote dissociation, then passed through a 70 mm cell strainer to obtain a single cell suspension.
Confined migration assay in a microfluidic device
Confined migration assays were performed using a microfluidic device as previously described57. The device consists of alternating parallel migration channels of either 5µm height and constrictions 1 to 2 µm width (confined migration) or 5 µm height and 15 µm width (unconfined migration). Devices were assembled as previously described and coated with a solution of 10µg/mL of fibronectin 24 hours prior to experiments23. Four hours prior to imaging, 3E4 MSCs or Cargocytes were stained with Hoechst 33342 (1:4000 v/v) then seeded into microfluidic devices and allowed to migrate along an FBS gradient (2% to 16.5%). Imaging was performed on a Zeiss LSM700 laser scanning confocal microscope with a 20x air objective. Cells and devices were imaged for 14 hours at 10-minute intervals in a temperature-controlled stage (37C°). The time required for cells to migrate through an individual constriction was quantified manually from images and was defined as starting at the first frame that the anterior portion of a cell entered a constriction and ending with the first frame after the cell posterior passed through the constriction.
LDV-FITC binding assay
Leu-1 N-2-[4-[[[(2-methylphenyl)amino]carbonyl]amino]phenyl]acetyl-, Lys-6 N-FITC conjugate (LDV-FITC) was purchased from R&D systems (#4577/1). The LDV-FITC binding assay was performed as previously described58. Briefly, MSCs or Cargocytes (1E6/100μl) suspended in HEPES buffer (140mM NaCl, 2.5mM KCL, 1.8mM CaCl2, 1mM MgCl2, 20mM HEPES, 0.5% FBS, pH=7.4) were incubated with 4nM LDV-FITC for 15 min at 37°C, were then stimulated with or without SDF-1μ (500ng/ml) for 2 min, and were immediately fixed for 5 min with 1ml pre-warmed 1% paraformaldehyde at 22°C to fix bound LDV-FITC. 1μM unconjugated LDV was used as a competitive control to show the binding specificity. Cells were washed with HEPES buffer for two times and then analyzed by the flow cytometry machine FACS Canto II (BD). The change of LDV-FITC binding was calculated as MFI ratio== (MFILDV-FITC+ SDF-1α - MFIunstained control)/ (MFILDV-FITC - MFIunstained control).
Endothelial cells (ECs) adhesion assay
EC adhesion assay was performed using primary HUVECs as previously described59. Briefly, HUVECs (passages 2–4) were grown to confluence in tissue-culture treated 6-well plates. HUVECs were pretreated with 10 ng/ml tumor necrosis factor-α (TNF-α, Peprotech, #300–01A) for 6 h and the upregulation of adhesion molecules were determined by flow cytometry analysis. MSCs and Cargocytes were stained with 5μM CFSE as previously described60. In SDF-1α stimulation group, after TNF-α stimulation, HUVECs were pretreated with 500ng/ml SDF-1α for 15 min. In antibody blocking groups, MSCs and Cargocytes were pre-incubated with 10 µg/ml antibodies (anti-VLA-4 or anti-PGSL-1, also see Supplementary Table 8) in CCM for 30 min at 37°C in 5% CO2. MSCs or Cargocytes suspended in 2 ml HEPES buffer (0.15 million/ml) per well were added to confluent EC and co-cultured for 15 min with occasionally shaking at 37°C with 5% CO2. After gently washing three times with HEPES buffer to remove nonadherent cells, adherent MSCs or Cargocytes were counted in 6 random fields (100X magnification) per well using Nikon Eclipse Ti epi-fluorescence microscope and analyzed with ImageJ Fiji software 1.53c.
