

Abstract
Cardiac troponin T (encoded by TNNT2) is involved in the contraction of cardiomyocytes during beating. The alternative splicing of TNNT2 results in four transcript variants with differential Ca2+ sensitivity. The splicing of TNNT2 involves phosphorylation of the splicing factor SRSF6 by DYRK1A. Altered TNNT2 splicing patterns have been identified in failing human hearts. There is a paucity of studies describing DYRK1A-SRSF6-TNNT2 interplays in human cardiomyocytes. Also, it is not known whether the sensitivity of cardiomyocytes to cardiotoxic anthracyclines is modified in the context of variable DYRK1A-TNNT2 expression. In this study, we investigated the impact of DYRK1A on the endogenous expression of TNNT2 splicing variants in iPSC-derived cardiomyocytes. We also examined whether DYRK1A expression modifies the sensitivity of cardiomyocytes to the cardiotoxic drug daunorubicin (DAU). DYRK1A over-expression increased the abundance of TNNT2 fetal variants by ~ 58% whereas the abundance of the adult cTnT3 variant decreased by ~ 27%. High DYRK1A expression increased the phosphorylation of SRSF6 by ~ 25–65%. DAU cytotoxicity was similar between cardiomyocytes with variable levels of DYRK1A expression. DYRK1A over-expression ameliorated the impact of DAU on beating frequency. This study lays the foundation to further investigate the contribution of variable DYRK1A-TNNT2 expression to Ca2+ handling and beating in human cardiomyocytes.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12012-022-09746-6.
Keywords: Human cardiomyocytes, Anthracyclines, Alternative splicing, Troponin, Heart, Cardiotoxicity
Introduction
Cardiomyocytes derived from human-induced pluripotent stem cells (iPSC cardiomyocytes) are an informative platform for the study of various cellular processes. iPSC cardiomyocytes express structural and functional genes that are key to myocardial function. Under specific culture conditions, iPSC cardiomyocytes form a spontaneously beating syncytium [1, 2]. Cardiac troponin T (cTnT), a protein encoded by the TNNT2 gene, is involved in the contraction of cardiomyocytes during beating. The alternative splicing of TNNT2 involving exons 4 and 5 results in four cTnT transcript variants namely cTnT1 (exon 5+, exon 4+), cTnT2 (exon 5+, exon 4−), cTnT3 (exon 5−, exon 4−) and cTnT4 (exon 5−, exon 4+). The splicing patterns of the TNNT2 gene change during embryonic and postnatal heart development. In human heart, cTnT1 is expressed at high levels in fetal heart, and cTnT2 is expressed at low levels throughout development. cTnT3 is the predominant variant in adult heart, and cTnT4 is expressed in fetal and in failing adult heart [3]. cTnT3 is the only variant expressed in mouse adult heart, and coexistence of two or more cTnT variants in transgenic mice results in decreased myocardial contractility [4]. cTnT isoforms confer differential Ca2+ sensitivity to cardiomyocytes. For example, TNNT2 splicing variants that retain exon 5 (i.e., cTnT1 and cTnT2) are mainly expressed in fetal and neonatal heart and result in myofibrils that are more sensitive to Ca2+ as determined by measurements of force development [5, 6]. In the adult heart, the expression of the fetal variants cTnT1 and cTnT2 is low or undetectable, and altered patterns of fetal and adult cTnT transcript variants have been identified in failing human hearts and in animals with dilated cardiomyopathy [7, 8].
DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A) is a protein kinase with a wide spectrum of targets [9, 10]. The DYRK1A gene is located in chromosome 21. Altered DYRK1A gene expression due to trisomy 21 (Down syndrome) has been linked to the pathogenesis of several conditions in Down syndrome including alterations in neuronal development, cognitive defects, heart defects, and hematological malignancies [11]. In cardiomyocytes, DYRK1A participates in cell proliferation and differentiation during fetal and early neonatal development [12]. DYRK1A has been identified as a negative regulator of cardiac hypertrophy through a mechanism involving the calcineurin/nuclear factor of activated T cells (NFAT) signaling pathway [13, 14]. Of note, increased DYRK1A expression in hearts from Ts65Dn mice, a mouse model of Down syndrome, impacts the splicing of TNNT2 and the relative proportions of cTnT transcript variants. In line, DYRK1A overexpression in human fetal kidney-derived HEK-293 cells increased the splicing of an artificial TNNT2 “mini-gene” construct [15]. Phosphorylation of the SR splicing factor SRSF6 by DYRK1A modulates exon inclusion in TNNT2 transcript variants [15]. Comparative analysis of the expression of the DYRK1A-SRSF6-TNNT2 pathway in myocardial tissue from individuals with and without Down syndrome revealed increased levels of phosphorylated SRSF6 and ~ 50% higher expression of fetal TNNT2 transcript variants in trisomic myocardium [16]. Whereas the role of DYRK1A during the alternative splicing of TNNT2 is becoming evident, there is still a paucity of data derived from observations in beating human cardiomyocytes.
Patients treated with anthracycline-based chemotherapeutic regimens may develop serious adverse side-effects, including cardiotoxicity. The cardiotoxicity exerted by anthracyclines spans a spectrum of signs and symptoms ranging from perturbations in cardiac rhythm and function to severe cardiomyopathy and congestive heart failure [17–20]. Interindividual variability between anthracycline exposure and cardiotoxic outcomes suggest that genetic factors could contribute to the risk for anthracycline-related cardiotoxicity [21]. The sensitivity of human cardiomyocytes to cardiotoxic anthracyclines in the context of variable TNNT2 and DYRK1A expression remains to be explored.
