

Abstract
Cell identity and function largely rely on the programming of transcriptomes during development and differentiation. Signature gene expression programs are orchestrated by regulatory circuits consisting of cis-acting promoters and enhancers, which respond to a plethora of cues via the action of transcription factors. In turn, transcription factors direct epigenetic modifications to revise chromatin landscapes, and drive contacts between distal promoter-enhancer combinations. In immune cells, regulatory circuits for effector genes are especially complex and flexible, utilizing distinct sets of transcription factors and enhancers, depending on the cues each cell type receives during an infection, after sensing cellular damage, or upon encountering a tumor. Here, we review major players in the coordination of gene regulatory programs within innate and adaptive immune cells, as well as integrative omics approaches that can be leveraged to decipher their underlying circuitry.
Keywords: epigenetics, innate lymphoid cells, adaptive lymphoid cells, chromatin, transcription factors, gene regulation
INTRODUCTION
The human body and its composite cells have been called living machines since the dawn of medicine. Like nearly any machine, at least those that run on electricity, each cell type relies on a complex circuitry, most of which is devoted to the control of gene expression. The switching on or off of these circuits begins at the earliest stages of development. In pluripotent cells, a small node of circuits controlled by factors like NANOG, SOX2, and OCT4 becomes active, but the vast majority of other gene expression circuits remain poised. As embryonic development proceeds, many of the poised circuits are shut off in each cell type, coinciding with a successive loss of pluripotency. However, each cell type, in turn, activates new circuit modules that are regulated by so-called master transcription factors (TFs) that cement their identities. These TFs control expression by binding cis-regulatory elements (CREs) in the genome, such as promoters or enhancers, the latter usually located within intronic or more distal noncoding regions. A similar scenario plays out during immune cell lineage decisions and during functional differentiation of mature myeloid and lymphoid cells. In this review, we focus on regulatory circuits and TF programs that govern entire modules of gene expression in innate and adaptive immunity, endowing cell identity and modulating the functional state of a given cell.
GENE REGULATORY CIRCUITS
Herein, we consider a regulatory circuit to be composed of the TFs, cis-acting elements, and requisite chromatin changes that coordinate the expression of a gene in a given cell type under certain conditions, or states. As such, the circuitry for a single gene is not always the same. A gene circuit can be rewired, perhaps using different sets of enhancers and TFs in different cell types or when the same cell type encounters distinct microenvironmental cues. One can appreciate the complexity of underlying regulatory logic for these circuits given the need to control expression of ~25,000 human genes in more than 200 cell types using the ~400,000 putative enhancers cataloged to date (1, 2). Indeed, genes whose activities are controlled in time-, location-, and stimulus-specific manners are often linked to multiple, even dozens, of enhancers, each contributing to the overall specificity of gene expression (3).
Gene promoters and enhancers, as well as other flavors of CREs, serve as conduits for TFs that, in turn, recruit other nuclear proteins to revise local epigenetic landscapes. These proteins write or erase DNA and histone modifications, alter chromatin accessibility, or stabilize interactions with the transcriptional machinery. The first identified epigenetic modification in mammals was DNA methylation at CpG dinucleotides, which forms 5meCpG (3a, 3b). These dinucleotides are enriched at promoters, where CpG islands enforce transcriptional silencing when methylated or activation when demethylated. In addition to this simple binary code, dozens of distinct modifications can be placed on the N-terminal tails of histones 3 and 4 (H3 and H4), forming a complex alphabet that is read to facilitate or repress transcription (4-6). The two most commonly studied histone modifications for transcriptional regulation are acetylation and methylation (mono-, di-, or trimethylation) of H3/4 lysine residues (Figure 1a,b).
Figure 3.
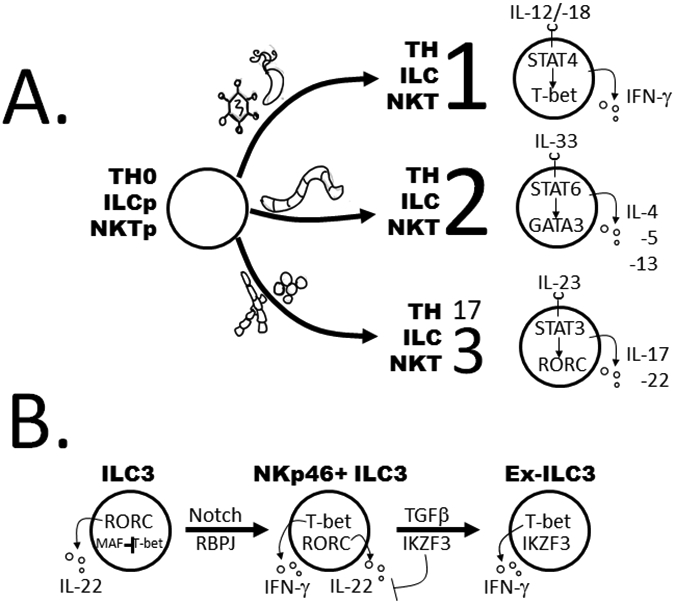
Regulatory programs for plasticity and functional polarization in helper lymphocytes. (a) Scheme for functional polarization within adaptive, innate, and innate-like helper lymphocytes. Upon activation, naive and progenitor lymphocytes (Th0, ILCp, or NKTp) will proceed along a polarization pathway based upon the type of infection and microenvironmental cues. Type 1, 2, and 3 immune cells are polarized in response to intracellular bacteria or viruses, helminths, and extracellular bacteria or fungi, respectively. Based upon danger signals and pathogen-associated molecular patterns, antigen-presenting cells release soluble cytokines (on top of cells) that activate JAK/STAT signaling through the relevant cytokine receptors. This is followed by activation of an LDTF by STAT, as shown, which drives initial stages of polarization. Signature TFs and cytokines for the three functional phenotypes are shown. (b) ILC3 plasticity. Indicated are cell types, transcription factors (inside cells), and soluble cytokines (outside cells) thought to be important for stepwise conversion of ILC3s to ILC1s. External signals that drive conversion events are indicated above the arrows. Abbreviations: ILC1, group 1 innate lymphoid cell; ILCp, ILC progenitor; LDTF, lineage-determining transcription factor; NKT, natural killer T; NKTp, NKT progenitor; Th, T helper; Th0, naive Th.
Figure 1.

The histone code. (a) Highlighted are the histone tail modifications mentioned in the text. Those below the histone tail are activation-associated modifications (green arrows are acetylation and circles are methylation), and those above are generally repressive (orange circles). For methylation, each circle represents one methyl group. (b) A key to the nomenclature and moieties. (c) Major modifications are shown that associate with different classes of accessible REs (i.e., ATAC+). Abbreviations: ATAC, assay for transposase-accessible chromatin using sequencing; RE, regulatory element.
Like any language, the histone code is established by (a) writers, enzymes that add chemical groups (e.g., histone acetyl transferases and histone methyltransferases); (b) erasers, enzymes that remove chemical groups [e.g., histone deacetylases (HDACs) and histone demethylases]; and (c) readers, protein modules that bind a chromatin modification and translate it into a specific biological function (e.g., bromodomains and chromodomains for acetylation and methylation, respectively) (4). Each of these word processors is recruited to a given locus through TF-bound REs or by binding directly to a set of preprinted histone modifications. In brief, active genes and chromatin regions are typically enriched for acetylation marks, such as H3K27ac, at active enhancers (7, 8), which are deposited by polymerase complex member P300 (9). For lysine methylation, H3K4me1, H3K4me3, and H3K36me3 are hallmarks of active enhancers, promoters, and transcription, respectively, whereas H3K27me3 and H3K9me2/3 are repressive marks associated with heterochromatin (8, 10-12) (Figure 1a-c).
DECIPHERING GENE REGULATORY CIRCUITS AND PROGRAMS
In the last decade, there have been major efforts to map the collection of CREs that participate in gene regulatory circuits, including initiatives by the ENCODE and IMMGEN consortia (13, 14). The emerging data have been especially valuable for deciphering another layer of biology—gene regulatory programs that guide immune cell development or their responses to a variety of stimuli. Herein, we refer to programs as the collection of regulatory circuits coordinated by key TFs that activate or inactivate cohorts of genes governing cell identity and function (also referred to as functional nodes or modules) (Figure 2a).
Figure 2.

Deciphering gene regulatory circuits and programs. (a) Regulatory programs for a given cell state. Shown are examples of the gene expression modules (blue and green) governed by two distinct TFs, which are two components of the regulatory program for a given cell type and functional state. The underlying regulatory circuits for these expression programs are not shown. The examples are assigned as modules that mediate stemness (TCF1-based) and homing (KLF2-based) within naive lymphocytes. (b) Identification of regulatory circuits for a given gene (white arrow indicating the direction of transcription) by chromatin profiling. Sorted cell types (X and Y) from a given sample (top) analyzed in two functional states (e.g., +/− agonist, normal versus disease, or two distinct tissues), as indicated by the numerals 1 and 2. Samples are initially subjected to chromatin profiling with ATAC-seq (accessibility) and ChIP-seq for H3K27ac (RE activity). Contacts between REs, including promoter-enhancer contacts, are determined by Hi-C or another conformation capture methodology. For cell type X, gene expression (transcription) in state 2 relies on a regulatory circuit consisting of the promoter (H3K27ac mark at 5′ end of the gene) contacting two active enhancers (two H3K27ac marks downstream of the gene). The middle enhancer is predicted to be irrelevant, converting from active to poised (state 1 to 2) with no promoter contacts in state 1. For cell type Y, distinct circuits are employed for either low-level (state 1; promoter–middle enhancer) or high expression (state 2; promoter–proximal + distal enhancers). (c) Workflow for motif analysis to identify important TFs. (Top) Enhancers are first classified based on activity profiles in distinct cell types or states. The DNA sequences corresponding to accessible regions (ATAC-seq peaks) are then extracted for each enhancer class. Sequence enrichment can be performed either de novo (i.e., any enriched sequence) or for known TF motifs (i.e., using a TF motif database). The resulting data set will predict TF families, rather than specific TFs, which can then be integrated with paired RNA-seq to predict specific TFs within a family. Predictions then can be validated by ChIP-seq or targeted perturbations. Abbreviations: ATAC-seq, assay for transposase-accessible chromatin sequencing; ChIP-seq, chromatin immunoprecipitation sequencing; Hi-C, high-resolution conformation capture; RE, regulatory element; TF, transcription factor.
