

Significance
Skeletal muscle is a mechanical organ that not only produces force but also uses mechanical stimuli as a signal to regulate cellular responses. Duchenne and Becker muscular dystrophy are lethal muscle wasting diseases that affect 1 in 3,500 boys and is caused by the absence or malfunction of dystrophin protein, respectively. There is a lack of understanding on how the integration of these mechanical signals is dysregulated in muscular dystrophy and how they may contribute to disease progression. In this study, we show that patient-relevant dystrophin mutations alter the mechanical signaling axis in muscle cells, leading to impaired migration. This work proposes dystrophin as a component of the cellular force-sensing machinery, furthering our knowledge in the pathomechanism of muscular dystrophy.
Keywords: mechanotransduction, muscular dystrophy, molecular tension sensors, BRET, cell migration
Abstract
Dystrophin is an essential muscle protein that contributes to cell membrane stability by mechanically linking the actin cytoskeleton to the extracellular matrix via an adhesion complex called the dystrophin–glycoprotein complex. The absence or impaired function of dystrophin causes muscular dystrophy. Focal adhesions (FAs) are also mechanosensitive adhesion complexes that connect the cytoskeleton to the extracellular matrix. However, the interplay between dystrophin and FA force transmission has not been investigated. Using a vinculin-based bioluminescent tension sensor, we measured FA tension in transgenic C2C12 myoblasts expressing wild-type (WT) dystrophin, a nonpathogenic single nucleotide polymorphism (SNP) (I232M), or two missense mutations associated with Duchenne (L54R), or Becker muscular dystrophy (L172H). Our data revealed cross talk between dystrophin and FAs, as the expression of WT or I232M dystrophin increased FA tension compared to dystrophin-less nontransgenic myoblasts. In contrast, the expression of L54R or L172H did not increase FA tension, indicating that these disease-causing mutations compromise the mechanical function of dystrophin as an FA allosteric regulator. Decreased FA tension caused by these mutations manifests as defective migration, as well as decreased Yes-associated protein 1 (YAP) activation, possibly by the disruption of the ability of FAs to transmit forces between the extracellular matrix and cytoskeleton. Our results indicate that dystrophin influences FA tension and suggest that dystrophin disease-causing missense mutations may disrupt a cellular tension-sensing pathway in dystrophic skeletal muscle.
Skeletal muscle is an organ under constant mechanical stress, even at rest. Dystrophin, a protein located beneath the muscle cell plasma membrane (sarcolemma) (1), is a key cytoskeletal protein that contributes to maintaining muscle integrity (2–4). Dystrophin maintains the structural stability of the sarcolemma by linking the actin cytoskeleton to the extracellular matrix (ECM) via a transmembrane protein adhesion complex known as the dystrophin–glycoprotein complex (DGC) (5–8). The DGC is enriched at the costameres in striated skeletal muscle and are subsarcolemmal protein assemblies that couple the force-generating sarcomeres to the sarcolemma (4, 9–11). Mutations in genes encoding different components of the DGC are the cause of various forms of muscular dystrophy, such as Duchenne and Becker muscular dystrophy (DMD and BMD, respectively). DMD is caused by the absence of dystrophin (3), while BMD is caused by decreased expression of a truncated and less functional dystrophin (12, 13). Partial to total loss of dystrophin leads to sarcolemmal fragility (14, 15) and myofiber death (16), which is outwardly manifested as muscle weakness (17).
The DGC adhesive complex interacts via the actin cytoskeleton with integrin-based focal adhesions (FAs) that connect the ECM to the cytoskeleton via talin and vinculin (18, 19). FAs transmit mechanical signals via the cytoskeleton to mechanotransducer proteins to ultimately modulate gene expression that regulates cell behavior, in a process known as mechanotransduction (20, 21). At tissue and cellular levels, previous studies showed that dystrophin contributes to the mechanical stability of muscle and that its absence leads to decreased tissue and cellular stiffness (22–24). Dysregulation of mechanical signals, force transmission between ECM and the cytoskeleton, and mechanotransducer activities have been shown to promote pathogenic phenotypes that promote muscular dystrophy (23, 25, 26), but it is unclear how disease-causing dystrophin mutations lead to aberrant mechanotransduction. We hypothesized that dystrophin enhances links between the ECM and cytoskeleton in muscle cells, thus influencing FA tension and in turn force transmission and regulation of mechanosensitive processes, such as migration. Dystrophin mutations, then, likely lead to abnormal FA tension and mechanotransduction.
Here, we demonstrate the use of a molecular tension sensor genetically encoded in the FA protein vinculin to study the impact of dystrophin on FA tension–sensing in C2C12 myoblasts transgenically expressing wild-type (WT) or mutant dystrophin. We show that the stable expression of WT dystrophin (WT-Dys) increases vinculin tension compared to dystrophin-less nontransgenic control cells. However, in the presence of DMD- or BMD-associated missense mutations, vinculin tensions remain low and comparable to the dystrophin-less control, even upon increasing expression levels of mutant dystrophins to WT levels. These results were confirmed by traction–force microscopy. Our results also demonstrate that the dystrophy-associated mutations manifest as impaired migration. To explore whether these migration defects were a result of aberrant mechanotransduction via altered FA tension transmission, we studied several known mechanosensitive signaling pathways and found that cells that had decreased FA tension also had reduced Yes-associated protein (YAP) and ERK1 mechanosignaling with little effect on RhoA signaling, filamentous actin morphology, and myosin II activity. These results suggest that missense mutations impair dystrophin’s ability to transmit cellular tension and provide a link between defects in mechanotransduction at the molecular level to muscle–level phenotypes that promote disease progression.
Results
Dystrophin Expression Impacts Vinculin Tension in Myoblasts.
To measure tensions in muscle cells harboring variable dystrophin levels and mutations, we used previously characterized transgenic C2C12 myoblast lines (27) that stably express WT-Dys or dystrophins with missense mutations associated with DMD and BMD (13, 28). These transgenic cell lines overcome the limitation of using C2C12 cell lines, which do not exhibit detectable levels of dystrophin until they are differentiated and cultured for long periods. To probe the ECM to cytoskeleton tensions at a molecular level using FA as a proxy, we used an established bioluminescence-based molecular tension sensor, VinTS, encoded in the FA protein vinculin (29, 30). We hypothesized that tensions sensed at FAs might be sensitive to the status of dystrophin via the linkage to actin that both adhesive complexes share (Fig. 1A and SI Appendix, Fig. S1A). Bioluminescence resonance energy transfer (BRET) ratios are modulated by vinculin tension, as explained in Fig. 1. A truncated version of the tension sensor that cannot experience tension (VinTL; Fig. 1A) was used as a control (30).