In this study, we investigated the impact of DYRK1A expression on the relative abundance of endogenous TNNT2 splicing variants in iPSC cardiomyocytes. Interplays between DYRK1A expression and SRSF6 phosphorylation were examined by the analysis of SRSF6 phosphorylation in the context of increased DYRK1A expression. We also determined whether increased DYRK1A expression modifies beating frequency and sensitivity to the cardiotoxic drug daunorubicin. This study contributes new information on the expression of endogenous TNNT2 transcript variants and daunorubicin-induced cardiotoxicity in the context of altered DYRK1A expression in a model of beating human cardiomyocytes.
Materials and Methods
Human iPSC Cardiomyocytes
The Institutional Review Board of the State University of New York at Buffalo (UB-IRB) approved this research. UB-IRB determined that this research is not research with human subjects. Human induced pluripotent stem cell-derived cardiomyocytes (iPSC cardiomyocytes) clone 11713 from Fujifilm Cellular Dynamics were used in this study. iPSC cardiomyocytes were cultured according to the provider’s specifications using the recommended plating and maintenance media (iCell Cardiomyocytes Kit, R1117, Fujifilm Cellular Dynamics). iPSC cardiomyocytes were seeded at a density of ~ 63,000 cardiomyocytes/cm2 in fibronectin-coated 96-well plates to promote the formation of a beating syncytium. iPSC cardiomyocytes were cultured in standard incubator conditions at 37 °C, 5% CO2, and 95% relative humidity for at least 6 days post initial plating.
Transfections and Drug Treatments
iPSC cardiomyocytes at day 6 post initial plating were transfected with 100 ng plasmid DNA using ViaFect transfection reagent (E4981, Promega) and the following human DYRK1A and SRSF6 expression constructs: DYRK1A human clone (NM_001396, SC314641, Origene), DYRK1A human tagged ORF Clone (NM_001396, RG212584, Origene), and SRSF6 cDNA ORF clone N-HA tag (HG19052-NY, Sinobiological). Control cultures were transfected with ~ 100 ng of empty PCMV6XL5 vector (Origene).
Drug treatments were initiated 24 h post-transfection with expression constructs. Daunorubicin (DAU, 14159, Cayman Chemical) and epigallocatechin gallate (EGCG, 709035, Cayman Chemical) were added to culture media for a total incubation time of 14 h. DMSO vehicle was added to controls. After treatments with DAU, iPSC cardiomyocytes were washed with PBS, and incubated in fresh culture medium, or medium supplemented with EGCG for 24 h. Cell viability was determined with the CellTiter-Glo luminescent viability kit (G7570, Promega).
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was isolated with Trizol (Thermo Fisher). The expression of TNNT2 transcript variants (cTnT1/cTnT2, cTnT3, and cTnT4) and DYRK1A mRNA was analysed with specific primers (Table S1) [16]. Total RNA (5 ng) was reverse transcribed and amplified with the iTaq Universal SYBR Green One-Step kit (Bio-Rad). DYRK1A, TNNT2 transcript variants, and the reference gene B2M were amplified in parallel in a CFX96 Touch Real-Time PCR detection system (Bio-Rad) with the following cycling parameters: 50 °C for 10 min (reverse transcription), 95 °C for 1 min, followed by 44 cycles of 95 °C for 10 s and 60.5 °C for 20 s. Calibration curves were prepared to analyse linearity and PCR efficiency. qRT-PCR data were analysed using the ΔΔCt method with the CFX manager software (Bio-Rad). The relative abundance of DYRK1A and TNNT2 transcript variants was determined with the ΔCt method. The proportion of TNNT2 variants was calculated as follows:
where v represents variants cTnT1/cTnT2, cTnT3, or cTnT4.
Live Cell Imaging and Image Analysis
At day 14 post initial plating, iPSC cardiomyocytes cultured in plates suitable for fluorescence microscopy were incubated with medium supplemented with 1 µM Fluo-4 AM (F14217, Molecular probes) for 30 min at 37 °C. After incubation, Fluo-4 was removed, replaced with fresh media, and cells were maintained in the incubator for up to 30 min before imaging. Cell imaging was performed at 37 °C, 5% CO2 in a humidified incubator using a Dragonfly spinning disk confocal microscope (Andor Technology Ltd.) attached to a DMi8 base (Leica Microsystems). Image stacks (16 bits, 0.15 μm per pixel) were obtained by imaging 240 frames in a total interval of 30 s with a Zyla 4.2 PLUS sCMOS camera using a PlanApo 40× 1.10 NA water immersion objective. Images from ~ 10 fields/well were randomly obtained. Independent incubations with Fluo-4 AM were performed in at least three wells for each condition.
Image analysis was performed with the Fiji (ImageJ) software [22]. Comparisons were performed by analyzing similar numbers of cellular regions of interest (ROIs) per condition with identical image processing parameters. ROIs comprised a circular section of 3811 µm2 created with the oval selection tool. For cardiomyocytes forming a synchronized contracting syncytium, the number of fluorescence peaks was similar for any ROIs comprised within a given image stack (Fig. S1). Based on this observation, one ROI per field of view was considered for analysis, unless unsynchronized beating was noticed within a given image stack. To quantify the number of beats per ROI, a list of fluorescence values vs time was obtained using the Plot Z-axis profile tool in Fiji. Values were saved as text files and exported to Excel 2016 (Microsoft Office) for further analysis. A macro was designed to find and count the minimum and maximum peaks of fluorescence for each ROI during a total time of 30 s. The number of beatings was defined as the total number of maximum peaks detected during 30 s.
