
Figure 3.
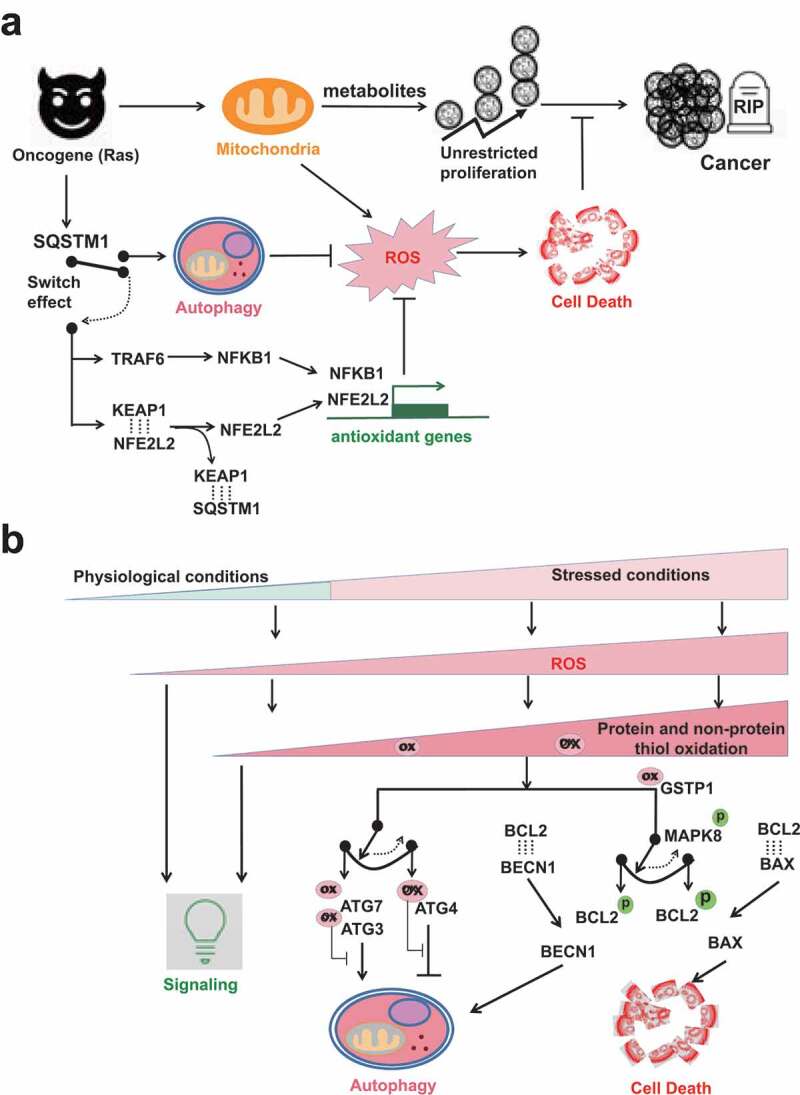
The ROS involved switch between autophagy and cell death. (A) SQSTM1 provides an alternative antioxidant pathway by functioning like an electric switch to connect either autophagy or NFE2L2-dependent antioxidant gene transcription. When autophagy is functional, SQSTM1 is a bona fide autophagic receptor protein. SQSTM1 itself is also recycled by autophagy. But when autophagy is impaired, SQSTM1 aggregates in cells and then switches to turn on NFE2L2- or NFKB1-dependent antioxidant gene transcriptions. Ras-driven oncogenesis over-expresses SQSTM1. The cancer cells in turn hijack SQSTM1 to activate either autophagy or antioxidant gene transcriptions to tame ROS, so that their unrestricted cancer cell proliferation will not be interfered by ROS-induced cell death. (B) ROS exist at different levels from physiological to pathological/stressed conditions. At low levels, ROS act as signaling messengers. Higher levels of ROS are restricted by the first-line protein thiols system and the second-line autophagy to maintain redox homeostasis. The rheostat effects of protein thiols in the regulation of autophagy inhibition/induction and in the regulation of autophagy/apoptosis are shown by two examples. The first switch is mediated by different susceptibilities of ATG3/7 vs. ATG4 to oxidation. ATG3/7 are more sensitive than ATG4 to oxidation, so that low levels of ROS oxidize ATG3/7 but spare ATG4. The second switch is mediated by different BCL2 complexes formation. MAPK8 phosphorylates BCL2 and thus interfere the association of BCL2 with binding partners. Notably, MAPK8 activation is affected by inhibitory binding with antioxidant GSTP1. Comparatively, the association of BCL2-BECN1 complex is weaker than that of BCL2-BAX complex. Thus, low levels of stress and GSTP1 oxidation cause transient MAPK8 activation and dissociation of BCL2-BECN1 complex but not BCL2-BAX complex. Dissociated BECN1 then turn on autophagy. But sustained MAPK8 activation frees BAX from the inhibitory complex BCL2-BAX and switches on apoptosis.