A typical starting point for deciphering gene regulatory programs is to sort the cell type of interest and, under the desired conditions, characterize its transcriptome and map its open chromatin regions (Figure 2b). The latter method tags genomic locations of poised or active REs with relatively high resolution. Traditionally, this has been accomplished using DNase-seq. However, the assay for transposase-accessible chromatin using sequencing (ATAC-seq) has emerged as the predominant method due to its ease of application in a range of cell types and its requirement for very low cell input (~104 for sorted populations or even a single cell; see below). These data yield accessible regulatory element (RE) maps but are not the optimal metric for relative RE activities, which are best monitored by chromatin immunoprecipitation sequencing (ChIP-seq) for specific histone modifications, especially H3K27ac for enhancers and K3K4me3 for promoters. Again, recent technical advancements have enabled analysis of histone modifications on samples with limited cell input (routinely ~104), including CUT-and-RUN (cleavage under targets and release using nuclease) sequencing and ultralow input (ULI)-ChIP-seq (15-18).
Integration of chromatin accessibility and histone modification maps in a given cell state permit classification of REs as active, poised, or silent. Coupling the information with transcriptome data, one can then proceed to parse out active versus inactive gene regulatory circuits and the factors that may govern their status (13). For example, we and others have developed algorithms to identify enriched DNA motifs in classes of REs for a given cell type (e.g., all active enhancers) or when comparing two cell types [e.g., T helper type 1 (Th1) cells versus group 1 innate lymphoid cells (ILC1s)] (15, 16). This approach will reveal TFs that target enriched motifs to activate (or repress) whole cohorts of REs and, presumably, their gene targets (Figure 2c).
Validation of a specific enhancer’s importance in a given regulatory circuit can now be achieved via a number of CRISPR-based approaches, including enhancer deletions, mutations, repression (CRISPRi), or activation (CRISPRa). However, pairing enhancers with their target genes is not always straightforward, as most chromosomal neighborhoods have collections of genes interspersed with arrays of enhancers. In general, enhancers relevant to the circuitry of a given gene and cell state make physical contacts with their target promoters. These genomic contacts can be captured by chromatin cross-linking, using so-called C (conformation-capture) techniques, which now can be performed at very high resolution on a genome-wide (Hi-C) scale (20, 21). Thus, by integrating transcriptome, chromatin, and conformational data for a given cell type and state, one can develop testable hypotheses regarding the important REs and TFs governing a gene regulatory circuit, or nodes within the expression program, for that cell state. However, when testing such hypotheses, one must also be aware that most expression circuits have built-in redundancies, which may muddle interpretation of validation experiments or CRISPR-based screens over complex regulatory landscapes.
Notwithstanding, integrative chromatin approaches have produced impressive advances in our understanding of gene regulatory circuits and programs that govern the development (22-24), receptor repertoire formation (25, 26), and function of our immune system in both health and disease (27). We now describe a subset of such advances, with a focus on the regulatory circuits that drive biological processes and cell identities in both the innate and adaptive arms of our immune defenses.
REGULATORY CIRCUITS AND PROGRAMS IN HUMORAL IMMUNITY
Historically, the molecular programs of B cells and humoral immune responses have been some of the most intensively studied with regard to cellular differentiation, effector functions, and memory, and they will serve as an appropriate springboard for our discussion.
Mature B cells emerging in the adult bone marrow undergo extensive selection to prune out autoreactive clones and generate the peripheral repertoire. Upon antigen encounter, naive follicular B cells differentiate in the germinal center (GC), where they proliferate and execute programs for isotype switching and affinity maturation. Activated B lymphocytes, as well as those in the GC (GCB cells), can differentiate into plasma cells (PCs) and memory B cells, subsets that are responsible for antibody secretion and long-term protection, respectively. These cell fate decisions are instructed mainly by the strength of signals received from B cell receptors (BCRs), pattern recognition receptors, and follicular T helper (Tfh) cells (CD40L and cytokines). Signal-responsive TFs then collaborate with other lineage-specific TFs to establish the regulatory programs for each cell fate.
In primary B lymphocytes, PAX5 and EBF1 are required for maintaining cellular identity (39). Master TFs also have been identified for the GCB and PC subsets, which include BCL6 and BLIMP1 (encoded by PRDM1), respectively. The memory B cell subset bears several striking resemblances to the naive program, including expression of BACH2 and KLF2, both of which maintain a quiescent state and prevent premature differentiation into PCs via suppression of PRDM1 (40, 41). Another TF, called HHEX, is critical for driving the GC B cell–memory B cell transition, where it is involved in a mutually repressive pathway with BCL6 (42).
One of the most extensively studied TF regulatory circuits in humoral immunity involves BLIMP1, IRF4, and BCL6, which largely govern GCB–PC programs. In naive or GCB cells, IRF4 is induced in proportion to signaling strengths from the BCR, and via CD40L on Tfh cells. A higher level of IRF4 (high-affinity BCR) induces expression of BLIMP1 and initiation of the PC program. In contrast, low to intermediate levels of IRF4 induce BCL6, which governs the GC program, including somatic mutation and selection to generate higher-affinity BCRs (43, 44). Both BLIMP1 and BCL6 are transcriptional repressors, directly inhibiting the expression of each other, which reinforces regulatory programs for PC and GCB cell fates, respectively (45, 46). In addition to this classical circuit, mathematical modeling unveiled the NF-κB subunit c-REL as a control for the switch between GCB proliferation and PC differentiation by antagonizing BLIMP1, which can otherwise directly repress c-REL expression (47).
As in most gene regulatory circuits, the collection of TFs that regulate B cell fate collaborate with epigenetic modifiers to revise chromatin and enhancer landscapes, generating cell type–specific transcriptomes. These relationships have been studied in detail using a T-independent activation model in which lipopolysaccharide (LPS)-stimulated B cells differentiate into PCs (48-50). While it is generally accepted that chromatin accessibility and H3K27ac densities positively correlate with RE activity, one critical challenge has been functional validation on a large scale. In this regard, a recent multi-omics study (3) identified the collection of active enhancers in LPS-stimulated B cells using a high-throughput screening method that assesses the transactivation potential of all accessible chromatin regions (51). Notably, active enhancer elements were often enriched for at least five to six TF motifs, compared with fewer in the inactive but accessible regions. Once the active RE cohort was defined, complementary Hi-C analysis revealed complex promoter-enhancer networks in the LPS-stimulated B cells, including promoters regulated by multiple enhancers (multi-enhancer genes), and enhancers that regulate multiple genes (multigenic enhancers). The latter were shown to coordinate metabolic and DNA or protein biogenesis pathways, whereas multi-enhancer genes are enriched in specialized pathways, including MHC-II presentation and the endoplasmic reticulum–associated degradation response. This study provides an exemplar for how integrative chromatin-transcriptome-conformational-functional analysis can lead to new biological insights and testable hypotheses.
In addition to chromatin accessibility, DNA methylation is an essential epigenetic mark controlling gene expression and cell differentiation. A systematic analysis of DNA methylomes during LPS-induced B cell differentiation to PCs revealed stepwise demethylation of CpGs in a broad collection of enhancers, which correlated with the extent of cell division (52). Motif analysis showed that early demethylated regions (DMRs) were enriched in NF-κB and bZIP (AP-1) motifs, while late DMRs were enriched in IRF and POU (Oct-2) motifs, patterns that were largely recapitulated in analyses of chromatin accessibility (53). Intriguingly, a majority of DMRs undergo hypomethylation and increased accessibility only in cells that have divided at least eight times (52, 53); many of these DMRs are associated with genes involved in mitosis and metabolism. These epigenetic processes potentially are mediated by TET (ten-eleven translocation) DNA 5-methylcytosine oxidases, which regulate B cell function and PC differentiation (54-56). Indeed, the link between cell division and demethylation is reminiscent of a recent finding that lymphocytes primarily demethylate DNA passively through TET-mediated DNA oxidation, which impedes CpG methylation during cell replication (57).
In vivo, it is clear that BCL6 serves as the master TF for GC reactions, not only governing the regulatory program of GCB cells, but also for the requisite T helper (Th) arm, Tfh cells (58-60), which rely on a similar BCL6-PRDM1 repressor-of-repressor circuit (61). In Tfh cells, BCL6 also induces expression of chemokine receptors, such as CXCR5 and SIPR2, to promote B-Tfh retention and congregation at the GC (62). In addition, BCL6 regulates metabolic programs amenable to the GC microenvironment, including decreased glycolysis, increased lipid metabolism, and upregulation of the cytidine diphosphate–ethanolamine pathway (63-65). Similar to gene expression, several epigenetic regulators appear to operate in both GCB cells and Tfh. For instance, TET2 represses the expansion of both cell types, consistent with its role in suppressing GCB cell– and Tfh cell–derived lymphomas (55, 66). TET2 also facilitates differentiation of GCB cells to PCs (54, 55), while the de novo methyltransferases DNMT3A and DNMT3B oppose PC differentiation (67). Speaking to its universal role in lymphoid cells, BCL6 appears to govern regulatory modules in suppressive T follicular regulatory (Tfr) cells, which exhibit hallmarks of GC cells at both the chromatin and transcriptome levels (68).