Western Blot and Dot-Blot
Protein samples were denatured with NuPAGE LDS sample buffer containing NuPAGE sample reducing agent and boiled at 70 °C for 10 min prior to use. Proteins were separated by gel electrophoresis using NuPAGE Novex 4–12% Bis-Tris precast gels and transferred onto PVDF membranes using the iBlot gel transfer device (Thermo Fisher Scientific). For dot-blot assays, protein samples were spotted on nitrocellulose membranes (88018, Thermo Fisher Scientific) and dried at room temperature. Membranes were blocked with 5% non-fat milk in 0.2% Tween 20 phosphate buffered saline (PBS) for 30 min at room temperature and probed overnight at 4 °C with the following primary antibodies: mouse monoclonal anti-phosphoepitope SR proteins (1:500, MABE50, Milipore), rabbit anti-HA Tag (1:250, SG77, Invitrogen), mouse monoclonal anti-DYRK1A (1:200, sc-100376, Santa Cruz). Next, membranes were washed and incubated with StarBright Blue 700 goat anti-mouse IgG secondary antibody (1:2500, 12004159, Bio-Rad), StarBright Blue 520 goat anti-rabbit IgG secondary antibody (1:2500, 12005869, Bio-Rad) and hFAB rhodamine anti-tubulin antibody (1:2500, 12004165, Bio-Rad) for 1 h at room temperature. Immunoreactive bands were visualized in a ChemiDoc MP gel imaging system (Bio-Rad). Densitometric analysis was performed using Fiji (ImageJ) software [22].
Immunoprecipitation
Immunoprecipitation (IP) assays were performed with an anti-HA immunoprecipitation kit (IP0010, Sigma-Aldrich). Briefly, transfected iPSC cardiomyocytes cultured in 24-well plates were lysed in CelLytic M reagent supplemented with protease inhibitor cocktail (Thermo Fisher Scientific) and Halt phosphatase inhibitor (Thermo Fisher Scientific). Cell lysates were incubated overnight at 4 °C with anti-HA-affinity gel in a mini-spin column. After wash, retained proteins were eluted from columns by incubation at 95 °C for 10 min with NuPAGE LDS sample buffer followed by centrifugation. The resulting IP samples were analyzed by Western blot and dot-blot as described above.
Phosphatase Treatment
Cell lysates from AC16 human cardiomyocytes (SCC109, Sigma-Aldrich) were incubated with 20 units of calf intestine alkaline phosphatase (CIAP, 18009, Invitrogen) in a 20 μl reaction volume containing 50 mM Tris/HCl (pH 9.3) for 1 h at 37 °C. AC16 cells were previously co-transfected with DYRK1A and SRSF6-HA tag expression constructs, as described above. Samples before and after CIAP treatment were analyzed by immunoblotting.
Data Processing and Statistical Analysis
Data processing was performed with Excel 2016 (Microsoft Office). Statistical analyses were performed with GraphPad Prism version 9. The D’Agostino & Pearson omnibus normality test was used to determine the normality of data sets. Comparisons between the means of two groups were performed with the Student’s t test or Mann–Whitney’s U test for sets with normal and non-normal distributions, respectively. All data were expressed as mean ± SD.
Results
Expression of TNNT2 Transcript Variants in the Context of DYRK1A Over-Expression
It is known that increased DYRK1A expression impacts the splicing of TNNT2 and the proportion of cTnT transcript variants in hearts from a mouse model of Down syndrome and in human non-cardiac cells [15]. Here, we examined whether DYRK1A over-expression modifies the pattern of TNNT2 splicing in human iPSC cardiomyocytes. The endogenous expression of the fetal variants cTnT1 and cTnT2, and the adult cTnT3 and cTnT4 transcript variants was examined with specific PCR primers (Fig. 1A, Table S1). DYRK1A over-expression (> 60-fold increase in DYRK1A mRNA) caused a ~ 58% increase in the proportion of cTnT1 and cTnT2 fetal variants (cTnT1/2DYRK1A: 0.30 ± 0.15, cTnT1/2EV: 0.19 ± 0.10) and a ~ 27% decrease in the proportion of the cTnT3 variant (cTnT3DYRK1A: 0.34 ± 0.11, cTnT3EV: 0.43 ± 0.08) in comparison to vehicle-transfected controls. There were no significant changes in the proportion of cTnT4 splicing variants (cTnT4DYRK1A: 0.35 ± 0.07, cTnT4EV: 0.36 ± 0.04) (Fig. 1B–D).
Fig. 1.

Impact of DYRK1A overexpression on the relative abundance of endogenous TNNT2 transcript variants in iPSC cardiomyocytes. A PCR amplification strategy for the analysis of fetal (cTnT1 and cTnT2) and adult (cTnT3 and cTnT4) TNNT2 transcript variants. F: forward primer, R: reverse primer. B Relative abundance of TNNT2 transcript variants in basal conditions (control) or in cells overexpressing DYRK1A (DYRK1A). C Relative DYRK1A mRNA fold expression in cells transfected with an empty vector (EV, control) or a DYRK1A expression construct (DYRK1A). D Relative abundance of fetal and adult TNNT2 transcript variants in basal conditions and in cardiomyocytes overexpressing DYRK1A. Each point represents the mean from independent transfections, with two determinations performed in triplicates. Horizontal bars show the mean ± SD. ***P < 0.001, *P < 0.05, ns not significant, Student’s t test
The expression of DYRK1A protein was assessed by fluorescence microscopy and immunoblotting in iPSC cardiomyocytes transfected with a DYRK1A-GFP expression construct (Fig. 2). The level of DYRK1A protein expression was very low to null in basal conditions (EV), and there was a relatively weak protein signal in DYRK1A-transfected cells (Fig. 2A). Moderate DYRK1A-GPF protein expression was observed by fluorescence microscopy in DYRK1A-transfected cells. DYRK1A was present in the nucleus and cytoplasm of cardiomyocytes. The distribution of DYRK1A showed a dotted pattern resembling nuclear speckles (Fig. 2B).
Fig. 2.