Fig. 1.

Vinculin BRET tension sensor in FAs. (A) Schematic of the BRET efficiencies of VinTS and VinTL in the presence and absence of forces. The addition of furimazine (Fz) excites the energy donor NanoLuc (NL). Efficiency of energy transfer to the acceptor NeonGreen (NG) is dependent on the load experienced by the tension sensor. BRET-tension sensor is localized between the head domain of vinculin − Vh − that binds to talin and the tail domain of vinculin − Vt − that binds to actin. (B) Representative images of apparent (app) BRET efficiency of VinTS and VinTL in myoblasts. (C) Distribution mean of app BRET efficiency (n = 3) for VinTS and VinTL in transfected myoblasts. All data shown are from WT-Dys myoblasts. (Scale bar, 20 μm.) Data analyzed via an unpaired two-tailed t test; ns, not significant (All error bars, SEM).
We first aimed to validate the activity of the tension sensor in C2C12 myoblasts and ensure that it did not disrupt FA formation. The VinTS Förster resonance energy transfer (FRET) and BRET sensors have been used in several cell types (29–32), but to our knowledge, it has not been used in C2C12 myoblasts. Our results confirm that the tension sensor does not alter vinculin localization or protein levels (SI Appendix, Figs. S1–S3), and myoblasts expressing VinTL formed larger FAs than VinTS (SI Appendix, Fig. S4), consistent with observations made by others (30). To assess whether VinTS sensed vinculin tension at FAs, we performed ratiometric BRET imaging on VinTS- and VinTL-encoded myoblasts expressing WT-Dys. As expected, VinTS-transfected cells showed significantly lower BRET ratios when compared to VinTL-transfected cells (Fig. 1 B and C), confirming that VinTS is under greater mechanical tension than VinTL. Interestingly, we did not observe tension gradients along FAs as we and others have previously reported (SI Appendix, Figs. S5 and S6) (29, 33, 34). Differences in measured BRET ratios between VinTS and VinTL cells cannot be explained by FA shape (aspect ratio) or cell morphology (SI Appendix, Figs. S4 and S7). Altogether, these results validate the use of VinTS as a tension sensor to study levels of tension transmitted across vinculin in the FA of C2C12 myoblasts.
To determine whether a tension sensor installed in an FA protein could detect the presence of the dystrophin linked to actin, we transfected VinTS and VinTL into nontransgenic cells (henceforth referred as NTg), which do not express dystrophin (Fig. 2A and SI Appendix, Fig. S8). When compared to NTg, transgenic myoblasts expressing WT-Dys exhibited lower VinTS BRET ratios (Fig. 2B) and therefore higher vinculin tension. As expected, force-insensitive VinTL BRET ratios were the same for NTg and WT-Dys myoblasts (Fig. 2B). As a control, we also characterized another transgenic myoblast line that expresses the nonpathogenic single nucleotide polymorphism (SNP) in dystrophin, I232M. The tension measured at the I232M adhesion complexes using VinTS did not differ from WT-Dys myoblasts (SI Appendix, Fig. S9A). These results show that stable dystrophin expression in myoblasts significantly impacts vinculin tension, increasing the tension sensed.
Fig. 2.

Dystrophin status influences FA tension. (A) Quantification of n = 3 separate cell lysates probed with green fluorescent protein (GFP)-dystrophin antibody (455 kDa) and normalized to actin (42 kDa) (Top). Each color represents the same independent experiment set. Representative Western blot of cell lysates (Bottom). (B) Apparent BRET efficiencies (n = 3) for myoblasts transfected with VinTS and VinTL. (C) Quantification of n = 3 separate lysates of cells incubated with DMSO or MG-132, probed with GFP-dystrophin antibody, and normalized to actin (Top). Each color represents the same independent experiment set. Representative Western blot of cell lysates (Bottom). (D) Apparent BRET efficiencies (n = 3) for myoblasts incubated with DMSO or MG-132, transfected with VinTS. (E) Representative phase–contrast images with traction field overlays (Left) and traction stress distribution map (Right) for WT-Dys and (F) L54R myoblasts, with their (G) mean strain energy quantifications in log scale (n = 4, L54R = 93 cells; WT = 88 cells) (Color bars, minimum and maximum amount of traction stress from the vectors). Overall, summed traction stress for representative L54R cell is 1.32 MPa and for WT cell is 1.42 MPa. (Scale bar, 5 μm.) Large dots denote the mean of independent experiments and smaller dots denote the individual mean BRET measurements. Each experiment and corresponding individual measurements are colored the same. Western blot measurements are relative to their respective WT sample. Cell line comparison data analyzed via one-way ANOVA; ns, not significant. MG-132 vs. DMSO vehicle control data analyzed via unpaired two-tailed t test. TFM data analyzed via two-tailed Mann–Whitney U test (All error bars, SEM).
Dystrophin Missense Mutations Decrease Vinculin Tension.
We next asked whether cell lines expressing dystrophin mutants showed similar effects on vinculin tension as WT-Dys. Missense mutations in dystrophin can lead to disease as severe as mutations causing complete loss of expression (28) through a pathomechanism that is not well understood. To test our hypothesis that forces between the ECM and the cytoskeleton of muscle cells are disrupted by dystrophin mutations, we first characterized the transgenic cell lines expressing dystrophins encoding pathogenic L54R and L172H mutations, which map to actin binding domain 1. L54R is expressed at 8% of WT and L172H at 130% (Fig. 2A) in myoblasts. As expected, non–load-bearing VinTL biosensors exhibited a relatively high BRET ratio, thus indicating low tension across all cell lines independent of dystrophin status (Fig. 2B). However, in contrast to the large decrease in BRET ratios observed in WT-Dys myoblasts, signifying higher tensions as productive adhesive contacts were added, the dystrophin missense mutant cells showed relatively small decreases in BRET ratios, more comparable to NTg (Fig. 2B and SI Appendix, Fig. S10). These data suggest that in contrast to WT-Dys, which acts to increase tension across vinculin, the L54R and L172H mutations compromise the mechanical function of dystrophin.
