
Figure 2. Deposition and detection of m6A.
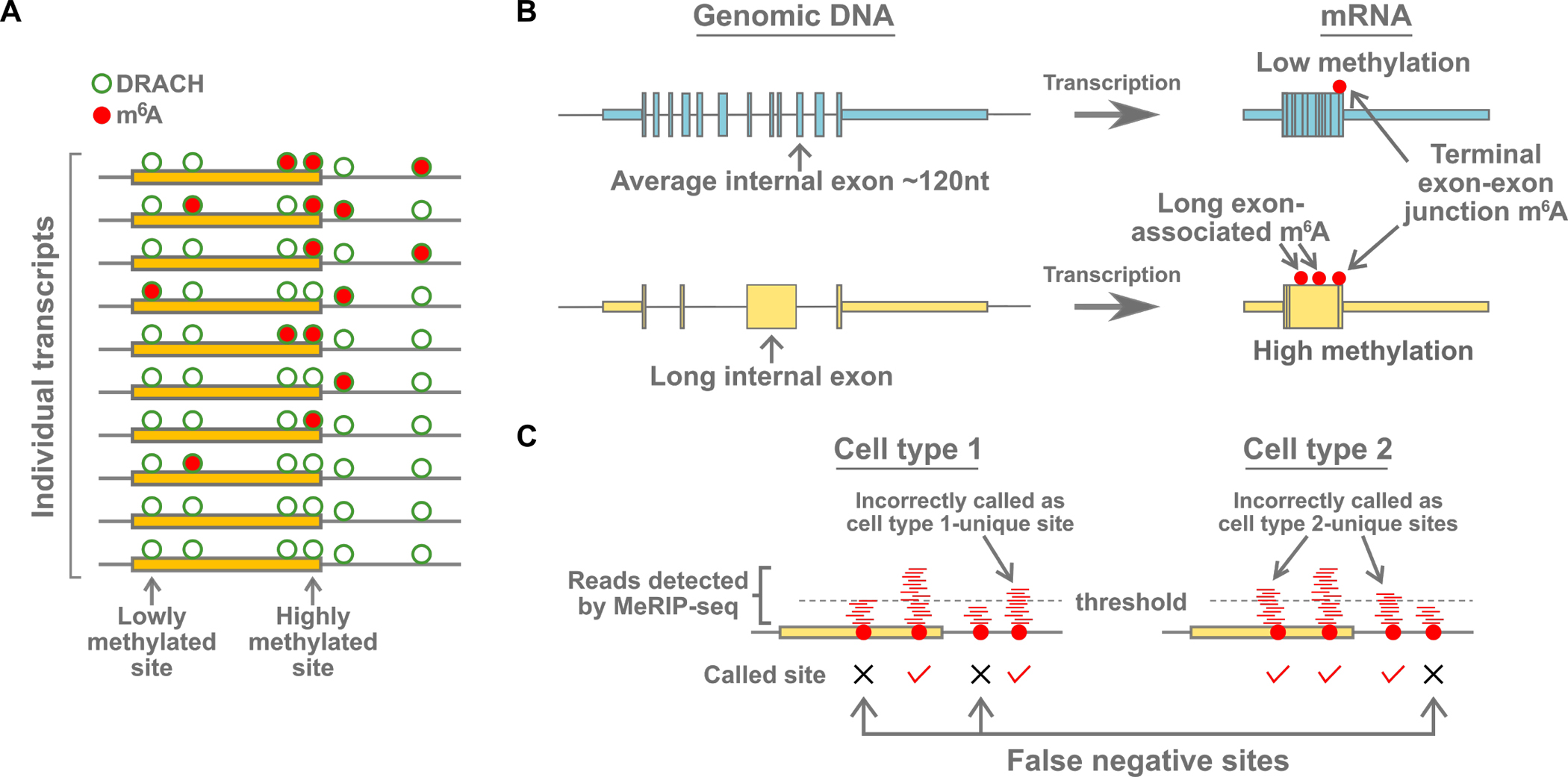
(A) m6A is a low stoichiometry modification. At any given m6A site in a specific mRNA, only a small number (usually less than 20%) of the transcript copies in the cell will have m6A at that site. Most of the DRACH m6A consensus sites are not methylated (green circle). Additionally, some DRACH sites are more methylated than others, and all DRACH sites are likely methylated to some degree, although the stoichiometry is likely very low. The molecular mechanism that causes some DRACH sites to be more methylated than others, even in the same transcript, are not fully understood. Although m6A mapping may reveal multiple m6A sites for a given mRNA, the individual transcripts that comprise the m6A annotation are typically methylated only at a subset of sites. Newer single-transcript analysis methods have revealed the stochastic nature of methylation on individual transcripts.
(B) Gene architecture influences m6A deposition. Shown (left) are examples of two genes, one containing a long internal exon. After transcription (right), m6A is preferentially formed on the regions of transcripts corresponding to long exons. m6A is also enriched near the terminal exon-exon junction, particularly in mRNAs with long 3’UTRs. The correlation of m6A with these genomic features suggests that the writer complex is regulated by other events that are responsive to gene architecture.
(C) Putative differences in m6A are sometimes artifacts of the methods used to call m6A sites from mapping data. Many studies have used mapping methods such as MeRIP-seq to map m6A and to compare transcriptome-wide distributions of m6A between two different cell conditions. These methods map reads (red lines) immunoprecipitated by an m6A-binding antibody. m6A sites are often called based on whether there are a sufficient number of reads above a threshold. However, the number of reads that map to any m6A sites can be very variable, even between replicates (McIntyre et al., 2020). Thus, the same m6A site in one sample might be called in one sample, but not the other, if the number of reads just passes or misses the threshold. For this reason, the differences in the transcriptome-wide distribution of m6A in many experiments may have been highly overestimated.