DYRK1A protein expression in iPSC cardiomyocytes. A Detection of DYRK1A with an anti-DYRK1A antibody (left panel). β-Tubulin was assayed as loading control (right panel). B Representative fluorescence microscopy of a complete field of view (upper panel) and in one individual cell (lower panels) showing the expression and subcellular distribution of DYRK1A-GFP (green). Cells were analyzed 72 h post-transfection with an empty vector (EV) or a DYRK1A-GFP expression construct
SRSF6 Phosphorylation and Expression of TNNT2 Transcript Variants
Phosphorylation of SRSF6 modulates exon inclusion in TNNT2 transcript variants [15]. To determine the impact of DYRK1A overexpression on SRSF6 phosphorylation, we expressed a hemagglutinin-tagged version of SRSF6 (SRSF6-HA). Expression of SRSF6-HA facilitates the detection of phosphorylation by combining HA-protein precipitation plus the antibody for detection of phosphoepitopes in SR proteins. First, we examined the specificity of the antibody that recognizes phosphoepitopes in SR proteins. Immunoblotting analysis of total protein extracts from AC16 cardiomyocytes expressing SRSF6-HA showed a decrease in band intensity in samples treated with calf intestine alkaline phosphatase (CAIP). This observation is consistent with recognition of SR phosphoepitopes by the anti-SR antibody (Fig. 3A). In iPSC cardiomyocytes overexpressing SRSF6, there was a strong signal at ~ 55 kDa consistent with detection of SRSF6-HA in cellular lysates and HA-immunoprecipitates (Fig. 3B, C). In the context of DYRK1A overexpression (DYRK1A mRNA expression > 39-fold), there was a trend towards ~ 25% increase in the amount of phosphorylated SRSF6 as determined by Western blotting and a ~ 65% increase as determined by dot-blot (Figs. 3C, D, S2).
Fig. 3.

SRSF6 phosphorylation in iPSC cardiomyocytes overexpressing DYRK1A. A Evaluation of the anti-phosphoepitope SR antibody in total protein extracts from AC16 cardiomyocytes treated with calf intestine alkaline phosphatase (CIAP). B Detection of phospho-SRSF6 and SRSF6 in iPSC cardiomyocytes transfected with empty vector (EV), SRSF6-HA expression construct (SRSF6) or co-transfected with DYRK1A and SRSF6-HA (SRSF6 + DYRK1A). β-Tubulin was assayed as loading control. Lower panel: densitometric analysis. Upper right panel: relative DYRK1A mRNA fold expression. (A.U.: arbitrary units). C Detection of phosphorylated SRSF6 in immunoprecipitated samples from iPSC cardiomyocytes. Right panel: densitometric analysis. Horizontal bars show the mean ± SD from determinations performed in quintuplicate. D Dot-blot analysis of phosphorylated SRSF6 in immunoprecipitated samples. Right panel: densitometric analysis. Horizontal bars show the mean ± SD from determinations performed in quintuplicate. ***P < 0.001, *P < 0.05, Student’s t test
Next, the expression of TNNT2 fetal and adult splicing variants was assessed in iPSC cardiomyocytes co-transfected with SRSF6 and DYRK1A expression constructs (Fig. 4A–C). Concomitant DYRK1A-SRSF6 overexpression caused a ~ 93% increase in the proportion of cTnT1 and cTnT2 (cTnT1/2 SRSF6-DYRK1A: 0.31 ± 0.08, cTnT1/2 SRSF6: 0.16 ± 0.04) and a ~ 56% decrease in cTnT3 in comparison to cells transfected with SRSF6 only (cTnT3 SRSF6-DYRK1A: 0.32 ± 0.06, cTnT3 SRSF6: 0.41 ± 0.07). Similar comparisons revealed no significant changes in the proportion of cTnT4 splicing variants (cTnT4SRSF6-DYRK1A: 0.40 ± 0.09, cTnT4SRSF6: 0.44 ± 0.04) (Fig. 4C). Finally, the impact of SRSF6 overexpression in the proportion of cTnT1 and cTnT2 variants was assessed in iPSC cardiomyocytes either expressing basal or increased levels of DYRK1A mRNA. Comparisons revealed no significant changes in the proportion of TNNT2 fetal splicing variants in SRSF6 transfected iPSC cardiomyocytes and non-SRSF6 transfected cells (Fig. 4D).
Fig. 4.

Impact of DYRK1A overexpression on the relative abundance of TNNT2 transcript variants in iPSC cardiomyocytes transfected with SRSF6. A Relative abundance of TNNT2 transcript variants in cells transfected with an SRSF6-HA (SRSF6) or co-transfected with DYRK1A-GFP and SRSF6 expression constructs (SRSF6-DYRK1A). B Relative DYRK1A mRNA fold expression in transfected cells. C Relative abundance of cTnT1 and cTnT2 (left), cTnT3 (center), and cTnT4 (right) transcript variants. Horizontal bars show the mean ± SD from three independent transfections per condition, with 2–3 determinations performed in triplicate. ***P < 0.001, *P < 0.05, ns not significant, Student’s t test. D Impact of SRSF6 overexpression on the relative abundance of cTnT1 and cTnT2 in cells with basal levels of DYRK1A (left), or overexpressing DYRK1A (right). **P < 0.01, *P < 0.05, ns not significant. One-way ANOVA (Tukey’s test)
DYRK1A Over-Expression and Sensitivity to Daunorubicin
We reasoned that variable DYRK1A expression may impact the sensitivity of iPSC cardiomyocytes to DAU. First, we examined whether treatment with epigallocatechin gallate (EGCG), a recognized DYRK1A inhibitor, modifies the cytotoxicity of the anthracycline daunorubicin (DAU) in iPSC cardiomyocytes. Cell viability was not significantly affected when EGCG was present during incubations with DAU (5 µM, 14 h) in cells with basal levels of DYRK1A expression (Fig. 5A). Increased DYRK1A expression did not modify the cytotoxicity of DAU (EV5 µM DAU: 30.41 ± 19.73%, DYRK1A5 µM DAU: 28.28 ± 17.04%) (Fig. 5B). In the context of DYRK1A over-expression, incubations with EGCG did not modify the cytotoxicity of DAU (EV5 µM DAU: 30.41 ± 19.73%, EV5 µM DAU+ 25 µM EGCG: 37.24 ± 23.10%; DYRK1A5 µM DAU: 28.28 ± 17.04%, DYRK1A 5 µM DAU+ 25 µM EGCG: 24.41 ± 10.99%) (Fig. 5B).