While the L172H mutant was expressed at similar levels as WT-Dys (Fig. 2A), L54R was expressed at significantly lower levels. To differentiate between the effect of the dystrophin mutations and dystrophin levels on the mechanical function of dystrophin, we treated WT- and L54R-Dys–expressing myoblasts with 1 μM of the proteasome inhibitor MG-132, which increased the expression of WT-Dys 4-fold and L54R-Dys 6.6-fold, relative to treatment with vehicle alone (Fig. 2C). Our BRET tension sensor measurements showed that even when L54R-Dys levels were raised to WT-Dys levels, there was no significant change in vinculin tension (Fig. 2D). Thus, the observed decrease in vinculin tension for L54R- and L172H-Dys mutants compared to WT-Dys (Fig. 2B) is likely due to effects of the missense mutations encoded in dystrophin and not different dystrophin expression levels. Moreover, using the same concentration of MG-132 on WT-Dys–expressing myoblasts demonstrated that increasing WT-Dys by a fourfold, relative to WT-Dys under vehicle conditions, also did not affect vinculin tension (Fig. 2 C and D), further reinforcing this conclusion. Vehicle alone did not alter the previously observed effects of WT-Dys and L54R on FA (Fig. 2B). These results corroborate what was observed for I232M myoblasts, where a benign SNP expressing 1.8-fold more dystrophin compared to WT-Dys levels does not alter vinculin tension (SI Appendix, Fig. S9 A and B). Furthermore, FA morphology and DGC protein levels did not differ between dystrophin-expressing cell lines (SI Appendix, Figs. S11–S13), indicating that dystrophin missense mutations alone are the main cause for decreased tension transmitted along the FA load-bearing protein, vinculin. Therefore, it is likely protein quality and not quantity that impacts the mechanical function of dystrophin in myoblasts.
To establish whether tensions measured across vinculin reflect overall FA tension, as vinculin tension does not always correlate with FA tensions (30), we employed traction-force microscopy to directly measure FA tension in WT and L54R myoblasts. Traction strain energies for WT were 33% higher than for L54R myoblasts (Fig. 2 E–G). Thus, vinculin tension reflects overall FA tension in our system.
Defective Migration in Myoblasts Expressing Mutant Dystrophins.
Cells migrate by transmitting contractile forces to their surrounding ECM by forming and detaching adhesion complexes at the membrane–ECM interface (35). Altered FA tension is accompanied by changes in cellular migration, although the relationship between traction force and migration is complex (36–38). We measured single-cell migration and proliferation in myoblasts expressing WT and mutant dystrophins. WT-Dys and I232M myoblasts had migration speeds comparable to that of to NTg, while L172H and L54R myoblasts were 27% and 46% slower, respectively, when compared to WT (Fig. 3 A and B and SI Appendix, Figs. S14 and S15). Directional persistence at the last point of trajectory was not significantly different for any of the cell lines, although there was correlation with cell speeds (Fig. 3C). However, when directionality was calculated discretely over time, NTg, WT-Dys, and I232M directionalities were the same while L54R and L172H directionalities were significantly lower (Fig. 3D). We also performed wound healing assays on confluent monolayers of cells plated on fibronectin and followed wound closure over time, and we obtained results similar to those of the random single–cell migration data (SI Appendix, Fig. S16).
Fig. 3.

Missense mutations cause myoblast migration defects. (A) Origin plots for all cell trajectories (n = 3, 15 trajectories each) of single migrating cells, from which (B) cell speed, (C) directionality ratio measured at the last point of trajectory, (D) directionality ratio over time, (E) mean square displacement, and (F) direction autocorrelation were calculated to characterize cell migration efficiency. Large triangles denote the mean of independent experiments, and smaller dots denote the mean of individual measurements. Each experiment and corresponding individual measurements are colored the same. Data analyzed via one-way ANOVA; ns, not significant (All error bars, SEM).
As another approach to estimate persistence, we calculated the mean square displacements (MSDs) and direction autocorrelation over increasing time intervals. As per our directionality ratio results, the MSD results showed that L54R and L172H myoblasts explored less area over time than NTg, WT, and I232M, all of which had comparable values among each other (Fig. 3E). Direction autocorrelation showed that the angles describing the trajectory of L54R and L172H cells were less aligned with each other over different time scales in comparison to NTg, WT, and I232M and were therefore less directional (Fig. 3F). MSD and direction autocorrelation were equivalent between L54R and L172H for almost all time intervals. These results demonstrate that missense dystrophin mutations associated with DMD and BMD cause myoblasts to migrate less efficiently, whereas not expressing dystrophin or expressing a benign SNP does not influence migration speed or directionality. Furthermore, these results highlight the pathogenicity of DMD-associated mutations. Although L54R myoblasts express 92% less dystrophin than WT-expressing myoblasts, the low levels of mutant dystrophin still severely impair migration.
Mechanoactivation Is Altered by DMD- and BMD-Associated Mutations.
To explore whether changes in vinculin tension caused by missense dystrophin mutants are related to altered cytoskeletal dynamics, we surveyed actin filament formation and myosin II activity because they are critical cellular force transmission components, as well as various established mechanosignaling pathways downstream of FA known to be altered in muscular dystrophy (23, 39–42). Dystrophin is a signaling hub that binds to filamentous actin (F-actin), not only to link the ECM to the cytoskeleton but to either directly modulate signaling or to do so via actin (43–45), possibly by regulating actin filament tension via FA tension. Actin filament morphology, number, globular actin to F-actin ratios, and myosin II activation were not significantly different between cell lines (SI Appendix, Figs. S17 and S18).