During the dynamic transitions between GCB cell subsets, TF networks and epigenomes undergo substantial reorganization. Traditionally, GCB cells are divided into light zone (LZ) and dark zone (DZ) cells, and they continuously cycle between zones, where they undergo selection and massive proliferation, respectively. A recent multi-omics study suggested addition of a new GCB classification: gray zone (GZ) B cells, a subset of DZ B cells that are actively dividing (69). While previous analysis of LZ and DZ B cells showed remarkable similarity in steady-state transcriptomes (13, 70), the integrative epigenome-transcriptome approach revealed distinct signatures when the GCB classification was divided into LZ, DZ, and GZ. For instance, ATAC-seq analysis showed that DZ-specific regions are enriched for CTCF, FOXK1, and ZFP691 motifs; LZ regions are enriched in SPIB, PU.1, and IRF8 motifs; and GZ regions are enriched in OCT2, SP1, E2A, and EBF1 motifs. Thus, integration of multiple data platforms provides critical information about how TF networks collaborate to carve epigenomes and orchestrate the GC reaction for optimizing humoral responses.
TRAINED INNATE IMMUNITY
Protection from pathogens and malignancies requires a multilayered, cooperative network of innate and adaptive immune responses. Functionally, the innate immune system recognizes conserved structures on pathogens, which enables a prompt but nonspecific response to the invader. By comparison, adaptive immunity is slower in eliminating the primary infection but develops an efficient, clonally selected, and antigen-specific response capable of producing long-lasting immune memory (71). Historically, innate immune cells have been thought to lack memory programs, responding anew with each pathogenic insult. However, we now appreciate that innate immune cells exhibit memory-like characteristics that manifest as a state of high alert against reinfection, a process referred to as trained immunity or innate memory (72).
A majority of studies have focused on trained immunity in macrophage and natural killer (NK) lineages, but there is evidence for memory in other cell types, including ILCs and epithelial stem cells (73, 74). In contrast with adaptive memory, trained immunity predisposes innate cells for an enhanced secondary response against the same, or even a different, pathogen (74-77). Both microbial [e.g., bacillus Calmette-Guérin (BCG), Candida albicans, β-glucan, Plasmodium falciparum, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)] and nonmicrobial (e.g., Western diet, oxidized low-density lipoprotein, uric acid) stimuli induce trained immunity in innate cells (78-81). Following SARS-CoV-2 infection, monocytes from convalescent individuals exhibit characteristics of trained immunity, with chromatin profiles implicating IRF1, SPIB, and NR4A1 as important components of the underlying regulatory program (81).
For macrophages, trained immunity has been studied most extensively using BCG vaccination or a β-glucan model. In primary, or naive innate cells, antimicrobial response loci are mostly blanketed by repressive chromatin, including methylated CpGs, enforcing an off state for these circuits (87, 88). Upon initial exposure to BCG or β-glucan, proinflammatory loci are activated for expression (e.g., genes for TNF-α, IL-6, and IL-1β), remain epigenetically poised after removal of the stimulus, and are more rapidly transcribed upon a subsequent challenge. Primary exposures are accompanied by the revision of histone modifications at trained loci, including deposition of H3K27ac, H3K4me3, and H3K4me1, which support a stable open chromatin state capable of rapidly binding upstream TFs upon subsequent induction (77, 79, 88a-92). Such epigenetic training is not restricted to mature effector cells but also can occur in developing progenitors. Upon vaccination and exposure to microbial ligands, hematopoietic stem cells (HSCs) remodel enhancer landscapes to generate licensed monocytes or macrophages that more potently respond to subsequent microbial challenges (93-95). While it is unlikely that a single key regulator exists for all trained immunity programs, important TFs have been described for select cell types. For example, polymorphisms in HNF1a and HNF1b correlate with BCG-induced training of human HSCs (95). C/EBPβ is another such factor that, when deleted in HSCs, results in a loss of LPS-induced open chromatin regions (96).
β-Glucan training of monocytes is a nice example of the interplay between cellular metabolism and epigenetic remodeling of gene regulatory circuits. Upon treatment with this fungal cell wall component, monocytes undergo a metabolic switch to aerobic glycolysis, which is dependent upon the dectin-1/Akt/mTOR/HIF-1α pathway (97). Tricarboxylic acid (TCA) cycle activity elevates fumarate levels in these trained monocytes, further inhibiting degradation of HIF-1α and the histone demethylase KDM5, which increases H3K4me3 and H3K27ac to augment the activities of select promoters and enhancers, respectively (98). In addition to metabolism, long noncoding RNAs (lncRNAs) also are predicted to play a role in β-glucan-induced trained immunity via epigenetic remodeling. Specifically, β-glucan induces an immune-priming lncRNA, UMLILO, which interacts with the WDR5-MLL1 complex, directing it to promoters of the innate response genes IL6, IL8, CSF1, and CXCLs, ultimately increasing H3K4me3 levels (90). Collectively, integrative chromatin/RNA profiling has provided new insights into how epigenetic reprogramming renders innate cells capable of rapid cytokine responses upon subsequent stimulation, a feature once regarded as unique to the adaptive immune system.
However, not all stimuli trigger a heightened inflammatory response upon a second encounter. For example, LPS treatment induces endotoxin tolerance in macrophages, which results in a lower inflammatory response upon subsequent exposure to this potent agonist (98a, 98b). Although the specific TFs governing LPS tolerance are still under investigation, this process is accompanied by a loss of active chromatin marks in monocytes at elements associated with key proinflammatory genes (88a, 98c, 98d).
POLARIZATION OF ADAPTIVE AND INNATE LYMPHOCYTES
Upon encountering a pathogen, antigen-presenting cells (APCs) generate an effector response by presenting peptides to adaptive CD8+ cytolytic T (Tc) and CD4+ Th cells. The Th response develops over several days, during which time the cells proliferate and are directed toward a functionally polarized fate by cues from APCs, pathogen-associated molecular patterns (PAMPs), and other danger signals in their microenvironment. Each polarized Th subset expresses a signature cocktail of cytokines and other immune response genes tailored to eradicate certain types of pathogens. In general, there are three major effector immune responses (Figure 3a). Type 1 immunity is generated when APCs recognize PAMPs common to viruses or intracellular pathogens, which triggers IL-12 and IL-18 production and, in turn, polarizes Th1 cells to produce IFN-γ. Type 2 responses are PAMP independent; instead, Th2 cells are produced upon recognition of signals common to tissue destruction in the wake of parasitic infections and allergens, which drive Th expression of IL-4, IL-5, and IL-13. Type 3 responses are initiated by fungal PAMPs, metabolic ligands, and APC-produced IL-23, which induce functional polarization to the Th17 fate, and secretion of IL-17 and/or IL-22.
The stable clonal expression patterns of each polarized Th subset are enforced via epigenetic modifications within regulatory circuits for genes induced or repressed by the corresponding microenvironmental cues (99, 100). In all functional subsets, chromatin revisions are initiated by cytokine-STAT signaling: IL-12-STAT4 for Th1, IL-4/IL-5/IL-13-STAT6 for Th2, and IL-23-STAT3 for Th17. Once in the nucleus, the dimeric STAT factors recruit activating histone acetyl transferases and P300 to key enhancers that control specific functional modules in each Th subset (101). The STAT-dependent wave of chromatin revision allows expression of lineage-determining transcription factors (LDTFs) that further activate gene modules within their respective polarization programs.
Although originally attributed only to CD4+ Th cells, polarization is now widely recognized as a biological process in other lymphoid lineages, including innate-like NK T (NKT) cells, γδ T cells, and ILCs. All of these subsets develop from common lymphoid progenitors but, unlike CD4+ Th, NKT, and γδ T cells, the ILCs lack clonally distributed antigen receptors. Instead, ILCs respond rapidly to their local cytokine milieu, producing profiles of effector molecules that largely parallel their Th cell functional counterparts. Indeed, the development and responses of ILC1/Th1, ILC2/Th2, and ILC3/Th17 counterparts each depend on a similar set of master TFs. Here, we focus on concepts that unify or distinguish the various Th and ILC regulatory programs.
A core aspect of their polarization programs is that ILC-Th counterparts must activate unique gene modules, specific to a functional subset, while repressing modules for alternative fates. These regulatory strategies employ similar sets of TFs and chromatin modifiers but rely on distinct cues and kinetics. The master TFs that drive each ILC-Th fate have been known for some time, as follows: ILC1s, NKT1 cells, and Th1 cells require Tbx21 (T-bet); ILC2s, NKT2 cells, and Th2 cells require GATA3; and ILC3s, NKT3 cells, and Th17 cells require RORC (reviewed in 101a), with production of IL-22 in the latter dependent upon aryl hydrocarbon receptor (AHR) (Figure 3a). Unlike ILCs, Th cells can differentiate into other functional subsets, including Foxp3+ regulatory T cells (Tregs) and Bcl6+ Tfh cells; TFs that do not appear to be expressed in any ILC subset (102, 103).