Whole mount clearing and imaging of the lung tissue
Tissue clearing was performed as previously described61. All incubation steps were done at 37°C shaking at 100 rpm. Decolorization and delipidation solutions were made with water. First, fix lung samples in 2% PFA on ice for 20 minutes. To decolorize the lungs, incubate in 25% Quadrol solution (Sigma, #122262) at 37°C for 2 days. For delipidation, incubate in 30% tert-butanol (Sigma-Aldrich 360538), 50% tert-butanol, and 70% tert-butanol for 4 hours, 6 hours, and 24 hours, respectively. For dehydration, lungs were incubated in a 70% tert-butanol/30% PEG-MMA-500 (Sigma, #447943) solution for 2 days. For the final clearing step lungs were submerged in a 75% benzyl benzoate (Sigma, B6630)/25% PEG-MMA-500 solution for one day. All solutions after the decolorization step contain 3% Quadrol by weight. The cleared lung tissue were directly imaged on Olympus FV1000 confocal microscope and analyzed with the Volocity 6.3 software.
Mouse model of acute inflammation in the ear
The LPS-induced model of dermal inflammation in the mouse pinna was established similar to previously described34, 62. Briefly, female BALB/cJ mice (8–12 weeks old) were anesthetized with isoflurane (VetOne, MWI 502017) and injected with 30 μg lipopolysaccharides (LPS, Sigma Aldrich, #L3024, in 30 μl saline) in the posterior/dorsal dermis of the right ear and 0.9% saline (Hospira NDC 0409–4888) in the control, contralateral ear. After 6 hours, mice were anesthetized for intravenous (i.v.) injection (tail-vein or retro-orbital) of 1E6 MSCs or Cargocytes, or PBS (vehicle control) in 100μl solution.
For flow cytometry analyses, MSCs or Cargocytes were stained with 10 μM Vybrant DiD (Invitrogen, #V22887) following manufacturer’s instructions prior to injection. Animals were euthanized 24 hours after MSC or Cargocyte injection. Ears were removed at the level of the base and the dorsal and ventral skin was peeled from the cartilage, then placed in 2 ml digestion buffer containing 0.1 mg/ml DNase I and 0.2 mg/ml Liberase TL (Sigma Aldrich, #5401020001) diluted in 1% FBS in RPMI media. After incubation at 37°C for 1 hour, ear skin was ground with a pestle in a 70μm cell strainer (Biopioneer, #DGN258368). When no large tissue pieces remained intact, strainers were washed with 2ml of 1% FBS, 2 mM EDTA in PBS. Cells were then treated with 1X RBC lysis buffer (Biolegend, # 420301) for 2 minutes, washed with PBS, and stained with PE anti-mouse F4/80 Antibody and 7-AAD. Cells were analyzed by flow cytometry machine FACS Canto II (BD).
To monitor the biodistribution of i.v. injected cells, MSCs or Cargocytes were first transfected with firefly luciferase mRNA (Fluc mRNA) as described above. Transfected MSCs or Cargocytes were plated in 96-well-plate (10,000 per well, 100 μl CCM media) and directly incubated with excessive VivoGlo™ -luciferin (1.5mg/ml, Promega, P1042) in CCM media for 10 mins at 37°C. Luciferase activity was then determined as relative luminescence units (RLU) using SpectraMax M2e plate reader (Molecular Devices). In the biodistribution assay, mice were first treated with LPS as described above. For 24 and 48 hr time points, MSCs/Cargocytes were i.v. injected immediately after mRNA transfection; for 2 hr time point, MSCs/Cargocytes were cultured in 3D spinner flask for 24 hr after mRNA transfection and then i.v. injected. Mice were i.p. injected with VivoGlo™-luciferin (1.5mg per mouse, Promega, P1042) at indicated time points after i.v. injection. Mouse organs were subjected to bioluminescence imaging 15 minutes after i.p. injection using IVIS Lumina Series III (PerkinElmer). Because the perfusion of luciferin to the inflamed ears was not very efficient by this method, the dorsal skin of the ears was then peeled from the underlying cartilage and the subcutaneous surfaces of the tissue were directly soaked in VivoGlo™-luciferin substrate (60μl at 15mg/ml, Promega, P1042) and immediately subjected to bioluminescence imaging with IVIS Lumina Series III. For all the IVIS imaging, the exposure time was 5 minutes, the binning was medium, the F/stop was 1.2, the excitation filter was blocked, and the emission filter was open.