Several mechanotransducer proteins function at the intersection of mechanotransduction nodes that have been linked to both FA and DGC adhesive complexes. As a start to understanding how mechanotransduction is disrupted by dystrophic mutations to lead to impaired cell migration, we probed RhoA and YAP mechanotransducer pathways as they are key mechanotransduction regulators for the G-protein and Hippo signaling cascades, respectively. The RhoA-actin-serum response factor pathway is dysregulated in dystrophin-deficient myotubes and in dystrophic mouse models (39) and also plays a role in FA size and dynamics (38). Our results showed that RhoA activation, measured as RhoA-guanosine triphosphate (RhoA-GTP) over total RhoA, was similar for WT and mutant dystrophin–expressing myoblasts (Fig. 4A and SI Appendix, Fig. S19). This finding was not that surprising since RhoA changes often accompany changes in FA size (38), which we did not observe in the mutant–dystrophin myoblasts. FA tension generally promotes the activation of the transcriptional coactivator YAP (46), which involves a change in localization from the cytoplasm to the nucleus, and complex changes in YAP activity have also been implicated in dystrophic contexts (40–42, 47, 48). We found that myoblasts expressing WT-Dys showed an increased nuclear/cytoplasmic YAP ratio compared to NTg, and myoblasts expressing dystrophin mutations had YAP ratios comparable to that of to NTg (Fig. 4 A and B and SI Appendix, Fig. S20). Thus, WT-Dys myoblasts have increased YAP activation compared to myoblasts expressing DMD- and BMD-associated mutations. Interestingly, the observed trends in YAP localization in WT versus dystrophic cells mirrored the trends in vinculin tension. We also found ERK1 to be more active in WT-Dys than the mutants (SI Appendix, Figs. S21 and S22), and the transcriptional coactivator with a PDZ-binding motif is a paralog of YAP; changes in its activity were not a subject of this study. Overall, our data suggest that in myoblasts, YAP is a mechanotransducer of dystrophin-mediated intracellular tension and a possible modulator of the observed migration phenotype for the dystrophin-expressing myoblasts. We also measured cell proliferation in the cell lines to rule out potential confounding effects (42) on migration caused by increased nuclear YAP, as its activation is also known to promote proliferation. However, we measured no differences between our cell lines in proliferation over 4 days (SI Appendix, Fig. S22). Altogether, decreased FA tension, impaired migration, and YAP and ERK1 decreased activation suggest that disease-associated missense mutations in dystrophin disrupt a link between tension transmission and mechanoactivation in myoblasts.
Fig. 4.
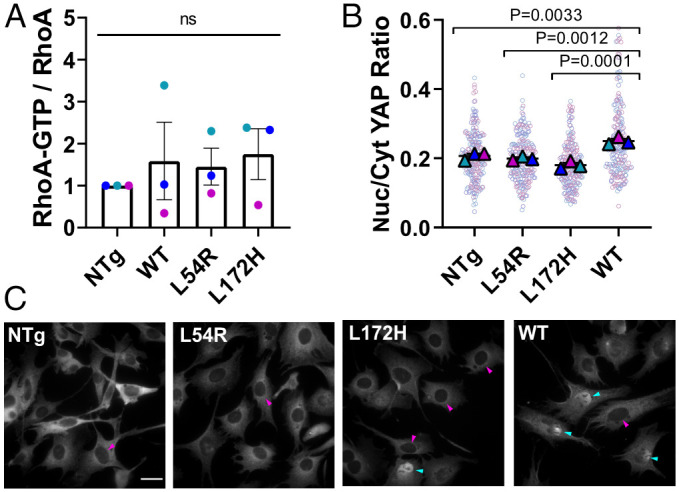
Dystrophin missense mutations alter YAP mechanoactivation. (A) RhoA activation as a ratio of RhoA-GTP over total RhoA, measured by a luminescent enzyme-linked immunosorbent assay–type assay and Western blot, respectively. (B) Nuclear-to-cytoplasmic YAP ratio quantification (n = 3). (C) Representative images of myoblasts immunostained for YAP. Cyan arrow heads show YAP-filled nuclei and magenta arrowheads show nuclei with low YAP staining. (Scale bar, 30 μm.) Large triangles denote the mean of independent experiments, and smaller dots indicate individual measurements. Each experiment and corresponding individual measurements are colored the same. Data are presented relative to corresponding NTg value. Data analyzed via one-way ANOVA; ns, not significant (All error bars, SEM).
Discussion
Force-transmitting complexes from the membrane to the nucleus allow for the transduction of physical cues into biochemical signals and the regulation of gene expression. In disease, the mechanoresponsiveness of these complexes and their connection to different elements of the mechanotransduction axis can be altered. A phenotype of muscle cells harboring abnormal dystrophin is a destabilized sarcolemma due to weaker cytoskeletal-ECM coupling (49), which leads to membrane breakage and increased compliance (14, 15, 22, 24). Remarkably, we show here that abnormal mechanical coupling manifests even at a molecular level as decreased vinculin tension within FAs, which disrupts cellular mechanotransduction pathways that may contribute to disease pathogenesis.
The key finding of this study (Fig. 5) is that two disease-associated missense mutations in the large dystrophin component of the DGC adhesive complex are sufficient to reduce vinculin tension at the FA complex compared to cells expressing WT-Dys. Our previous studies indicated that these DMD- and BMD-associated mutations lead to protein instability, aggregation, and later degradation by the proteasome (27, 50) as perhaps they are “poisonous” to the cell. Our findings here show that these dystrophin mutants are less functional and lead to pathogenic phenotypes, supporting our previous hypothesis of the toxic gain-of-function nature of these mutations and providing insight to the evolutionary basis of why cells dispose of these proteins. The mutations map to the actin-binding domain of dystrophin, suggesting that defective binding to actin could weaken the coupling between the ECM and actin. Indeed, the actin-binding affinity of dystrophin is moderately decreased by L54R, although this is not the case for L172H (50). Mutations could potentially alter dystrophin mechanical properties (51, 52) and thus dampen the response of the actomyosin machinery. Alternatively, phosphorylation of the dystrophin homolog utrophin in spectrin repeat 9 has been shown to stabilize the binding of its C-terminal domain with β-dystroglycan at the membrane (53), which could alter coupling to the ECM. Future studies will address whether disease-associated missense mutations impact dystrophin’s effect on tension sensing via similar long-range allostery.
Fig. 5.

Dystrophin as a mechanical focal adhesion allosteric regulator. Proposed model of dystrophin expression effects on myoblast mechanotransduction via FA. Expression of WT or benign I232M dystrophin allosterically regulates FA function by increasing vinculin tension and overall cellular traction forces (Ftrac). This cross talk between the DGC and FA leads to increased force transmission via the actin cytoskeleton (Insets), which deforms the nucleus-promoting nuclear YAP translocation. Dystrophin encoding L54R and L172H mutations have impaired mechanical function and cannot regulate FA function via the DGC. This weakened connection between the DGC and FA decreases FA tension and cellular traction, possibly reducing force transmitted toward the nucleus by actin and decreasing force-induced YAP nuclear translocation. Alteration of FA tension caused by disease-associated mutants ultimately manifests as defects in migration.