A second distinction between the ILC-Th counterparts is that epigenetic programs at key cytokine and effector loci are acquired early in ILC development. Thus, in the absence of infection, naive Th cells and mature ILC counterparts display significantly different chromatin landscapes at signature regulatory circuits (104). However, the ILC-Th circuitry and gene expression patterns converge upon responses to acute infection. The convergence of regulatory programs is exemplified by elegant studies that examined accessible chromatin landscapes of ILC2s and Th2 cells in mice infected with a type 2 pathogen, Nippostrongylus brasiliensis (104). Specifically, Th2 cells developed chromatin accessibility at regions opened during ILC2 development, based on either unsupervised clustering or principle component analysis of ATAC-seq data. A similar convergence is observed in inflamed human mucosae, wherein signature cytokine loci exhibit nearly identical patterns of distal enhancer activity in Th1-ILC1 or Th17-ILC3 counterparts (16). At a molecular level, motif imprinting predicts that the acquisition of shared regulatory programs is dependent upon LDTFs, such as GATA3, RORC, and T-bet, and TFs that activate numerous cytokine loci (e.g., Runx1 and Runx3). Moreover, enhancers uniquely active in Th cells, not in ILCs, are enriched for motifs that bind TFs downstream of the TCR, including the AP-1 factor FOS (16). Thus, while Th cells and ILCs share a core set of TFs, ILCs can activate a similar set of enhancers during their development, independent of TCR signaling. The precise mechanisms by which these regulatory programs are activated in ILC progenitors remain unknown.
Regulatory Programs for ILC-Th Plasticity
When examined ex vivo, functional polarization appears to be an irreversible fate decision coinciding with (a) a loss of chromatin bivalency (Figure 1), (b) acquisition of permissive chromatin at subset-specific regulatory circuits, and (c) deposition of repressive chromatin modifications at alternative fate loci, thus suppressing functional plasticity. However, certain innate and adaptive lymphoid cells retain at least a degree of functional plasticity in vivo. In these cases, the immature helper-type cell, upon receiving specific signals, can revise its chromatin landscape to that of another subset, conferring the immune system a degree of functional flexibility. Fate-mapping studies using LDTF reporter loci reveal conversion within some ILC or Th subsets, usually promoted by specific proinflammatory conditions. For example, single-cell chromatin profiling revealed that immature skin ILC2s retain a degree of accessibility at some enhancers containing RORC motifs, normally a characteristic of ILC3s (105). In an IL-23 injection model of psoriasis, these enhancers acquire additional accessibility, while other ILC2 enhancers lose accessibility. The converted ILC2s ultimately contribute to ~10% of the ILC3 population in psoriatic lesions (105).
Type 3 lymphoid cells also retain at least some degree of functional plasticity. As an example, in mice with sustained inflammation, most T-bet+ Th1 cells were fate mapped as once being RORC+ Th17 cells (106-109), indicative of a Th17-to-Th1 conversion. Similarly, ILC3s can produce a spectrum of T-bet+ cells in response to their tissue microenvironments (110, 111), indicating a capacity for ILC3s to become ILC1s. One function of helper cell plasticity may be to control immunological tone within a tissue, which, when dysregulated, renders animals susceptible to disease and autoimmunity. For example, IFN-γ+ ILC3s are enriched in the inflamed colon of Crohn disease patients (112, 113) and promote IFNγ-dependent colitis during Campylobacter jejuni infection or anti-CD40 administration (114, 115).
Epigenetic profiling has been an important approach for deciphering the precise mechanisms that govern Th17-to-Th1 and ILC3-to-ILC1 conversion (Figure 3b). In one example, we used identification of superenhancers (15)—contiguous regions decorated by H3K27ac and harboring multiple powerful enhancer elements (116)—to find genes that help establish ILC and Th fates. Superenhancers associated with two genes, the surface receptor gene CD300LF and the TF gene IKZF3 (encodes Aiolos), emerged as potential markers for cells transitioning from ILC3s (CD300LF+Aiolos−) to ILC1s (CD300LF−Aiolos+) (16, 117). In follow-up studies, we found that Aiolos binds putative IL22 enhancers, reducing H3K27ac levels during the transition from ILC3 to ILC1 (117, 118).
Although sharing some TFs, the regulatory programs that govern functional plasticity in Th17 cells and ILC3s are characterized by distinct molecular targets. Both ILC3s and Th17 cells utilize RBPJ, a TF downstream of Notch signaling, and cMAF, as plasticity determinants. Within ILC3, cMAF and RBPJ suppress T-bet expression (119-122). In contrast, Th17 cells utilize these TFs to adopt an IL-10+ regulatory phenotype (123, 124), which has not been identified reproducibly in ILC3s (102). One outstanding question that remains is why type 1 cells, unlike their type 2 and 3 counterparts, appear to lack functional plasticity. The key to resolving this question from mechanistic and biological standpoints will lie in more directed profiling of regulatory programs and their perturbation in Th1 cells, ILC1s, and NK cells.
Shared NK-CD8 Regulatory Programs
Similar to ILC-Th parallels, in many regards NK cells can be viewed as Tc cells that lack a TCR-dependent mode of activation. Upon initial encounter with antigen, Tc cells transition to memory and effector phenotypes, which ensures the generation of both self-renewing cells and highly cytolytic killers, respectively. A similar phenotypic divide occurs during the course of NK differentiation, in which these innate lymphocytes initially exhibit features common to naive or central memory CD8+ cells (e.g., lymphatic homing and self-renewal) but also can differentiate into potent killers resembling effector Tc cells.
For CD8+ cells, the naive stage is characterized by bivalent chromatin marks at promoters of effector genes, which are marked by both repressive (e.g., H3K27me3) and permissive (e.g., H3K4me3) histone modifications (126). Upon infection, these promoters lose their repressive marks, but nearby enhancers gain accessibility and H3K4me1/2, which allows epigenetic commitment to memory and effector fates (126). As expected, TCR and coreceptor stimulation leads to activation of enhancers bearing AP-1 and NFAT motifs (127, 128). A subset of TCR-mediated expression changes are epigenetically unstable, reverting upon antigen withdrawal. However, the stimulated CD8+ cells stably activate a cohort of enhancers containing RUNX and T-box motifs, which drives gene modules for memory and effector functions (129). Importantly, RUNX3, along with T-box factors EOMES and T-bet, is critical for sculpting regulatory programs in both mature CD8+ and NK cells, generating REs associated with memory loci that are both accessible and marked with H3K27ac (130-132).
Integrative chromatin-transcriptome analyses of human lymphoid cells revealed additional classes of enhancers in NK and CD8 regulatory programs that distinguish self-renewing and dedicated effector cells (15, 133, 134) (Figure 4). In self-renewing CD8+ cells (naive and central memory) or phenotypically equivalent NK cells (CD27+CD11b− in mice and CD56bright in humans), TCF1, EOMES, ZEB1, and MYC are expressed, and target enhancers are enriched for their representative motifs (15, 120, 136, 138, 139). Within the NK cell lineage, TCF1 promotes the expression of cytokine receptor genes, including Il7r, and restrains effector functions, preventing NK cell maturation and granzyme expression (140). However, inactivation of the TCF7 gene (encoding TCF1) in CD8+ or NK cells produces relatively minor phenotypes during acute infection, likely because TCF1 has a close functional paralog called LEF1 that is expressed at lower levels (141). Notwithstanding, TCF1/LEF1 factors clearly play a pioneering role in activating REs that maintain self-renewal and helper phenotypes in CD8+ and NK cells, but they may also contribute to repression of effector-linked REs via intrinsic HDAC activity (23, 142).
Figure 4.

Regulatory programs conferring effector versus memory phenotypes, and tissue residency in NK and CD8+ T cells. Diagram of tissue migration patterns and TFs shared by NK cells and CD8+ T cells. (Left) CD56bright NK cells (CD27+ NK cells in mice) and naive and memory CD8+ T cells, which have a reduced potential for cytotoxicity compared to effectors. These cells home to the lymph nodes and express TCF1. (Right) CD56dim NK cells (CD11b+ NK cells in mice) and effector CD8+ T cells express high levels of cytotoxic effector molecules, are abundant in the circulation, and express the TF BLIMP1. Other TFs that mediate regulatory programs for cellular phenotypes, or mutually repress alternative phenotypes, are listed in the center. Select TFs contributing to tissue residency (e.g., mucosal lining) of memory CD8+ T cells and ILC1-like NK cells are shown at the bottom. Abbreviations: ILC1, group 1 innate lymphoid cell; NK, natural killer; TF, transcription factor.
Upon activation and differentiation, effector CD8+ and NK cells upregulate a new set of TFs, including T-bet, ZEB2, and BLIMP1 (143, 144). These components of the effector program cooperatively activate new genes and silence TCF7 and its downstream self-renewal modules. The latter requires DNMT3-dependent methylation of TCF7 and is accompanied by H3K9me3 chromatin deposition across SELL (encoding CD62L, which mediates lymphatic homing) and IL7R (mediates self-renewal) (145, 146). Accordingly, CD8+ cells deficient in the histone methyltransferase SUV39H1, which imparts H3K9me3, fail to suppress memory programs (145). In addition to the CD8-NK axis, it is important to note that the TCF1-BLIMP1 counterbalance in programs associated with self-renewal versus quiescence is highly conserved through evolution (mouse to macaques to humans) and is applied in numerous lymphoid populations. The latter include Th1 versus Tfh CD4+ cells, memory versus exhausted cells in several lineages, and tissue-resident versus memory populations of lymphoid cells (147).