For whole mount immunostaining of mouse ears, inflammation was initiated and treated in groups as described above. Mice were euthanized 24 hours after i.v. injection of CargocyteTri-E, and ears were shaved, removed at the level of the base, and carefully dissected by peeling the dorsal skin surface from the underlying cartilage. Skin was fixed in 2% PFA on ice for 10 minutes, permeabilized in cold 100% methanol for 10 minutes and blocked in PBS with 20% FBS, 20% NGS (Normal goat serum, Jackson Immuno Research, # 005–000-121) and 0.2% Triton X-100 for 2 hours. The ears were then incubated with dye-conjugated primary antibodies (Supplementary Table 8) in PBS with 10% FBS, 10% NGS and 0.2% Triton X-100 overnight. After washing in PBS with 0.2% Trion X-100 for at least 2 hours, the ears were post-fixed with 4% PFA and mounted in VECTASHIELD® Vibrance™ Antifade Mounting Medium (Vector Laboratories, # H-1700). Confocal images were taken in an Olympus FV-1000 microscope and maximum projection of Z stacks of fluorescent images were generated in ImageJ Fiji.
To test human IL-10 cytokine delivery efficiency, cells and Cargocytes were transfected with human IL-10 mRNA and i.v. injected into the mice treated with LPS as described above. Animals were euthanized after 24 hours, and ears were minced and snap frozen in liquid nitrogen for ELISA analysis. A separate batch of animals treated in the same manner were euthanized at 48 hours after LPS injection. Mouse ears were either fixed in 4% PFA and submitted to the histology core of Moores Cancer Center for tissue sections and H&E (Hematoxylin and Eosin) staining, or mouse ears were harvested and snap frozen in liquid nitrogen for Real time RT-PCR analysis. The ear thickness was also measured in triplicate with a digital micrometer (Mitutoyo, # 293–831-30) prior to LPS injection and at the time of euthanasia (48 hours post LPS injection), and the change of ear thickness was used to monitor the inflammation status.
Mouse model of acute pancreatitis
The Caerulein-induced mouse acute pancreatitis model was established as previously described41. Briefly, BALB/cJ mice (8–12 weeks old, 22–26g) were fasted overnight (16–18 hours) before the experiments but permitted water ad libitum. Acute pancreatitis (AP) in mouse was induced by seven i.p. injection of Caerulein (Sigma, #C9026, 75 μg/kg bw) in 0.9% saline at hourly intervals over 6 hours. Control mice received same volume of saline without Caerulein. Indicated MSCs or Cargocytes were i.v. injected 2 hours after the last Caerulein injection.
For homing analyses, MSCs or Cargocytes were stained with 10μM Vybrant DiD prior to the i.v. injection as previously described. Animals were euthanized 16 hours after MSC or Cargocyte injection, and mouse tissues were then harvested and dissociated with collagenase I solution (0.5 mg/ml collagenase (Sigma Aldrich, #C9891–100MG), 20 μg/ml DNase, 5% FBS in PBS). After incubation at 37°C for 1 hour, mouse tissues were ground with a pestle in a 70μm cell strainer. When no large tissue pieces remained intact, strainers were washed with 2 ml of 1% FBS, 2 mM EDTA in PBS. Cells were then treated with 1X RBC lysis buffer (Biolegend, # 420301) for 5 minutes, washed with PBS, and stained with PE anti-mouse F4/80 Antibody and 7-AAD. Cells were analyzed by flow cytometry machine FACS Canto II (BD).