We next found that decreased tensions resulting from dystrophin disease-associated mutations were accompanied by migration defects. Interestingly, the addition of WT-Dys to C2C12 cells did not alter cell migration speeds appreciably despite drastically increasing FA tension. On the other hand, the mutants significantly reduced migration speeds while also reducing tension. The relationship between cell migration and traction force is complex and depends on many factors. Indeed, disease-relevant missense mutations in keratin 5 or 14, which disrupt the cellular keratin network to cause epidermolysis bullosa simplex (38), also decrease cell traction forces and reduce nuclear YAP yet result in an increase in migration speeds. Computational models such as the motor clutch model (54, 55) and extensions such as the cell migration simulator (56, 57) can be useful in interpreting changes in migration and traction force that accompany alterations in adhesion complexes connecting the ECM and cytoskeleton, and this analysis constitutes important future work. Even so, the traction force data alone clearly show a dystrophin disease mutation effect on overall force generation, suggesting that this is relevant to the disease phenotype.
Finally, the data clearly demonstrate cross talk between FAs and dystrophin, most likely via common binding to filamentous actin. We found that YAP and ERK mechanotransduction contributed to cross talk between adhesive complexes to couple mutations in dystrophin with altered FA tension while RhoA signaling did not, which is consistent with no obvious changes in FA size or aspect ratio (SI Appendix, Fig. S12) (38). The YAP transcriptional coactivator integrates signals from many pathways that control its shuttling to the nucleus, including Hippo signaling, cell–cell adhesions, cell polarity, extracellular forces exerted by the cell microenvironment, metabolic pathways, and extracellular growth factors, in contrast to classic signal transduction pathways that are controlled by a dedicated ligand (e.g., Notch, Wnt). The Hippo pathway regulates YAP nuclear translocation by phosphorylating YAP on S127, promoting its cytoplasmic retention. All cell lines exhibited similar ratios of P-YAP S127 over total YAP (SI Appendix, Fig. S20C), although this ratio trends to lower values for WT compared to mutants; thus the Hippo pathway may contribute to YAP regulation. In contrast, our study showed that YAP localization is directly correlated to FA tension; higher tensions lead to more YAP in the nucleus, suggesting that a major contributor to the regulation of YAP activity in our system may be FA tension. Indeed, FA tension has been shown to deform the nucleus (stretch and flatten) and promote YAP nuclear translocation, via the enlarging of nuclear pores to increase YAP import rates (46, 58). Several studies have supported the concept that altered YAP signaling contributes to pathogenesis in dystrophin-deficient muscle (40–42, 48, 59), although the mechanism of dysregulation is uncertain. There are various conflicting reports regarding YAP signaling in dystrophic muscle, possibly due to the complex influences of mechanical stress, muscle regeneration, developmental stage, inflammation, and cytokines on YAP activity in muscle and nonmuscle cells (48). For example, YAP phosphorylation increases during muscle differentiation while YAP expression declines (48). In cardiac muscle, the Hippo pathway was reported to play a significant role in the regulation of YAP through direct involvement of the DGC (42). However, Morikawa et al. (42) reported that treatment of C2C12 myotubes with siRNA to DMD led to decreased sarcolemmal and cytoplasmic YAP and increased YAP nuclear localization. In our control C2C12 myoblasts, however, we could not detect an endogenous DMD transcript (SI Appendix, Fig. S8) or dystrophin protein (Fig. 2A), which supports our conclusion that exogenous dystrophin expression directly impacts YAP signaling and that this function of dystrophin is impaired by two different missense mutations associated with DMD or BMD.
Our findings pave the way for future studies to address whether other disease-associated mutations in dystrophin and other members of the DGC impact tension–sensing via similar long-range allostery mechanisms to contribute to muscular dystrophy pathogenesis. Moreover, dystrophin truncations and mutations have recently been identified in many types of cancer (60, 61), indicating that the DGC can function as a tumor suppressor, so our findings may be relevant to defects in mechanotransduction associated with cancer. Our studies using molecular tension sensors in C2C12 myoblasts provides a proof of concept for investigating mechanotransduction during differentiation of myoblasts, in other 2-dimensional and 3-dimensional cell culture models of muscular dystrophy, and potentially even in animal models of muscular dystrophy. Finally, our studies provide a starting point for further investigations into molecular mechanisms of mechanotransduction and potentially provide therapeutic approaches targeting mechanotransduction as a treatment for muscular dystrophy.
Materials and Methods
Cell Culture.
C2C12 (ATCC; CRL-1772) and transgenic C2C12 cell lines were cultured and grown at 37 °C, 5% CO2 in DMEM (Gibco) supplemented with 10% FBS (ATCC), 1% penicillin/streptomycin mixture (HyClone), and 0.1% fungizone (Gibco). Cells were cultured on either plastic cell dishes or Matek glass-bottom plates previously coated with 12.5 ng/mL fibronectin solution in PBS 1× (Corning).
BRET Imaging.
Cells cultured on fibronectin-coated glass-bottom plates were washed with PBS and the DMEM culture media was replaced with warm FluoroBrite media (Gibco) supplemented with 10% FBS and 1:1,000 Nano-Glo luciferase assay substrate (Furimazine, Promega). First, the donor and acceptor emissions were imaged at 100× using an IX83 inverted microscope (Olympus) equipped with an iXon Ultra 888 EM-CCD camera (Andor), using a filter wheel with Semrock light filters FF01-460/60 (NanoLuc, donor) and FF01-536/40 (mNeonGreen, acceptor). Integration time was 45 s for both acquisitions. Next, cells were imaged for mNeonGreen using an LED lighting source and a standard FITC cube to obtain a regular fluorescent image of the FA.
Traction Force Experiments.