CD8-NK Memory and Tissue-Residency Programs
Conventional NK cells are short-lived terminal effectors, incapable of rapid expansion and, in several important ways, are epigenetically distinct from CD8+ cells. However, during murine cytomegalovirus (CMV) infection, adaptive-like NK cells bearing the Ly49H activation receptor are generated alongside their CD8+ T cell counterparts (147a). This NK population exhibits many CD8-like characteristics, including clonal-like expansion. In humans, CMV infection produces a similar NK subset, identified by upregulation of the NKG2C receptor (148), though it is unclear whether NKG2C participates as an activation receptor in recognition of human CMV–infected cells, akin to Ly49H recognition of murine CMV–infected cells in mice. Consistent with their phenotypic overlap, transcriptomes and regulomes of adaptive-like NK and CD8+ effector cells are highly similar, in humans and mice (133).
Clonal expansion programs in CD8+ and adaptive NK cells depend on signaling downstream of the proinflammatory cytokine IL-12, in which STAT4 serves as a pioneer factor to open chromatin at a shared cohort of REs (149), including those controlling additional TFs (150-152). The key TFs in this shared, adaptive NK-CD8 program have been identified as T-bet and EOMES, which function as master regulators of NK and CD8+ cells; IRF8 and RUNX3, which control NK and CD8+ cell proliferative bursts, and ZBTB32, which regulates proliferation and killing capacity (153-156). Only the latter TF appears to have some opposing roles in CD8+ versus NK cells. ZBTB32 limits CD8+, but not NK, cell expansion, possibly due to ZBTB32-mediated repression of BLIMP1 in NK cells (153, 157). Post-CMV infection, epigenetically altered NK cells return to a TCF1− basal state, unlike CD8+ memory cells, which become long-lived TCF1+ cells (151). Indeed, memory and memory-like populations produced from the two lymphoid lineages are clearly distinct, consistent with the observation that ILCs and T cells share some, but not all functional circuitry.
Similar to effector and memory programs, chromatin landscapes of tissue-resident CD8+ and NK cells are established by RUNX3, TCF1, and BLIMP1, or their paralogs (Figure 4). Upon exposure to TGF-β and IL-15, effectors in both lineages will convert into tissue-resident cells that are poorly cytotoxic but have a metabolic platform supporting robustness in mucosal or tissue microenvironments (158, 159). For NK cells, the converted populations are commonly called ILC1s, although a precise nomenclature remains controversial due to an absence of unambiguous NK versus ILC1 markers in humans (158). Active enhancers within ILC1s from oral mucosae are enriched for a cohort of TF motifs corresponding to SMADs, AP-1, and RUNX family members, while being depleted for BLIMP1 motifs (16). This TF profile likely corresponds to a generalized pathway in which strong TGF-β signaling triggers noncanonical SMADs to activate the appropriate adhesion genes. Conversely, repressors, such as BLIMP1, or its homolog HOBIT, silence REs that coordinate expression modules for circulation and cytotoxicity. However, one must be careful not to universalize these regulatory models given the wide range of variation when CD8+ and NK cells reside in different tissues (160, 161). A possible exception to this cautionary note may be RUNX3, which has been shown critical for establishing chromatin accessibility in resident CD8+ T cells across five tissues (162). These programs are also evident within tumor-associated CD8+ cells, wherein RUNX3 controls the activation of residency-associated enhancer elements (162). However, tumor-resident Tc cells must contend with chronic stimulation, which engages additional regulatory programs that strike the delicate balance between effector functions and exhaustion.
Regulatory Programming of Exhaustion
After acute exposure, a subset of antigen-experienced CD8+ T cells dedifferentiate into long-lived TCF1+ memory cells (163, 164). However, during chronic viral infections or cancer, this pathway is perturbed and the cells enter an alternative state, often referred to as exhausted or dysfunctional. CD8 exhaustion is characterized by a progressive decline in effector functions, reduced proliferation potential, and upregulation of inhibitory receptors, including PD1, TIM3, CTLA4, TIGIT, and LAG3. Integrated chromatin-transcriptome analysis revealed that exhausted CD8+ T (Tex) cells have a landscape distinct from that of their effector and memory counterparts (165-170a).
Acquisition of a fully exhausted phenotype is progressive, transitioning through several identifiable subpopulations, including (a) stem-like progenitors (PD1+Ly108+TCF1+) that retain proliferation potential and effector functions, (b) intermediate Tex cells (PD1+CX3CR1+T-bethi) that are highly cytolytic; and (c) fully exhausted Tex cells (PD1+TIM3+TCF1−T-betlo) characterized by a complete loss of proliferative potential and effector cytokine production (171-171b). Progression through this pathway is driven by underlying changes in TFs and epigenetic programs, which become more irreversible, ultimately enforcing a heritable dysfunctional state in terminally differentiated Tex cells. Indeed, profiling of the open chromatin landscape within Tex subsets revealed a significant reduction of chromatin accessibility at genes regulating effector function and self-renewal capacity (e.g., Ifng, Tbx21, Tcf7, and Lef1) during progression to the fully exhausted state (171-171b).
A recent study showed that de novo DNA methylation, mediated by the DNMT3A enzyme, is a critical component of the commitment to terminal T cell exhaustion (166). Accordingly, lymphocytic choriomeningitis virus (LCMV)-specific CD8+ T cells from Dnmt3a conditional knockout mice fail to lose effector functions or proliferation capacity, despite persistent antigen stimulation during chronic infection. Instead, Dnmt3a-deficient CD8+ T cells remain in a reprogrammable, TCF-1+ stem-like state (166). This epigenetically reprogrammable state is of considerable therapeutic interest, since stem-like progenitor and transitory Tex cells are the main subsets that respond to immune checkpoint blockade used in cancer treatments. Thus, an in-depth understanding of molecular mechanisms that preserve functional plasticity in Tex cells, preventing their terminally exhausted fate, may enhance current checkpoint regimens or the efficacy of adoptive cell therapies [e.g., chimeric antigen receptor (CAR) T cells].
Master regulators of T cell exhaustion remain largely unknown. Although transcriptional profiling has implicated the differential expression of multiple TFs in Tex subsets (e.g., T-bet, EOMES, TCF1, IRF4, BATF, TOX, NR4A), some of these factors also are essential for differentiation of functional effector and memory CD8+ cells (156, 172-181). Indeed, single-cell analysis revealed a striking degree of similarity in the transcriptional signatures of T cell exhaustion and activation (182). These paradoxical findings indicate that transcriptional analysis alone cannot fully dissect master regulators of lineage commitment versus exhaustion.
As one example, TCR-induced NFAT drives opposing transcriptional programs in T cells. In the absence of AP-1 cooperation, NFAT initiates regulatory programs leading to a hyporesponsive state (27, 167, 168). The activation of NFAT also drives expression of the high mobility group box (HMG-box) TF TOX, which primes chromatin opening as effector T cells transition to TCF1+ progenitor Tex cells (183-185). However, once established, TOX expression and its downstream programs are maintained in an NFAT-independent manner (185). The sustained upregulation of TOX in antigen-specific CD8+ T cells during chronic viral infections is likely enforced by a unique DNA demethylation landscape at the Tox locus (184). Importantly, recent studies showed that genetic deletion of Tox initially enhances effector cytokine production in chronically stimulated CD8+ T cells but eventually induces a massive loss of Tex cells (183-185). Notably, in TOX-deficient T cells, chromatin accessibility is increased at a cohort of T effector genes, such as Gzmb, Klrg1, Zeb2, and Nr4a1, with reduced accessibility observed at genes associated with Tex progenitor programs, including Tcf7, Bach2, and Ikzf2 (185). These data indicate that targeting TOX may not be an effective therapeutic approach, given its counteractive functions in exhaustion and survival.
In addition to its role in modulating TOX, NFAT induces expression of the NR4A nuclear receptor family of TFs in hyporesponsive T cells. Although the molecular mechanisms integrating these regulatory activities are still unfolding, published studies highlight the potential for altering TF expression to unleash a T cell response against cancer. Illustrating this point, deletion of NR4A in CAR T cell models limited tumor growth, presumably via its ability to enhance chromatin accessibility at T effector REs enriched for bZIP and Rel/NF-κB motifs (186, 187). On the flip side, during chronic LCMV infection, BACH2 epigenetically represses regulatory programs for terminal exhaustion while promoting those for stem-like CD8+ T cell differentiation (188).
Intriguingly, the concept of dysfunctionality or exhaustion is no longer confined to T cells, with similar states reported for NK cells and ILC2s, albeit the underlying molecular mechanisms remain obscure (189, 190). Chronic viral infection and tumor studies have highlighted that dysfunctional NK cells have limited cytotoxic activity, elevated expression of inhibitory molecules, lower levels of activating receptors, and impaired cytokine production (189, 191). Similar to T cell dysfunction, these effects rely upon epigenetic reprogramming. Genome-wide profiles of human NK cells isolated from CMV-infected individuals, which were chronically stimulated ex vivo with NKG2C activating antibodies, revealed dramatic changes in DNA methylation. Notably, the promoters of LAG3, PDCD1, and TIGIT, which encode inhibitory molecules and are associated with the PD-1 signaling pathway, and genes like IKZF3, NFATC2, TOX, and ZBTB38, encoding TFs and epigenetic regulators, were hypomethylated compared with control NK cells from CMV-negative sources (189). Similarly, Tex cells maintain a demethylated state at the PDCD1 locus during chronic viral infections, including LCMV and HIV infections (192). Even after diminishing viral load, PDCD1 remains unmethylated, allowing Tex cells to rapidly reexpress high levels of PD-1 upon TCR restimulation. Given the growing importance of NK and CD8+ T cells in cancer therapies, continued studies to identify key regulatory circuits controlling exhaustion-associated genes may provide new opportunities for improving their efficacies.