In another experiment, 8μg/kg bw recombinant human IL-10 protein, BM-MSCs (1E6 per mouse), B-MSC exosomes (2E10 per mouse), MSCTri E C19 (1E6 per mouse), and CargocyteTri E C19 (1E6 per mouse) with or without human IL-10 mRNA transfection were i.v. injected into mice with induced-AP. Animals were euthanized after 16 hours, and the tissues were harvested for subsequent analyses. The human IL-10 level in the plasma and pancreas were analyzed by ELISA as previously described. The inflammatory markers (TNF-α, IL-1β, IL-6 and Ccl2) in mouse pancreas were analyzed by Real time RT-PCR. The amylase activity in the serum was determined by the ACP Diagnostic Services Laboratory at UCSD, and the lipase activity in the serum was determined by the kit (Sigma, #MAK046) according to manufacturer’s instructions. The harvested tissues were also fixed in 4% PFA and submitted to the histology core of Moores Cancer Center for tissue sections, H&E staining and anti-CD45 immunohistochemistry staining. The H&E stained pancreas sections were blindly scored by the licensed pathologist (C.N.A) to evaluate the tissue damage and inflammation. The scoring criteria: edema was graded from 0–3 (0: absent; 1: focally increased between lobules; 2: diffusely increased between lobules; 3: acini disrupted and separated), and acinar necrosis was graded as 0–3 (0: absent; 1: periductal necrosis, <5%; 2: focal necrosis, 5∼20%; 3: diffuse parenchymal necrosis, 20∼50%44. The CD45 positive stained infiltrated leukocytes in the pancreas sections were blindly counted by the licensed pathologist (C.N.A). Ten high powered fields (400x) were observed per pancreas by light microscopy, starting with the area of highest number of positive cells. Number of cells with membranous immunoreactivity per consecutive field (in which at least 80% of the field contained tissue) were counted, exclusive of cells within blood vessels, in adjacent adipose tissue, or lymphoid follicles of lymph nodes. Immunoreactive cells were predominantly located at tissue periphery or in the connective tissue between lobules, and rarely within parenchyma.
Statistical analysis
GraphPad Prism8 was used to analyze the data and to determine statistical significance of differences between groups by applying unpaired Student’s t test, one-way or two-way ANOVA with post-hoc multiple comparisons test as indicated. Adjusted P values <0.05 were considered significant.
Supplementary Material
{kind=link}
Acknowledgements
This work was supported by the National Institutes of Health grants (R01 CA097022, CA184594 and CA182495 [R.L.K.] and R01 HL082792, R01 GM137605 and U54 CA210184 [J.L]), the National Science Foundation (CAREER Award CBET-1254846 to J.L), the Hartwell Foundation (J.D.B), and Center for Drug Discovery and Innovation at UC San Diego (R.L.K). C.N.A. was supported by NIH 5T32 OD17863–4. Imaging and analysis were in part performed at the UCSD School of Medicine Microscopy Core (NS047101) and UCSD Moores Cancer Center Tissue Technology Shared Resource Core (CCSG Grant P30CA23100). Human primary bone marrow MSCs were obtained from Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine (NIH P40OD011050). This work was performed in part at the Cornell NanoScale Science & Technology Facility (CNF), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant NNCI-2025233). We would like to thank Dr. Weian Zhao and Dr. Aude Ségaliny of UC Irvine, and Diane Thomas, Dr. Lage de Siqueira Neto, Dr. Maureen Ruchhoeft and Dr. Linjie Zhao from UCSD for thoughtful discussion.
Footnotes
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interest
Cargocyte is the trademark of Cytonus Therapeutics. R.L.K is the co-founder and equity holder of Cytonus Therapeutics. H.W and R.L.K are co-inventors of a related patent (WO 2019/032628 A1, US 10,927,349 B2) and H.W., W.P., and R.L.K. are co-inventors of another related patent (PCT/US21/016,919). Both patents were filed by UCSD.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-01X-XXXX-X.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.