Briefly, 4.6 kPa polyacrylamide gels (PAGs) were generated by mixing 40% acrylamide solution (Fisher), 2% bis-acrylamide solution (Fisher), 1 M 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (Hepes, Sigma) solution, and 200 nm crimson fluorospheres (Invitrogen). To polymerize the prepolymer solution, 1% ammonium persulfate (Bio-Rad) solution and 0.4% (vol/vol)N,N,N′,N′-TEMED (Fisher) were added to the solution and then were casted into silanized Matek glass-bottom plates. Once polymerized, PAGs were activated with 0.5 mg/mL 1-sulfo-SANPAH (Thermo) and coated with 12.5 ng/mL fibronectin solution in PBS overnight. Before plating, dishes were washed with PBS 3 times and exposed to ultraviolet light for 15 min. Approximately 10 × 103 C2C12 cells were plated onto the gel in 1 mL media and incubated at 37 °C for 4 to 5 h. Imaging was performed using a Plan Apo 40 ×/0.95 NA objective. For fluorescence, an mCherry/EGFP filter set was used. Phase contrast and fluorescent images were taken. Afterward, media was removed and 0.05% trypsin with EDTA in Hanks’ balanced salt solution (Gibco) was added to remove cellular adhesions, allowing the gel to go back to a relaxed state. Phase contrast and fluorescent images were taken once more. The traction and displacement fields were analyzed using a previously used MATLAB code (57).
Immunostaining.
C2C12 cells were cultured on fibronectin-coated glass-bottom plates. After 24 h incubation, cells were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) for 13 min at room temperature (RT) and immediately rinsed with RT PBS twice. Fixed cells were permeabilized and blocked with a PBS + 2% BSA (Thermo Fisher Scientific) + 0.05% Triton X-100 (Fisher Scientific) blocking solution for 10 min at RT. Blocked cells were labeled with YAP1 (1:100, Abnova; 2F12) and paxillin (1:100, Abcam; Y113) antibodies in blocking buffer for 1 h at RT, then washed 3 times with blocking buffer. The cells were then incubated for 45 min with a secondary antibody diluted in blocking buffer (1:500, Alexa Fluor 568, Molecular Probes) and washed twice with blocking buffer and once with PBS. Finally, labeled cells were mounted with ProLong Diamond antifade with DAPI (Thermo Fisher Scientific).
Immunostained cells were imaged on an IX83 inverted microscope (Olympus) equipped with an iXon Ultra 888 EM-CCD camera (Andor), using an LED lighting source and standard FITC, TRITC, and DAPI filter cubes. Image overlays were created using ImageJ (version 1.53h).
Image Analysis.
Acquired emission images were analyzed on ImageJ (version 1.53h). Stacked images were aligned using the Linear Stack with Alignment with an SIFT plugin and then analyzed using the BRET-Analyzer plugin (62). mNeonGreen images were used to generate FA masks using a machine learning tool, Ilastik (version 1.3.2). Ratiometric images generated by the BRET-Analyzer were masked, and BRET distributions were obtained and visualized as 16-color LUT. To analyze FA tension gradients from the BRET ratiometric images, line scans were done across individual FAs by manually using a line tool, from the edge of the cell toward the centroid. Obtained intensity profiles were normalized to FA length. Cell and FA morphology analysis were done using the Analyze Particles Plugin.
Immunostained images were masked for the complete cell and for the nucleus; masks were generated using Ilastik. Using ImageJ, YAP cellular and nuclear intensity was measured, and then nuclear intensity was subtracted from cellular intensity to calculate cytoplasmic YAP intensity. From this data, nuclear-to-cytoplasmic-YAP ratios were calculated.
Western Blot Analysis.
Harvested cells were weighted and lysed with a volume of lysis buffer proportional to the cell pellet mass. The lysis buffer used was a radioimmunoprecipitation assay buffer (10 mM Tris⋅Cl [pH 8.0], 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) with added protease inhibitors (100 nM aprotinin, 10 μM E-64, 10 μM leupeptin, 1 mM pepstatin A, 1 mM PMSF). Lysates sampled with phospho-specific antibodies were also lysed with phosphatase inhibitors (phosSTOP, Roche). Lysates were then centrifuged at 20,000 g, and equal volumes of collected supernatant were separated on SDS polyacrylamide gel at 100 V for 30 min and 150 V for 1 h and transferred to a PVDF membrane at 0.8A for 1 h. Membranes were blocked for 1 h in either 5% milk in PBST or 5% BSA in TBST, depending on the primary antibody used. Blocked membranes were incubated with primary antibodies overnight at 4 °C. The primary antibodies diluted with milk PBST were green fluorescent protein (GFP) (1:1,000, Cell Signaling; 2956), pan-actin C4 (1:5,000 Seven Hills Bioreagents; LMAB-C4), α-tubulin (1:1,000, Sigma-Aldrich; DM1A), utrophin (1:50, DHSB; Mancho3), and YAP (1:500, Abnova; 2F12). The primary antibodies diluted with BSA TBST were vinculin (1:1,000, Sigma-Aldrich; V9131), paxillin (1:100, Abcam; Y113), α-dystroglycan (1:500, Millipore; 05–298), β-dystroglycan (1:1,000, Thermo Fischer Scientific), and P-YAP S127 (1:2,000, Abcam; EP1675Y). Membranes were washed with PBST or TBST and then incubated with a secondary antibody diluted in the appropriate blocking buffer. The secondary antibodies used were anti-mouse or anti-rabbit IgG Dylight 800 or IgG Dylight 700 (1:10,000, Cell Signaling). The secondary antibody signal was visualized on LI-COR’s Odyssey CLx Imaging System, and band density was calculated with Image Studio Software.
RT-qPCR.
Cells were collected, and their RNA was extracted using the Aurum Total RNA Mini Kit (Bio-Rad; 732–6820). Total RNA concentration and purity were determined using a nanodrop spectrophotometer (Thermo Scientific). A standardized amount of RNA was transcribed into complementary DNA (cDNA) using the iScript Advanced cDNA Synthesis Kit (Bio-Rad; 170–8843), and cDNA was then amplified using the primers GFP forward (TCCGCCATGCCCGAAGGCTA) and reverse (CCGTTCACCAGGGTGTCGCC) to detect transgenic dystrophin (GFP-dystrophin). Measurements were done relative to Hprt using the primers forward (CCCTGGTTAAGCAGTACAGCCCC) and reverse (GGCCTGTATCCAACACTTCGAGAGG). cDNA was amplified using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad; 172–5270) on the C1000 Touch Thermal Cycler (Bio-Rad) and was then analyzed with the CFX Manager software (Bio-Rad).
Wound Healing Assay.
Confluent monolayers of C2C12 cells plated on fibronectin were gently washed with PBS and then scratched with a 200 μL micropipette tip to create a wound (cell-free area) all along the center of the well. Then, cells were washed once with PBS and warm FluoroBrite media supplemented with 10% FBS was added. Bright-field images of the scratched cells were taken every hour over a period of 9 h, using an IX83 inverted microscope (Olympus) equipped with an iXon Ultra 888 EM-CCD camera (Andor) in a top-stage incubator (STX, Tokai Hit) at 37 °C and 5% CO2.