LOOKING TO THE FUTURE
The advent of single-cell expression and chromatin-based assays is revolutionizing how we understand immune cell identity and heterogeneity. These techniques were originally limited to transcriptome studies, but single-cell microfluidics have recently been commercialized as easy-to-use kits, which has democratized the utility of this approach. Processed data from single-cell approaches, be they transcriptomes or epigenomes, can computationally identify new cell populations, impute branch points in cell fates, and deconvolute heterogeneity as it arises during development or disease, in terms of both discrete cell types and distinct states of a given cell type (some refer to the latter as metacells) (134).
In this context, single-cell transcriptomics and accessibility landscapes have fundamentally challenged the notion of discrete functional lymphoid subsets. For example, while Th cell subsets generated ex vivo by strong polarizing cytokines resolve as distinct clusters, when Th cells are characterized in Salmonella (type 1 polarizing), Nippostrongylus (type 2 polarizing), or Citrobacter (type 3 polarizing) pathogen models by expression or motif imprinting of their TFs, single-cell RNA sequencing (scRNA-seq) and scATAC-seq reveal a continuum of helper cell populations along each polarization pathway (193). Within this functional spectrum exist cells that have regulatory landscapes and programs merging all three Th subsets. In addition to mouse infection models, experimental asthma produces a continuum of Th1 and Th2 programs within lungs, and analysis of inflamed human gut resolves ILC1s and ILC3s as a gradient of programs (117, 194). One caveat to acknowledge when considering the lack of clear functional subsets is the inherent biases of single-cell omics, stemming from low cell numbers in scarce states or incomplete representation of all genes in expression data. However, proteome-based analyses, like cytometry by time of flight (CyTOF), of T effectors generated in large numbers during in vivo infections again yield a continuum of cells (106). Thus, it appears that infection and inflammation can direct regulatory programs for cells within a given microenvironment to converge on a functional phenotype, presumably to either clear an infection or repair inflammation-induced damage.
In terms of deciphering gene regulatory circuits within cell states, the most exciting approaches to emerge are multi-omics, which combine expression and chromatin profiling at the single-cell level of resolution. For example, cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) combines RNA and protein expression (via sequence-tagged antibodies), which is useful to categorize cell types based on signature protein markers whose mRNAs are expressed at minimal steady-state levels and often drop out at the lower depth of scRNA-seq (195). One of the most commonly used multi-omics platform is now scRNA-ATAC, which will be extremely helpful for dissecting regulatory circuits in rare cell types or states, as well as identifying enhancers that are important for transitions between states (196, 197). As the transposase-based CUT-and-Tag (cleavage under targets and tagmentation) technique improves, it may one day be possible to examine chromatin binding profiles of nuclear factors at a single-cell level (198). In this context, a determination of the TF networks that cement cell states or facilitate transitions between them will benefit from data that emerge from perturb-seq approaches, which use CRISPR methodologies, coupled with either scRNA-seq or ATAC-seq (195, 199). Of course, the complexity of data sets from these techniques, and their analyses, is increasing dramatically. Continual progress in machine learning methods will go hand-in-hand with technical advances as we continue our journey to unravel the intricacies of circuits that drive expression programs in our immune systems, especially those that provide the requisite flexibility to face an ever-evolving universe of pathogens. Such insights will drive advances in the engineering of cellular therapeutics to activate or repress expression modules for more efficacious treatments of cancer, infectious diseases, and chronic inflammation.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) grants RO1 AI134035, CA188286, AI130231, and AI118852 (E.M.O.) and R21 AI156411 (P.L.C.); The Ohio State University Pelotonia Fellowship Program (A.S.), and National Cancer Institute K22 Career Transition Award K22CA241290 (C.-W.J.L.). Given the space constraints, we apologize to our colleagues whose work we have not mentioned, especially the many seminal studies on immune cell development, which have been covered in several recent reviews (28–38).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Sartorelli V, Lauberth SM. 2020. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol 27(6):521–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, et al. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaudhri VK, Dienger-Stambaugh K, Wu Z, Shrestha M, Singh H. 2020. Charting the cis-regulome of activated B cells by coupling structural and functional genomics. Nat. Immunol 21(2):210–20 [DOI] [PubMed] [Google Scholar]
- 3a.Holliday R, Pugh JE. 1975. DNA modification mechanisms and gene activity during development. Science 187(4173):226–32 [PubMed] [Google Scholar]
- 3b.Riggs AD. 1975. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 14(1):9–25 [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403(6765):41–45 [DOI] [PubMed] [Google Scholar]
- 5.Rothbart SB, Strahl BD. 2014. Interpreting the language of histone and DNA modifications. Biochem. Biophys. Acta Gene Reg. Mech 1839(8):627–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293(5532):1074–80 [DOI] [PubMed] [Google Scholar]
- 7.Myers FA, Evans DR, Clayton AL, Thorne AW, Crane-Robinson C. 2001. Targeted and extended acetylation of histones H4 and H3 at active and inactive genes in chicken embryo erythrocytes. J. Biol. Chem 276(23):20197–205 [DOI] [PubMed] [Google Scholar]
- 8.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. PNAS 107(50):21931–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, et al. 2018. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 24(7):1722–29 [DOI] [PubMed] [Google Scholar]
- 10.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. 2003. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. PNAS 100(4):1820–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang G, Lin JCY, Wei V, Yoo C, Cheng JC, et al. 2004. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. PNAS 101(19):7357–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrera LO, Li Z, Smith AD, Arden KC, Cavenee WK, et al. 2008. Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome Res. 18(1):46–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, et al. 2019. The cis-regulatory atlas of the mouse immune system. Cell 176(4):897–912.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abascal F, Acosta R, Addleman NJ, Adrian J, Afzal V, et al. 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583(7818):699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins PL, Cella M, Porter SI, Li S, Gurewitz GL, et al. 2019. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell 176(1–2):348–60.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koues OI, Collins PL, Cella M, Robinette ML, Porter SI, et al. 2016. Distinct gene regulatory pathways for human innate versus adaptive lymphoid cells. Cell 165(5):1134–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brind’Amour J, Liu S, Hudson M, Chen C, Karimi MM, Lorincz MC. 2015. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun 6:6033. [DOI] [PubMed] [Google Scholar]
- 18.Skene PJ, Henikoff JG, Henikoff S. 2018. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13(5):1006–19 [DOI] [PubMed] [Google Scholar]
- 19.Deleted in proof
- 20.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7):1665–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kempfer R, Pombo A. 2020. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet 21(4):207–26 [DOI] [PubMed] [Google Scholar]
- 22.Roels J, Kuchmiy A, De Decker M, Strubbe S, Lavaert M, et al. 2020. Distinct and temporary-restricted epigenetic mechanisms regulate human αβ and γδ T cell development. Nat. Immunol 21(10):1280–92 [DOI] [PubMed] [Google Scholar]
- 23.Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, et al. 2018. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity 48(2):243–57.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ungerbäck J, Hosokawa H, Wang X, Strid T, Williams BA, et al. 2018. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res. 28(10):1508–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majumder K, Koues OI, Chan EAW, Kyle KE, Horowitz JE, et al. 2015. Lineage-specific compaction of Tcrb requires a chromatin barrier to protect the function of a long-range tethering element. J. Exp. Med 212(1):107–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopalakrishnan S, Majumder K, Predeus A, Huang Y, Koues OI, et al. 2013. Unifying model for molecular determinants of the preselection Vβ repertoire. PNAS 110(34):E3206–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, et al. 2015. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42(2):265–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nutt SL, Kee BL. 2007. The transcriptional regulation of B cell lineage commitment. Immunity 26(6):715–25 [DOI] [PubMed] [Google Scholar]
- 40.Hart GT, Hogquist KA, Jameson SC. 2012. Krüppel-like factors in lymphocyte biology. J. Immunol 188(2):521–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Igarashi K, Ochiai K, Itoh-Nakadai A, Muto A. 2014. Orchestration of plasma cell differentiation by Bach2 and its gene regulatory network. Immunol. Rev 261(1):116–25 [DOI] [PubMed] [Google Scholar]
- 42.Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. 2020. The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat. Immunol 21(9):1082–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, et al. 2013. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38(5):918–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. 2006. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25(2):225–36 [DOI] [PubMed] [Google Scholar]
- 45.Crotty S, Johnston RJ, Schoenberger SP. 2010. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol 11(2):114–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi J, Crotty S. 2021. Bcl6-mediated transcriptional regulation of follicular helper T cells (TFH). Trends Immunol. 42(4):336–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy K, Mitchell S, Liu Y, Ohta S, Lin Y-S, et al. 2019. A regulatory circuit controlling the dynamics of NFκB cRel transitions B cells from proliferation to plasma cell differentiation. Immunity 50(3):616–28.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patterson DG, Kania AK, Zuo Z, Scharer CD, Boss JM. 2021. Epigenetic gene regulation in plasma cells. Immunol. Rev 303(1):8–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manakkat Vijay GK, Singh H. 2021. Cell fate dynamics and genomic programming of plasma cell precursors. Immunol. Rev 303(1):62–71 [DOI] [PubMed] [Google Scholar]
- 50.Trezise S, Nutt SL. 2021. The gene regulatory network controlling plasma cell function. Immunol. Rev 303(1):23–34 [DOI] [PubMed] [Google Scholar]
- 51.Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339(6123):1074–77 [DOI] [PubMed] [Google Scholar]
- 52.Barwick BG, Scharer CD, Bally APR, Boss JM. 2016. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat. Immunol 17(10):1216–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scharer CD, Barwick BG, Guo M, Bally APR, Boss JM. 2018. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat. Commun 9(1):1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujii K, Tanaka S, Hasegawa T, Narazaki M, Kumanogoh A, et al. 2020. Tet DNA demethylase is required for plasma cell differentiation by controlling expression levels of IRF4. Int. Immunol 32(10):683–90 [DOI] [PubMed] [Google Scholar]