References
- 1.Allen TM & Cullis PR Drug delivery systems: entering the mainstream. Science 303, 1818–1822 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Yoo JW, Irvine DJ, Discher DE & Mitragotri S Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov 10, 521–535 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Lyerly HK, Osada T & Hartman ZC Right Time and Place for IL12: Targeted Delivery Stimulates Immune Therapy. Clin Cancer Res 25, 9–11 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Fioranelli M & Roccia MG Twenty-five years of studies and trials for the therapeutic application of IL-10 immunomodulating properties. From high doses administration to low dose medicine new paradigm. J Integr Cardiol 1, 2–6 (2014). [Google Scholar]
- 5.Samanta S et al. Exosomes: new molecular targets of diseases. Acta Pharmacol Sin 39, 501–513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han X, Wang C & Liu Z Red Blood Cells as Smart Delivery Systems. Bioconjugate Chemistry 29, 852–860 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Fang RH, Kroll AV, Gao W & Zhang L Cell Membrane Coating Nanotechnology. Adv Mater 30, e1706759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thanuja MY, Anupama C & Ranganath SH Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: So near and yet so far. Adv Drug Deliv Rev 132, 57–80 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Labusca L, Herea DD & Mashayekhi K Stem cells as delivery vehicles for regenerative medicine-challenges and perspectives. World J Stem Cells 10, 43–56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sackstein R The lymphocyte homing receptors: gatekeepers of the multistep paradigm. Curr Opin Hematol 12, 444–450 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Nitzsche F et al. Concise Review: MSC Adhesion Cascade-Insights into Homing and Transendothelial Migration. Stem Cells 35, 1446–1460 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Ullah M, Liu DD & Thakor AS Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 15, 421–438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer UM et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem cells and development 18, 683–692 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karp JM & Leng Teo GS Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell 4, 206–216 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Galipeau J & Sensebe L Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 22, 824–833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks PW, Witten CM & Califf RM Clarifying Stem-Cell Therapy’s Benefits and Risks. N Engl J Med 376, 1007–1009 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Wigler MH & Weinstein IB A preparative method for obtaining enucleated mammalian cells. Biochem Biophys Res Commun 63, 669–674 (1975). [DOI] [PubMed] [Google Scholar]
- 18.Shay JW Cell enucleation, cybrids, reconstituted cells, and nuclear hybrids. Methods Enzymol 151, 221–237 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Coimbra VC et al. Enucleated L929 cells support invasion, differentiation, and multiplication of Trypanosoma cruzi parasites. Infect Immun 75, 3700–3706 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham DM et al. Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J Cell Biol 217, 895–914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolbank S et al. Telomerase immortalized human amnion- and adipose-derived mesenchymal stem cells: maintenance of differentiation and immunomodulatory characteristics. Tissue Eng Part A 15, 1843–1854 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malawista SE, Van Blaricom G & Breitenstein MG Cryopreservable neutrophil surrogates. Stored cytoplasts from human polymorphonuclear leukocytes retain chemotactic, phagocytic, and microbicidal function. J Clin Invest 83, 728–732 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keys J, Windsor A & Lammerding J Assembly and Use of a Microfluidic Device to Study Cell Migration in Confined Environments. Methods Mol Biol 1840, 101–118 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Lammerding J Mechanics of the nucleus. Compr Physiol 1, 783–807 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guilak F, Tedrow JR & Burgkart R Viscoelastic properties of the cell nucleus. Biochem Biophys Res Commun 269, 781–786 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Stewart-Hutchinson PJ, Hale CM, Wirtz D & Hodzic D Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp Cell Res 314, 1892–1905 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caille N, Thoumine O, Tardy Y & Meister JJ Contribution of the nucleus to the mechanical properties of endothelial cells. J Biomech 35, 177–187 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Marquez-Curtis LA & Janowska-Wieczorek A Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res Int 2013, 561098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandolfi F et al. Integrins: Integrating the Biology and Therapy of Cell-cell Interactions. Clin Ther 39, 2420–2436 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Chigaev A et al. Real time analysis of the affinity regulation of alpha 4-integrin. The physiologically activated receptor is intermediate in affinity between resting and Mn(2+) or antibody activation. J Biol Chem 276, 48670–48678 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Boltze J et al. The Dark Side of the Force - Constraints and Complications of Cell Therapies for Stroke. Front Neurol 6, 155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung JW et al. Familial occurrence of pulmonary embolism after intravenous, adipose tissue-derived stem cell therapy. Yonsei Med J 54, 1293–1296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartosh TJ et al. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc Natl Acad Sci U S A 107, 13724–13729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy O et al. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood 122, e23–32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon S et al. Antigen markers of macrophage differentiation in murine tissues. Curr Top Microbiol Immunol 181, 1–37 (1992). [DOI] [PubMed] [Google Scholar]