Then, the % wound closure over time was plotted and the linear portion of the data was fitted to a y = mx linear fit, where x is the slope of the fitted line.
The inferred edge “speed” of wound closure was calculated as:
As the migration of the sheet of cells can effectively be considered one directional, only toward the center of the wound, the vertical velocity was assumed to be zero. Therefore, the area of the wound only changes by its width and not length (L), which is twice as slow as the change in wound size because both cell sheets contribute to wound closure.
The wound closure speed (μm2/time) was calculated by multiplying the fitted slope from the % wound closure over time with the initial wound size, as:
where I is the wound size, k is the slope obtained from the % wound closure fit, and t is time.
Live Cell Imaging.
We cultured 5 × 103 C2C12 cells on a Nunc glass base dish (Thermo Scientific) coated with fibronectin and cultured overnight. Cells were then washed with PBS, and media was replaced with warm FluoroBrite supplemented with FBS that was incubated overnight at 37 °C, 5% CO2. Cells were imaged on a Delta Vision personal DV (GE Technologies), using a 10×/NA0.25 objective, in a closed environmental chamber at 37 °C, with phase–contrast illumination, for 4 h at 10 min intervals. Migration was tracked using the ImageJ Manual Tracking plugin, and the xy track data were analyzed using the DiPer excel plugin (63). Cells that contacted other cells, came in and out of frame, or visibly divided during imaging were excluded for analysis.
Proliferation Assay.
We plated 8 × 102 C2C12 cells on 96 wells in triplicate for each measurement day (4 d) and cultured overnight with DMEM. The next day, CellTiter-Glo substrate, media, and cell plate were left to equilibrate at RT. Cells were lysed with a 1:1 solution of substrate and DMEM for 10 min. The cell and substrate mixture were then transferred to an opaque white 96-well plate, and luminescence was measured using the LMax II 384 Luminometer (Molecular Devices). A blank of DMEM was included and was subtracted from the values acquired for each measurement day.
Statistical Analysis.
All statistical calculations were performed using the GraphPad Prism 8 software. All data are presented as mean ± SEM. Plots showing layered data were created as superplots (64). An unpaired two-tailed t test was performed to compare the means of variables between VinTL and VinTS cells (same cell line), performed at α = 0.05. An unpaired one-way ANOVA was performed to compare the means of three or more populations, with α = 0.05 to determine significance. Significance was confirmed with a Tukey’s posthoc test, performed at α = 0.05. An unpaired two-way ANOVA was performed to determine the significant differences between populations to a dependent variable (e.g., time), at α = 0.05.
Supplementary Material
Acknowledgments
We thank Karen Evans for manuscript revision, Polina Alexandrovich for assistance with data compilation, and Yahor Savich for helpful discussion. We acknowledge funding from the NIH (R01AR042423, R01AR049899- J.M.E.; R35GM119483-W.R.G.; U54 CA210190, P01 CA254849, U54 CA268069-D.J.O.) and the Muscular Dystrophy Association. W.R.G. is a Pew Biomedical Scholar. M.P.R. acknowledges funding from the University of Minnesota Frieda M. Kunze and Doctoral Dissertation Fellowships; M.J.M.A. acknowledges the Medical Scientist Training Program (T32GM008244); M.D.K. acknowledges a Cancer Center Training grant (T32CA009138); and L.J.S. acknowledges the National Institute on Aging Training Program for Functional Proteomics of Aging (T32AG029796).
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2205536119/-/DCSupplemental.
Data Availability
All study data are included in the article and/or SI Appendix.
References
- 1.Zubrzycka-Gaarn E. E., et al. , The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 333, 466–469 (1988). [DOI] [PubMed] [Google Scholar]
- 2.Ervasti J. M., Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 1772, 108–117 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Hoffman E. P., Brown R. H. Jr., Kunkel L. M., Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987). [DOI] [PubMed] [Google Scholar]
- 4.Rybakova I. N., Patel J. R., Ervasti J. M., The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 150, 1209–1214 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ervasti J. M., Campbell K. P., Membrane organization of the dystrophin-glycoprotein complex. Cell 66, 1121–1131 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Ervasti J. M., Ohlendieck K., Kahl S. D., Gaver M. G., Campbell K. P., Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315–319 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Ohlendieck K., Ervasti J. M., Snook J. B., Campbell K. P., Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J. Cell Biol. 112, 135–148 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams M. W., Bloch R. J., Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. J. Cell Biol. 144, 1259–1270 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ervasti J. M., Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 278, 13591–13594 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Pardo J. V., Siliciano J. D., Craig S. W., A vinculin-containing cortical lattice in skeletal muscle: Transverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma. Proc. Natl. Acad. Sci. U.S.A. 80, 1008–1012 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Straub V., Bittner R. E., Léger J. J., Voit T., Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol. 119, 1183–1191 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beggs A. H., et al. , Exploring the molecular basis for variability among patients with Becker muscular dystrophy: Dystrophin gene and protein studies. Am. J. Hum. Genet. 49, 54–67 (1991). [PMC free article] [PubMed] [Google Scholar]
- 13.Hamed S., Sutherland-Smith A., Gorospe J., Kendrick-Jones J., Hoffman E., DNA sequence analysis for structure/function and mutation studies in Becker muscular dystrophy. Clin. Genet. 68, 69–79 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Menke A., Jockusch H., Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 349, 69–71 (1991). [DOI] [PubMed] [Google Scholar]
- 15.Mokri B., Engel A. G., Duchenne dystrophy: Electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 25, 1111–1120 (1975). [DOI] [PubMed] [Google Scholar]
- 16.Tidball J. G., Albrecht D. E., Lokensgard B. E., Spencer M. J., Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci. 108, 2197–2204 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Goldstein J. A., McNally E. M., Mechanisms of muscle weakness in muscular dystrophy. J. Gen. Physiol. 136, 29–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burridge K., Chrzanowska-Wodnicka M., Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12, 463–518 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Wozniak M. A., Modzelewska K., Kwong L., Keely P. J., Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 1692, 103–119 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Chen C. S., Mechanotransduction—A field pulling together? J. Cell Sci. 121, 3285–3292 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Jaalouk D. E., Lammerding J., Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 10, 63–73 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.García-Pelagio K. P., Bloch R. J., Ortega A., González-Serratos H., Biomechanics of the sarcolemma and costameres in single skeletal muscle fibers from normal and dystrophin-null mice. J. Muscle Res. Cell Motil. 31, 323–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A., Khandelwal N., Malya R., Reid M. B., Boriek A. M., Loss of dystrophin causes aberrant mechanotransduction in skeletal muscle fibers. FASEB J. 18, 102–113 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Lopez M. A., et al. , Mechanics of dystrophin deficient skeletal muscles in very young mice and effects of age. Am. J. Physiol. Cell Physiol. 321, C230–C246 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham Z. A., Gallagher P. M., Cardozo C. P., Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil. 36, 305–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boppart M. D., Mahmassani Z. S., Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 317, C629–C641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talsness D. M., Belanto J. J., Ervasti J. M., Disease-proportional proteasomal degradation of missense dystrophins. Proc. Natl. Acad. Sci. U.S.A. 112, 12414–12419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prior T. W., et al. , A missense mutation in the dystrophin gene in a Duchenne muscular dystrophy patient. Nat. Genet. 4, 357–360 (1993). [DOI] [PubMed] [Google Scholar]