Fig. 5.
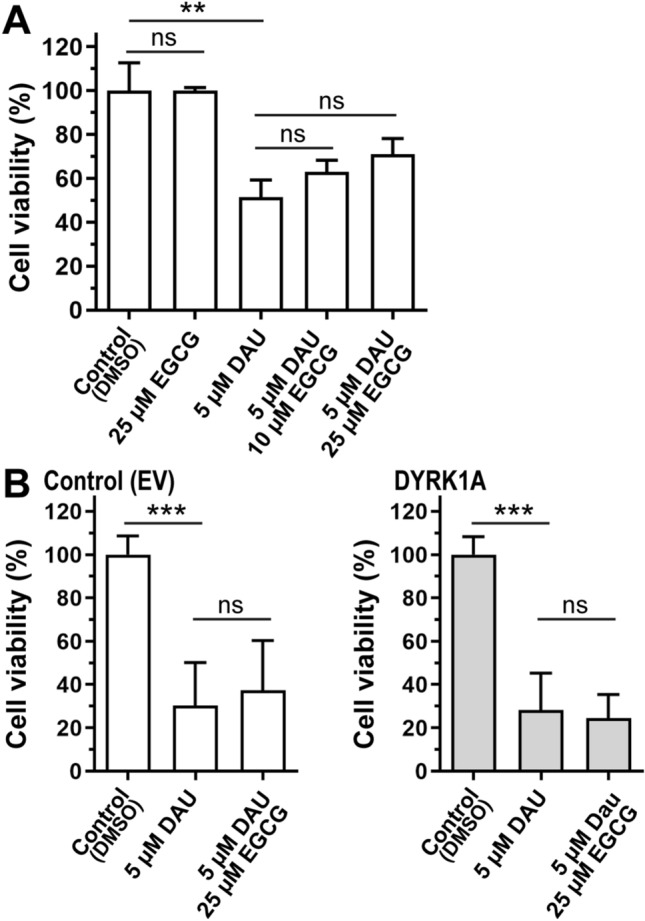
Impact of DYRK1A expression and drug treatments on the viability of iPSC cardiomyocytes. A Cellular viability after incubations with daunorubicin (DAU) and epigallocatechin gallate (EGCG). B Viability in controls and DYRK1A-overexpressing cells. Mean ± SD from two determinations performed in triplicates. ***P < 0.001, **P < 0.01, ns not significant, One-way ANOVA (Tukey’s test) and Student’s t test
Next, we examined whether DYRK1A overexpression in combination with DAU treatment modifies the beating frequency of cardiomyocytes. Beating frequency was determined by live-cell imaging with a calcium-sensitive fluorescent dye (Fig. 6, Videos 1, 2, 3 and 4). Assays were performed at day 14 post initial plating and 48 h post-transfection. Under these conditions, iPSC cardiomyocytes formed a synchronized contracting syncytium (Videos 1 and 2). In control conditions, DYRK1A overexpression (DYRK1A mRNA expression > 45-fold) did not modify the beating frequency of cardiomyocytes (EV: 24.5 ± 8.4 beats/30 s, DYRK1A: 23.7 ± 11.2 beats/30 s). In cardiomyocytes over-expressing DYRK1A, incubation with DAU decreased beating frequency by ~ 47% vs a ~ 66% decrease in controls transfected with empty vector (EV5 µM DAU: 8.3 ± 5.3, DYRK1A5 µM DAU: 12.5 ± 11.1) (Fig. 6A, B, and Videos 3 and 4).
Fig. 6.

Analysis of iPSC cardiomyocytes contractility. A Beating rate in cardiomyocytes overexpressing DYRK1A in control conditions (left) and after exposure to daunorubicin (DAU) (middle). Right: relative DYRK1A fold expression in transfected cells. Each point represents individual measurements in a region of interest (ROI) (n = 45–55 ROIs per condition). Each bar represents the mean ± SD of three measurements performed in triplicates for each condition. *P < 0.05, ns not significant, Student’s t test. B Representative fluorograms showing beating patterns from controls and treated cells. C Representative live-confocal microscopy images of iPSC cardiomyocytes showing changes in Fluo-4 AM intensity (green) over time. Scale bar: 50 µm
Discussion
This study examined DYRK1A-SRSF6-TNNT2 interplays in beating iPSC cardiomyocytes. Increased DYRK1A expression in cardiomyocytes resulted in a ~ 58% increase in the endogenous relative abundance of the fetal variants cTnT1 and cTnT2, and a decrease of ~ 27% in the abundance of the cTnT3 variant. The relative abundance of the cTnT4 variant remained unchanged (Figs. 1, 4). In line with our observations, a previous study in immortalized human embryonic kidney HEK-293 cells showed that DYRK1A overexpression increased the retention of exon 5 in PCR products derived from the splicing of an artificial TNNT2 “mini-gene”. In the same study, the authors showed that increased cardiac DYRK1A expression in Ts65Dn mice, a model of Down syndrome, delayed the exclusion of exon 5 during the neonatal period [15]. Results from our previous analysis of myocardial tissue samples from donors with Down syndrome suggest that the higher relative abundance of the fetal troponin variants cTnT1 and cTnT2 seems to persist during adulthood (i.e., ~ 50% increase in comparison to myocardial samples from subjects without Down syndrome) [16].