- 55.Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, et al. 2018. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov. 8(12):1633–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lio CWJ, Shukla V, Samaniego-Castruita D, González-Avalos E, Chakraborty A, et al. 2019. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci. Immunol 4(34):eaau7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Onodera A, González-Avalos E, Lio C-WJ, Georges RO, Bellacosa A, et al. 2021. Roles of TET and TDG in DNA demethylation in proliferating and non-proliferating immune cells. Genome Biol. 22(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, et al. 2009. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325(5943):1006–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu D, Rao S, Tsai LM, Lee SK, He Y, et al. 2009. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31(3):457–68 [DOI] [PubMed] [Google Scholar]
- 60.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, et al. 2009. Bcl6 mediates the development of T follicular helper cells. Science 325(5943):1001–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi J, Diao H, Faliti CE, Truong J, Rossi M, et al. 2020. Bcl-6 is the nexus transcription factor of T follicular helper cells via repressor-of-repressor circuits. Nat. Immunol 21(7):777–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moriyama S, Takahashi N, Green JA, Hori S, Kubo M, et al. 2014. Sphingosine-1-phosphate receptor 2 is critical for follicular helper T cell retention in germinal centers. J. Exp. Med 211(7):1297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu G, Guy CS, Chapman NM, Palacios G, Wei J, et al. 2021. Metabolic control of TFH cells and humoral immunity by phosphatidylethanolamine. Nature 595(7869):724–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, et al. 2020. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol 21(3):331–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, et al. 2014. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol 15(10):957–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lemonnier F, Couronné L, Parrens M, Jaïs JP, Travert M, et al. 2012. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 120(7):1466–69 [DOI] [PubMed] [Google Scholar]
- 67.Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, et al. 2018. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat. Commun 9(1):1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sage PT, Sharpe AH. 2016. T follicular regulatory cells. Immunol. Rev 271(1):246–59 [DOI] [PubMed] [Google Scholar]
- 69.Kennedy DE, Okoreeh MK, Maienschein-Cline M, Ai J, Veselits M, et al. 2020. Novel specialized cell state and spatial compartments within the germinal center. Nat. Immunol 21(6):660–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, et al. 2010. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143(4):592–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Danilova N. 2012. The evolution of adaptive immunity. Adv. Exp. Med. Biol 738:218–35 [DOI] [PubMed] [Google Scholar]
- 72.Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, et al. 2016. Trained immunity: a program of innate immune memory in health and disease. Science 352(6284):aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, et al. 2017. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550(7677):475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Jacobs C, et al. 2014. BCG-induced trained immunity in NK cells: role for non-specific protection to infection. Clin. Immunol 155(2):213–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, et al. 2021. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat. Immunol 22(1):2–6. Erratum. 2021. Nat. Immunol. 22:928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Netea MG, van der Meer JWM. 2017. Trained immunity: an ancient way of remembering. Cell Host Microbe 21(3):297–300 [DOI] [PubMed] [Google Scholar]
- 77.Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang SY, et al. 2018. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 23(1):89–100.e5 [DOI] [PubMed] [Google Scholar]
- 78.Schnack L, Sohrabi Y, Lagache SMM, Kahles F, Bruemmer D, et al. 2019. Mechanisms of trained innate immunity in oxLDL primed human coronary smooth muscle cells. Front. Immunol 10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schrum JE, Crabtree JN, Dobbs KR, Kiritsy MC, Reed GW, et al. 2018. Cutting edge: Plasmodium falciparum induces trained innate immunity. J. Immunol 200(4):1243–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, et al. 2018. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 172(1–2):162–75.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.You M, Chen L, Zhang D, Zhao P, Chen Z, et al. 2021. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol 23(6):620–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fang TC, Schaefer U, Mecklenbrauker I, Stienen A, Dewell S, et al. 2012. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med 209(4):661–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. 2007. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of Polycomb-mediated gene silencing. Cell 130(6):1083–94 [DOI] [PubMed] [Google Scholar]
- 88a.Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, et al. 2014. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buffen K, Oosting M, Quintin J, Ng A, Kleinnijenhuis J, et al. 2014. Autophagy controls BCG-induced trained immunity and the response to intravesical BCG therapy for bladder cancer. PLOS Pathog. 10(10):e1004485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fanucchi S, Fok ET, Dalla E, Shibayama Y, Börner K, et al. 2019. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat. Genet 51(1):138–50 [DOI] [PubMed] [Google Scholar]
- 91.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, et al. 2012. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12(2):223–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, et al. 2012. Bacille Calmette-Guérin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. PNAS 109(43):17537–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, et al. 2018. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172(1–2):176–90.e19 [DOI] [PubMed] [Google Scholar]
- 94.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, et al. 2018. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172(1–2):147–61.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, et al. 2020. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe 28(2):322–34.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Laval B, Maurizio J, Kandalla PK, Brisou G, Simonnet L, et al. 2020. C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell 26(5):657–74.e8 [DOI] [PubMed] [Google Scholar]
- 97.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, et al. 2014. MTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345(6204):1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arts RJW, Novakovic B, ter Horst R, Carvalho A, Bekkering S, et al. 2016. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 24(6):807–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98a.Fan H, Cook JA. 2004. Molecular mechanism of endotoxin tolerance. J. Endotoxin Res 10(2):71–84 [DOI] [PubMed] [Google Scholar]
- 98b.West MA, Heagy W. 2002. Endotoxin tolerance: A review. Crit. Care Med 30(1 Suppl.):S64–73 [PubMed] [Google Scholar]
- 98c.DiNardo AR, Netea MG, Musher DM. 2021. Postinfectious epigenetic immune modifications—a double-edged sword. N. Engl. J. Med 384(3):261–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98d.Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, et al. 2016. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167(5):1354–68.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, et al. 2009. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30(1):155–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hawkins RD, Larjo A, Tripathi SK, Wagner U, Luu Y, et al. 2013. Global chromatin state analysis reveals lineage-specific enhancers during the initiation of human T helper 1 and T helper 2 cell polarization. Immunity 38(6):1271–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, et al. 2012. STATs shape the active enhancer landscape of T cell populations. Cell 151(5):981–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101a.Dong C. 2021. Cytokine regulation and function in T cells. Annu. Rev. Immunol 39:51–76 [DOI] [PubMed] [Google Scholar]
- 102.Bando JK, Gilfillan S, Di Luccia B, Fachi JL, Sécca C, et al. 2020. ILC2s are the predominant source of intestinal ILC-derived IL-10. J. Exp. Med 217(2):e20191520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, et al. 2018. Innate lymphoid cells: 10 years on. Cell 174(5):1054–66 [DOI] [PubMed] [Google Scholar]
- 104.Shih HY, Sciumè G, Mikami Y, Guo L, Sun HW, et al. 2016. Developmental acquisition of regulomes underlies innate lymphoid cell functionality. Cell 165(5):1120–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bielecki P, Riesenfeld SJ, Hütter JC, Torlai Triglia E, Kowalczyk MS, et al. 2021. Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature 592(7852):128–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tortola L, Jacobs A, Pohlmeier L, Obermair FJ, Ampenberger F, et al. 2020. High-dimensional T helper cell profiling reveals a broad diversity of stably committed effector states and uncovers interlineage relationships. Immunity 53(3):597–613.e6 [DOI] [PubMed] [Google Scholar]
- 107.Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. 2015. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. PNAS 112(22):7061–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, et al. 2009. Late developmental plasticity in the T helper 17 lineage. Immunity 30(1):92–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, et al. 2011. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol 12(3):255–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nussbaum K, Burkhard SH, Ohs I, Mair F, Klose CSN, et al. 2017. Tissue microenvironment dictates the fate and tumorsuppressive function of type 3 ILCs. J. Exp. Med 214(8):2331–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mazzoni A, Maggi L, Liotta F, Cosmi L, Annunziato F. 2019. Biological and clinical significance of T helper 17 cell plasticity. Immunology 158(4):287–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, et al. 2013. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38(4):769–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernink JH, Peters CP, Munneke M, Te Velde AA, Meijer SL, et al. 2013. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol 14(3):221–29 [DOI] [PubMed] [Google Scholar]
- 114.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, et al. 2006. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25(2):309–18 [DOI] [PubMed] [Google Scholar]
- 115.Muraoka WT, Korchagina AA, Xia Q, Shein SA, Jing X, et al. 2021. Campylobacter infection promotes IFNγ-dependent intestinal pathology via ILC3 to ILC1 conversion. Mucosal Immunol. 14(3):703–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SCJ, et al. 2015. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520(7548):558–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cella M, Gamini R, Sécca C, Collins PL, Zhao S, et al. 2019. Subsets of ILC3–ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol 20(8):980–91. Erratum. 2019. Nat. Immunol. 20(10):1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mazzurana L, Forkel M, Rao A, Van Acker A, Kokkinou E, et al. 2019. Suppression of Aiolos and Ikaros expression by lenalidomide reduces human ILC3–ILC1/NK cell transdifferentiation. Eur. J. Immunol 49(9):1344–55 [DOI] [PubMed] [Google Scholar]
- 119.Chea S, Perchet T, Petit M, Verrier T, Guy-Grand D, et al. 2016. Notch signaling in group 3 innate lymphoid cells modulates their plasticity. Sci. Signal 9(426):ra45. [DOI] [PubMed] [Google Scholar]