- 29.Aird E. J., Tompkins K. J., Ramirez M. P., Gordon W. R., Enhanced molecular tension sensor based on bioluminescence resonance energy transfer (BRET). ACS Sens. 5, 34–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grashoff C., et al. , Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466, 263–266 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaCroix A. S., Lynch A. D., Berginski M. E., Hoffman B. D., Tunable molecular tension sensors reveal extension-based control of vinculin loading. eLife 7, e33927 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F., et al. , Vinculin force sensor detects tumor-osteocyte interactions. Sci. Rep. 9, 5615 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothenberg K. E., Scott D. W., Christoforou N., Hoffman B. D., Vinculin force-sensitive dynamics at focal adhesions enable effective directed cell migration. Biophys. J. 114, 1680–1694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarangi B. R., et al. , Coordination between intra- and extracellular forces regulates focal adhesion dynamics. Nano Lett. 17, 399–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trepat X., et al. , Physical forces during collective cell migration. Nat. Phys. 5, 426–430 (2009). [Google Scholar]
- 36.De Pascalis C., Etienne-Manneville S., Single and collective cell migration: The mechanics of adhesions. Mol. Biol. Cell 28, 1833–1846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martino F., Perestrelo A. R., Vinarský V., Pagliari S., Forte G., Cellular mechanotransduction: from tension to function. Front. Physiol. 9, 824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujiwara S., Deguchi S., Magin T. M., Disease-associated keratin mutations reduce traction forces and compromise adhesion and collective migration. J. Cell Sci. 133, jcs243956 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Betts C. A., et al. , Dystrophin involvement in peripheral circadian SRF signalling. Life Sci. Alliance 4, e202101014 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer S. R., et al. , Differential YAP nuclear signaling in healthy and dystrophic skeletal muscle. Am. J. Physiol. Cell Physiol. 317, C48–C57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vita G. L., et al. , Hippo signaling pathway is altered in Duchenne muscular dystrophy. PLoS One 13, e0205514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morikawa Y., Heallen T., Leach J., Xiao Y., Martin J. F., Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 547, 227–231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oak S. A., Zhou Y. W., Jarrett H. W., Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J. Biol. Chem. 278, 39287–39295 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Nelson D. M., Ervasti J. M., “Structural proteins: Dystrophin: A multifaceted protein critical for muscle health” in Encyclopedia of Biological Chemistry, J. Jez, Ed. (Elsevier, 2021, ed. 3), vol. 3, pp. 625–638. [Google Scholar]
- 45.Valera I. C., et al. , Essential roles of the dystrophin-glycoprotein complex in different cardiac pathologies. Adv. Med. Sci. 66, 52–71 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Dasgupta I., McCollum D., Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 294, 17693–17706 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dupont S., et al. , Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Fischer M., Rikeit P., Knaus P., Coirault C., YAP-mediated mechanotransduction in skeletal muscle. Front. Physiol. 7, 41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lapidos K. A., Kakkar R., McNally E. M., The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 94, 1023–1031 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Henderson D. M., Lee A., Ervasti J. M., Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl. Acad. Sci. U.S.A. 107, 9632–9637 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhasin N., et al. , Molecular extensibility of mini-dystrophins and a dystrophin rod construct. J. Mol. Biol. 352, 795–806 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Le S., et al. , Dystrophin as a molecular shock absorber. ACS Nano 12, 12140–12148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamashita K., et al. , The 8th and 9th tandem spectrin-like repeats of utrophin cooperatively form a functional unit to interact with polarity-regulating kinase PAR-1b. Biochem. Biophys. Res. Commun. 391, 812–817 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Chan C. E., Odde D. J., Traction dynamics of filopodia on compliant substrates. Science 322, 1687–1691 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Bangasser B. L., Rosenfeld S. S., Odde D. J., Determinants of maximal force transmission in a motor-clutch model of cell traction in a compliant microenvironment. Biophys. J. 105, 581–592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klank R. L., et al. , Biphasic dependence of glioma survival and cell migration on CD44 expression level. Cell Rep. 18, 23–31 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bangasser B. L., et al. , Shifting the optimal stiffness for cell migration. Nat. Commun. 8, 15313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elosegui-Artola A., et al. , Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171, 1397–1410.e14 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Hulmi J. J., et al. , Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am. J. Physiol. Endocrinol. Metab. 304, E41–E50 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Wang Y., et al. , Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 46, 601–606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Körner H., et al. , Digital karyotyping reveals frequent inactivation of the dystrophin/DMD gene in malignant melanoma. Cell Cycle 6, 189–198 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Chastagnier Y., Moutin E., Hemonnot A.-L., Perroy J., Image processing for bioluminescence resonance energy transfer measurement-BRET-analyzer. Front. Comput. Neurosci. 11, 118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gorelik R., Gautreau A., Quantitative and unbiased analysis of directional persistence in cell migration. Nat. Protoc. 9, 1931–1943 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Lord S. J., Velle K. B., Mullins R. D., Fritz-Laylin L. K., SuperPlots: Communicating reproducibility and variability in cell biology. J. Cell Biol. 219, e202001064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or SI Appendix.