DYRK1A modulates the splicing activity of SR proteins mainly through selective phosphorylation of their proline-rich domain, and also the RS1 domain [23]. Phosphorylation of SRSF6 by DYRK1A promotes the inclusion of exon 5 into TNNT2 transcripts, and increased levels of phosphorylated SRSF6 were detected in myocardium from subjects with Down syndrome [15, 16]. Because detection of post-translational modifications is usually challenging and requires large amounts of material, we examined the phosphorylation status of HA-tagged SRSF6. In the context of DYRK1A over-expression, SRSF6 phosphorylation was ~ 25–65% higher in SRSF6-HA immunoprecipitated samples in comparison to controls (Fig. 3). In line with our observations, Yin et al. reported a 20% decrease in the phosphorylation of SRSF6 by DYRK1A after deletion of the proline-rich and RS1 domains of SRSF6 [23]. The overexpression of SRSF6 alone did not impact the relative abundance of TNNT2 transcripts variants (Fig. 4D). Thus, the observed shift in TNNT2 transcripts variants in the context of elevated DYRK1A expression may be mediated by an increase in SRSF6 phosphorylation by DYRK1A.
Some pediatric patients with acute myeloid leukemia and Down syndrome develop anthracycline-related cardiotoxicity [24, 25]. It is unclear why some patients treated with anthracyclines develop cardiotoxicity whereas others identically treated do not. The expression of myocardial DYRK1A in diploid and trisomic myocardium is variable. We documented higher expression of fetal TNNT2 variants in myocardial tissue from donors with Down syndrome in comparison to samples from donors without Down syndrome [16]. We reasoned that increases in the relative abundance of fetal TNNT2 transcript variants resulting from DYRK1A overexpression may render cardiomyocytes more sensitive to DAU cytotoxicity. The cytotoxic activity of DAU was similar between iPSC cardiomyocytes with basal and high levels of DYRK1A expression (Fig. 5). Cardiomyocytes expressing basal levels of DYRK1A showed a non-statistically significant trend towards increased cell viability after concomitant incubations with the DYRK1A inhibitor ECGC and DAU (Fig. 5A, B). The inhibition of DYRK1A by ECGC has been examined in preclinical studies with the aim of improving cognitive functions in Down syndrome [26–28]. ECGC has antioxidant properties and target other proteins besides DYRK1A [29, 30]. For example, the ECGC derivative Y6 decreases the expression of the anthracycline reductase CBR1 and the synthesis of daunorubicinol which in turn reduces DAU cardiotoxicity [31]. Our results suggest that in the context of DYRK1A overexpression, ECGC does not protect cardiomyocytes against DAU cytotoxicity (Fig. 5B). Cardiac myofibrils containing fetal troponin isoforms are more sensitive to Ca2+ which influences the contractile properties of myocardium [5, 32, 33]. iPSC cardiomyocytes with basal or increased levels of DYRK1A exhibited similar beating frequencies as determined by confocal live-cell imaging with a Ca2+ sensitive dye (Fig. 6A). Incubations with DAU reduced the beating frequency of cardiomyocytes by ~ 56%. The overexpression of DYRK1A ameliorated the impact of DAU on beating frequency (Fig. 6 and Videos 1, 2, 3 and 4). Increased expression of the fetal isoforms cTnT1/cTnT2 has been detected in failing hearts [7, 34]. Evidence suggests that the expression of fetal cTnT isoforms in the failing heart could represent a cardioprotective response [35].
One of the main limitations of this study is that iPSC cardiomyocytes were transiently transfected, and only a fraction of the cell population showed increased DYRK1A expression. Thus, it is possible to speculate that the observed differences in TNNT2 splicing and beating in response to DAU could be more pronounced in cultures with higher efficiencies of cellular transfection. Thus, it would be of interest to further examine whether alterations in DYRK1A-TNNT2 expression in vivo or in more nuanced cellular scenarios such as iPSC cardiomyocytes with trisomy 21 or cells with various degrees of stable DYRK1A over-expression modify Ca2+ handling and beating in response to cardiotoxic insults including anthracyclines. Another limitation is that we did not examine absolute amounts of each TNNT2 mRNA splicing variant in cardiomyocytes. Our methodology is similar to the one used by Lu and Yin for the examination of TNNT2 splicing patterns, and our observations further support the notion that increased DYRK1A expression impacts the relative expression of TNNT2 splicing variants [15].
Although iPSC-derived cardiomyocytes have limited relevance to model the biology of the adult heart, they provide a more suitable platform for the study of drug metabolism and disposition than transformed cell lines with abnormal karyotypes and deregulated transcriptional networks [1]. A recent systematic study showing major limitations of cardiac cells revealed that gene expression in iPSC-derived cardiomyocytes is more similar to adult cardiac tissue compared to the cardiac cell lines AC16, H9C2 and HL-1. Nevertheless, the expression of elements of the contractile machinery was decreased in human AC16 and iPSC-derived cardiomyocytes compared to healthy adult cardiac samples [36]. In this context, the translational relevance of DYRK1A expression on TNNT2 splicing and anthracycline toxicity remains to be determined. Points that merit further consideration are (1) analysis of beating frequency and sensitivity to DAU in cells overexpressing fetal cTnT isoforms, (2) evaluation of the impact of alternative concentrations of DAU and other anthracyclines, (3) study of the contribution of DYRK1A phosphorylation to DAU response and SRSF6 activity in iPSC cardiomyocytes, and (4) quantitative analysis of TNNT2 mRNA splicing variants and other markers of cytotoxicity (e.g., LDH activity) in cardiomyocytes exposed to anthracyclines.