- 36.Devine MJ, Mierisch CM, Jang E, Anderson PC & Balian G Transplanted bone marrow cells localize to fracture callus in a mouse model. J Orthop Res 20, 1232–1239 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Ignowski JM & Schaffer DV Kinetic analysis and modeling of firefly luciferase as a quantitative reporter gene in live mammalian cells. Biotechnol Bioeng 86, 827–834 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Alvarez HM et al. Effects of PEGylation and immune complex formation on the pharmacokinetics and biodistribution of recombinant interleukin 10 in mice. Drug Metab Dispos 40, 360–373 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Greenberg JA et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 59, 128–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forsmark CE, Vege SS & Wilcox CM Acute Pancreatitis. N Engl J Med 375, 1972–1981 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Niederau C, Ferrell LD & Grendell JH Caerulein-induced acute necrotizing pancreatitis in mice: protective effects of proglumide, benzotript, and secretin. Gastroenterology 88, 1192–1204 (1985). [DOI] [PubMed] [Google Scholar]
- 42.Su KH, Cuthbertson C & Christophi C Review of experimental animal models of acute pancreatitis. HPB (Oxford) 8, 264–286 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rongione AJ et al. Interleukin 10 reduces the severity of acute pancreatitis in rats. Gastroenterology 112, 960–967 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Van Laethem JL et al. Interleukin 10 prevents necrosis in murine experimental acute pancreatitis. Gastroenterology 108, 1917–1922 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Fedorak RN et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology 119, 1473–1482 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Nowakowski A, Andrzejewska A, Janowski M, Walczak P & Lukomska B Genetic engineering of stem cells for enhanced therapy. Acta Neurobiol Exp (Wars) 73, 1–18 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Cui LL et al. The cerebral embolism evoked by intra-arterial delivery of allogeneic bone marrow mesenchymal stem cells in rats is related to cell dose and infusion velocity. Stem Cell Res Ther 6, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krueger TEG, Thorek DLJ, Denmeade SR, Isaacs JT & Brennen WN Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. Stem Cells Transl Med 7, 651–663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yin JQ, Zhu J & Ankrum JA Manufacturing of primed mesenchymal stromal cells for therapy. Nat Biomed Eng 3, 90–104 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Ungerechts G et al. Moving oncolytic viruses into the clinic: clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol Ther Methods Clin Dev 3, 16018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Marino S & Ho CY 97. Large Scale Purification of AAV with Continuous Flow Ultracentrifugation. Molecular Therapy 24, S42 (2016). [Google Scholar]
- 52.Vazquez-Lombardi R, Roome B & Christ D Molecular Engineering of Therapeutic Cytokines. Antibodies 2, 426–451 (2013). [Google Scholar]
- 53.Wigler MH, Neugut AI & Weinstein IB Enucleation of mammalian cells in suspension. Methods Cell Biol 14, 87–93 (1976). [DOI] [PubMed] [Google Scholar]
- 54.Bartosh TJ & Ylostalo JH Preparation of anti-inflammatory mesenchymal stem/precursor cells (MSCs) through sphere formation using hanging-drop culture technique. Curr Protoc Stem Cell Biol 28, Unit 2B 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frith JE, Thomson B & Genever PG Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng Part C Methods 16, 735–749 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Egger D, Tripisciano C, Weber V, Dominici M & Kasper C Dynamic Cultivation of Mesenchymal Stem Cell Aggregates. Bioengineering (Basel) 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davidson PM, Sliz J, Isermann P, Denais C & Lammerding J Design of a microfluidic device to quantify dynamic intra-nuclear deformation during cell migration through confining environments. Integr Biol (Camb) 7, 1534–1546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hyduk SJ et al. Talin-1 and kindlin-3 regulate alpha4beta1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J Immunol 187, 4360–4368 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Semon JA et al. Integrin expression and integrin-mediated adhesion in vitro of human multipotent stromal cells (MSCs) to endothelial cells from various blood vessels. Cell Tissue Res 341, 147–158 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Quah BJ, Warren HS & Parish CR Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc 2, 2049–2056 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Jing D et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Res 28, 803–818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corradetti B et al. Hyaluronic acid coatings as a simple and efficient approach to improve MSC homing toward the site of inflammation. Sci Rep 7, 7991 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.