In conclusion, this study contributes new information on the expression of endogenous TNNT2 transcript variants and SRSF6 phosphorylation in the context of altered DYRK1A expression in a relevant model of human beating cardiomyocytes. This work lays the foundation to further investigate the contribution of variable DYRK1A-TNNT2 expression to drug-induced cardiotoxicity in human cardiomyocytes and in vivo models.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We acknowledge the excellent assistance and advise of Dr. Andrew McCall from the Optical Imaging and Analysis Facility at the-School of Dental Medicine, SUNY Buffalo.
Author Contributions
RBC designed the study, performed experiments, analyzed data, and wrote the manuscript. MT-B performed experiments and analyzed data. FJE designed the macros for data analysis. JGB conceptualized research, designed the study, revised, and edited the manuscript, provided resources, and acquired funding. All authors reviewed the manuscript.
Funding
This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Award R21 HD089053), the National Cancer Institute (Award R21 CA245067), and the National Institute of General Medical Sciences (Award R01 GM073646).
Data Availability
The datasets generated for this study are available from the corresponding author upon request.
Declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors have no conflict of interests to declare that are relevant to the content of this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Meseguer-Ripolles J, Khetani SR, Blanco JG, Iredale M, Hay DC. Pluripotent stem cell-derived human tissue: Platforms to evaluate drug metabolism and safety. American Association of Pharmaceutical Scientists Journal. 2017;20(1):20. doi: 10.1208/s12248-017-0171-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circulation Research. 2015;117(1):80–88. doi: 10.1161/CIRCRESAHA.117.305365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson PA, Greig A, Mark TM, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK. Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circulation Research. 1995;76(4):681–686. doi: 10.1161/01.RES.76.4.681. [DOI] [PubMed] [Google Scholar]
- 4.Feng HZ, Jin JP. Coexistence of cardiac troponin T variants reduces heart efficiency. American Journal of Physiology. Heart and Circulatory Physiology. 2010;299(1):H97–H105. doi: 10.1152/ajpheart.01105.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes AV, Guzman G, Zhao J, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development Insights into the role of troponin T isoforms in the heart. Journal of Biological Chemistry. 2002;277(38):35341–9. doi: 10.1074/jbc.M204118200. [DOI] [PubMed] [Google Scholar]
- 6.Wei B, Jin JP. TNNT1, TNNT2, and TNNT3: Isoform genes, regulation, and structure-function relationships. Gene. 2016;582(1):1–13. doi: 10.1016/j.gene.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biesiadecki BJ, Elder BD, Yu ZB, Jin JP. Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. Journal of Biological Chemistry. 2002;277(52):50275–50285. doi: 10.1074/jbc.M206369200. [DOI] [PubMed] [Google Scholar]
- 8.Mesnard-Rouiller L, Mercadier JJ, Butler-Browne G, Heimburger M, Logeart D, Allen PD, Samson F. Troponin T mRNA and protein isoforms in the human left ventricle: Pattern of expression in failing and control hearts. Journal of Molecular and Cellular Cardiology. 1997;29(11):3043–3055. doi: 10.1006/jmcc.1997.0519. [DOI] [PubMed] [Google Scholar]
- 9.He J, Yao J, Sheng H, Zhu J. Involvement of the dual-specificity tyrosine phosphorylation-regulated kinase 1A-alternative splicing factor-calcium/calmodulin-dependent protein kinase IIdelta signaling pathway in myocardial infarction-induced heart failure of rats. Journal of Cardiac Failure. 2015;21(9):751–760. doi: 10.1016/j.cardfail.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Martinez P, Zahonero C, Sanchez-Gomez P. DYRK1A: The double-edged kinase as a protagonist in cell growth and tumorigenesis. Molecular & Cellular Oncology. 2015;2(1):e970048. doi: 10.4161/23723548.2014.970048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laham AJ, Saber-Ayad M, El-Awady R. DYRK1A: A down syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cellular and Molecular Life Sciences. 2021;78(2):603–619. doi: 10.1007/s00018-020-03626-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hille S, Dierck F, Kühl C, Sosna J, Adam-Klages S, Adam D, Lüllmann-Rauch R, Frey N, Kuhn C. Dyrk1a regulates the cardiomyocyte cell cycle via D-cyclin-dependent Rb/E2f-signalling. Cardiovascular Research. 2016;110(3):381–394. doi: 10.1093/cvr/cvw074. [DOI] [PubMed] [Google Scholar]
- 13.Kuhn C, Frank D, Will R, Jaschinski C, Frauen R, Katus HA, Frey N. DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy. Journal of Biological Chemistry. 2009;284(25):17320–17327. doi: 10.1074/jbc.M109.006759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441(7093):595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 15.Lu S, Yin X. Overexpression of Dyrk1A regulates cardiac troponin T splicing in cells and mice. Biochemical and Biophysical Research Communications. 2016;473(4):993–998. doi: 10.1016/j.bbrc.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Quinones-Lombrana A, Blanco JG. Comparative analysis of the DYRK1A-SRSF6-TNNT2 pathway in myocardial tissue from individuals with and without Down syndrome. Experimental and Molecular Pathology. 2019;110:104268. doi: 10.1016/j.yexmp.2019.104268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipshultz SE, Cochran TR, Franco VI, Miller TL. Treatment-related cardiotoxicity in survivors of childhood cancer. Nature Reviews Clinical Oncology. 2013;10(12):697–710. doi: 10.1038/nrclinonc.2013.195. [DOI] [PubMed] [Google Scholar]
- 18.Lipshultz SE, Karnik R, Sambatakos P, Franco VI, Ross SW, Miller TL. Anthracycline-related cardiotoxicity in childhood cancer survivors. Current Opinion in Cardiology. 2014;29(1):103–112. doi: 10.1097/HCO.0000000000000034. [DOI] [PubMed] [Google Scholar]
- 19.Menna P, Gonzalez Paz O, Chello M, Covino E, Salvatorelli E, Minotti G. Anthracycline cardiotoxicity. Expert Opinion on Drug Safety. 2012;11(Suppl 1):S21–36. doi: 10.1517/14740338.2011.589834. [DOI] [PubMed] [Google Scholar]
- 20.Brown SA, Sandhu N, Herrmann J. Systems biology approaches to adverse drug effects: The example of cardio-oncology. Nature Reviews Clinical Oncology. 2015;12(12):718–731. doi: 10.1038/nrclinonc.2015.168. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Sun CL, Quiñones-Lombraña A, Singh P, Landier W, Hageman L, Mather M, Rotter JI, Taylor KD, Chen YD, Armenian SH. CELF4 variant and anthracycline-related cardiomyopathy: A Children’s Oncology Group genome-wide association study. Journal of Clinical Oncology. 2016;34(8):863–870. doi: 10.1200/JCO.2015.63.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY. Fiji: An open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin X, Jin N, Gu J, Shi J, Zhou J, Gong CX, Iqbal K, Grundke-Iqbal I, Liu F. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) modulates serine/arginine-rich protein 55 (SRp55)-promoted Tau exon 10 inclusion. Journal of Biological Chemistry. 2012;287(36):30497–30506. doi: 10.1074/jbc.M112.355412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hefti E, Blanco JG. Pharmacokinetics of chemotherapeutic drugs in pediatric patients with Down syndrome and leukemia. Journal of Pediatric Hematology/Oncology. 2016;38(4):283–287. doi: 10.1097/MPH.0000000000000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hefti E, Blanco JG. Anthracycline-related cardiotoxicity in patients with acute myeloid leukemia and Down syndrome: A literature review. Cardiovascular Toxicology. 2016;16(1):5–13. doi: 10.1007/s12012-015-9307-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guedj F, Sébrié C, Rivals I, Ledru A, Paly E, Bizot JC, Smith D, Rubin E, Gillet B, Arbones M, Delabar JM. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS ONE. 2009;4(2):e4606. doi: 10.1371/journal.pone.0004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De la Torre R, De Sola S, Pons M, Duchon A, de Lagran MM, Farré M, Fitó M, Benejam B, Langohr K, Rodriguez J, Pujadas M. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Molecular Nutrition & Food Research. 2014;58(2):278–288. doi: 10.1002/mnfr.201300325. [DOI] [PubMed] [Google Scholar]
- 28.Catuara-Solarz S, Espinosa-Carrasco J, Erb I, Langohr K, Notredame C, Gonzalez JR, Dierssen M. Principal component analysis of the effects of environmental enrichment and (-)-epigallocatechin-3-gallate on age-associated learning deficits in a mouse model of Down syndrome. Frontiers in Behavioral Neuroscience. 2015;9:330. doi: 10.3389/fnbeh.2015.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nature Reviews Cancer. 2009;9(6):429–439. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao SY, Zhao CN, Gan RY, Xu XY, Wei XL, Corke H, Atanasov AG, Li HB. Effects and mechanisms of tea and its bioactive compounds for the prevention and treatment of cardiovascular diseases: An updated review. Antioxidants (Basel) 2019;8(6):166. doi: 10.3390/antiox8060166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou H, Fu LX, Li L, Chen YY, Zhu HQ, Zhou JL, Lv MX, Gan RZ, Zhang XX, Liang G. The epigallocatechin gallate derivative Y6 reduces the cardiotoxicity and enhances the efficacy of daunorubicin against human hepatocellular carcinoma by inhibiting carbonyl reductase 1 expression. Journal of Ethnopharmacology. 2020;261:113118. doi: 10.1016/j.jep.2020.113118. [DOI] [PubMed] [Google Scholar]
- 32.Briggs MM, Maready M, Schmidt JM, Schachat F. Identification of a fetal exon in the human fast troponin T gene. FEBS Letters. 1994;350(1):37–40. doi: 10.1016/0014-5793(94)00729-2. [DOI] [PubMed] [Google Scholar]
- 33.Nassar R, Malouf NN, Mao L, Rockman HA, Oakeley AE, Frye JR, Herlong JR, Sanders SP, Anderson PA. cTnT1, a cardiac troponin T isoform, decreases myofilament tension and affects the left ventricular pressure waveform. American Journal of Physiology-Heart and Circulatory Physiology. 2005;288(3):H1147–H1156. doi: 10.1152/ajpheart.00140.2004. [DOI] [PubMed] [Google Scholar]
- 34.Anderson PA, Malouf NN, Oakeley AE, Pagani ED, Allen PD. Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circulation Research. 1991;69(5):1226–33. doi: 10.1161/01.RES.69.5.1226. [DOI] [PubMed] [Google Scholar]
- 35.Pinto JR, Yang SW, Hitz MP, Parvatiyar MS, Jones MA, Liang J, Kokta V, Talajic M, Tremblay N, Jaeggi M, Andelfinger G. Fetal cardiac troponin isoforms rescue the increased Ca2+ sensitivity produced by a novel double deletion in cardiac troponin T linked to restrictive cardiomyopathy: A clinical, genetic, and functional approach. Journal of Biological Chemistry. 2011;286(23):20901–20912. doi: 10.1074/jbc.M111.234336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onodi Z, Visnovitz T, Kiss B, Hambalkó S, Koncz A, Ágg B, Váradi B, Tóth VÉ, Nagy RN, Gergely TG, Gergő D. Systematic transcriptomic and phenotypic characterization of human and murine cardiac myocyte cell lines and primary cardiomyocytes reveals serious limitations and low resemblances to adult cardiac phenotype. Journal of Molecular and Cellular Cardiology. 2022;165:19–30. doi: 10.1016/j.yjmcc.2021.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for this study are available from the corresponding author upon request.