- 120.Pokrovskii M, Hall JA, Ochayon DE, Yi R, Chaimowitz NS, et al. 2019. Characterization of transcriptional regulatory networks that promote and restrict identities and functions of intestinal innate lymphoid cells. Immunity 51(1):185–97.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Parker ME, Barrera A, Wheaton JD, Zuberbuehler MK, Allan DSJ, et al. 2020. c-Maf regulates the plasticity of group 3 innate lymphoid cells by restraining the type 1 program. J. Exp. Med 217(1):e20191030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tizian C, Lahmann A, Hölsken O, Cosovanu C, Kofoed-Branzk M, et al. 2020. c-Maf restrains T-bet-driven programming of CCR6-negative group 3 innate lymphoid cells. eLife 9:e52549. Erratum. 2020. eLife 9:e56774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu J, Yang Y, Qiu G, Lal G, Wu Z, et al. 2009. c-Maf regulates IL-10 expression during Th17 polarization. J. Immunol 182(10):6226–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Meyer zu Horste G, Wu C, Wang C, Cong L, Pawlak M, et al. 2016. RBPJ controls development of pathogenic Th17 cells by regulating IL-23 receptor expression. Cell Rep. 16(2):392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Deleted in proof
- 126.Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, et al. 2014. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8+ T cell differentiation. Immunity 41(5):853–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yukawa M, Jagannathan S, Vallabh S, Kartashov AV, Chen X, et al. 2020. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med 217(1):e20182009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bevington SL, Cauchy P, Piper J, Bertrand E, Lalli N, et al. 2016. Inducible chromatin priming is associated with the establishment of immunological memory in T cells. EMBO J. 35(5):515–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.van der Veeken J, Zhong Y, Sharma R, Mazutis L, Dao P, et al. 2019. Natural genetic variation reveals key features of epigenetic and transcriptional memory in virus-specific CD8 T cells. Immunity 50(5):1202–17.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Istaces N, Splittgerber M, Lima Silva V, Nguyen M, Thomas S, et al. 2019. EOMES interacts with RUNX3 and BRG1 to promote innate memory cell formation through epigenetic reprogramming. Nat. Commun 10(1):3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Levanon D, Negreanu V, Lotem J, Bone KR, Brenner O, et al. 2014. Transcription factor Runx3 regulates interleukin-15-dependent natural killer cell activation. Mol. Cell. Biol 34(6):1158–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang D, Diao H, Getzler AJ, Rogal W, Frederick MA, et al. 2018. The transcription factor Runx3 establishes chromatin accessibility of cis-regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity 48(4):659–74.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Holmes TD, Pandey RV, Helm EY, Schlums H, Han H, et al. 2021. The transcription factor Bcl11b promotes both canonical and adaptive NK cell differentiation. Sci. Immunol 6(57):eabc9801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McFarland AP, Yalin A, Wang SY, Cortez VS, Landsberger T, et al. 2021. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity 54(6):1320–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Deleted in proof
- 136.Zook EC, Li ZY, Xu Y, de Pooter RF, Verykokakis M, et al. 2018. Transcription factor ID2 prevents E proteins from enforcing a naïve T lymphocyte gene program during NK cell development. Sci. Immunol 3(22):eaao2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Deleted in proof
- 138.Wu JQ, Seay M, Schulz VP, Hariharan M, Tuck D, et al. 2012. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLOS Genet. 8(3):e1002565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lin WHW, Nish SA, Yen B, Chen YH, Adams WC, et al. 2016. CD8+ T lymphocyte self-renewal during effector cell determination. Cell Rep. 17(7):1773–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li ZY, Morman RE, Hegermiller E, Sun M, Bartom ET, et al. 2021. The transcriptional repressor ID2 supports natural killer cell maturation by controlling TCF1 amplitude. J. Exp. Med 218(6):e20202032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yu S, Zhou X, Steinke FC, Liu C, Chen SC, et al. 2012. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 37(5):813–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xing S, Li F, Zeng Z, Zhao Y, Yu S, et al. 2016. Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nat. Immunol 17(6):695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu T, Shin HM, Moseman EA, Ji Y, Huang B, et al. 2015. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep. 12(12):2099–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, et al. 2015. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med 212(12):2027–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, et al. 2018. The epigenetic control of stemness in CD8+ T cell fate commitment. Science 359(6372):177–86 [DOI] [PubMed] [Google Scholar]
- 146.Danilo M, Chennupati V, Silva JG, Siegert S, Held W. 2018. Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep. 22(8):2107–17 [DOI] [PubMed] [Google Scholar]
- 147.Shao P, Li F, Wang J, Chen X, Liu C, Xue H-H. 2019. Cutting edge: Tcf1 instructs T follicular helper cell differentiation by repressing Blimp1 in response to acute viral infection. J. Immunol 203(4):801–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147a.Sun JC, Beilke JN, Lanier LL. 2009. Adaptive immune features of natural killer cells. Nature 457(7229):557–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lopez-Vergès S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, et al. 2011. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. PNAS 108(36):14725–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wiedemann GM, Santosa EK, Grassmann S, Sheppard S, Le Luduec JB, et al. 2021. Deconvoluting global cytokine signaling networks in natural killer cells. Nat. Immunol 22(5):627–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sciumè G, Mikami Y, Jankovic D, Nagashima H, Villarino AV, et al. 2020. Rapid enhancer remodeling and transcription factor repurposing enable high magnitude gene induction upon acute activation of NK cells. Immunity 53(4):745–58.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lau CM, Adams NM, Geary CD, Weizman O, Rapp M, et al. 2018. Epigenetic control of innate and adaptive immune memory. Nat. Immunol 19(9):963–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. 2012. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J. Exp. Med 209(5):947–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Beaulieu AM, Zawislak CL, Nakayama T, Sun JC. 2014. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat. Immunol 15(6):546–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rapp M, Lau CM, Adams NM, Weizman O, O’Sullivan TE, et al. 2017. Core-binding factor and Runx transcription factors promote adaptive natural killer cell responses. Sci. Immunol 2(18):eaan3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Adams NM, Lau CM, Fan X, Rapp M, Geary CD, et al. 2018. Transcription factor IRF8 orchestrates the adaptive natural killer cell response. Immunity 48(6):1172–82.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, et al. 2005. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol 6(12):1236–44 [DOI] [PubMed] [Google Scholar]
- 157.Shin HM, Kapoor VN, Kim G, Li P, Kim HR, et al. 2017. Transient expression of ZBTB32 in anti-viral CD8+ T cells limits the magnitude of the effector response and the generation of memory. PLOS Pathog. 13(8):e1006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Colonna M. 2018. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity 48(6):1104–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Verbist KC, Cole CJ, Field MB, Klonowski KD. 2011. A role for IL-15 in the migration of effector CD8 T cells to the lung airways following influenza infection. J. Immunol 186(1):174–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, et al. 2016. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352(6284):459–63 [DOI] [PubMed] [Google Scholar]
- 161.Behr FM, Kragten NAM, Wesselink TH, Nota B, Van Lier RAW, et al. 2019. Blimp-1 rather than Hobit drives the formation of tissue-resident memory CD8+ T cells in the lungs. Front. Immunol 10:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, et al. 2017. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552(7684):253–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, et al. 2017. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552(7685):404–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, et al. 2017. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552(7685):362–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Abdel-Hakeem MS, Manne S, Beltra J-C, Stelekati E, Chen Z, et al. 2021. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol 22(8):1008–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, et al. 2017. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170(1):142–57.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Scott-Browne JP, López-Moyado IF, Trifari S, Wong V, Chavez L, et al. 2016. Dynamic changes in chromatin accessibility occur in CD8+ T cells responding to viral infection. Immunity 45(6):1327–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, et al. 2017. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. PNAS 114(13):E2776–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. 2012. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37(6):1130–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, et al. 2016. The epigenetic landscape of T cell exhaustion. Science 354(6316):1165–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170a.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, et al. 2016. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354:1160–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, et al. 2020. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52(5):825–41.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171a.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, et al. 2019. Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity 51:1043–58.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171b.Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, et al. 2021. BATF regulates progenitor to cytolytic effector CD8+ T cell transition during chronic viral infection. Nat. Immunol 22: 996–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Page N, Lemeille S, Vincenti I, Klimek B, Mariotte A, et al. 2021. Persistence of self-reactive CD8+ T cells in the CNS requires TOX-dependent chromatin remodeling. Nat. Commun 12(1):1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, et al. 2012. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338(6111):1220–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. 2010. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 33(2):229–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, et al. 2003. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302(5647):1041–43 [DOI] [PubMed] [Google Scholar]
- 176.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, et al. 2007. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27(2):281–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mann TH, Kaech SM. 2019. Tick-TOX, it’s time for T cell exhaustion. Nat. Immunol 20(9):1092–94 [DOI] [PubMed] [Google Scholar]
- 178.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, et al. 2017. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity 47(6):1129–41.e5 [DOI] [PubMed] [Google Scholar]
- 179.Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, et al. 2010. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med 16(10):1147–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, et al. 2016. T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45(2):415–27 [DOI] [PubMed] [Google Scholar]
- 181.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, et al. 2016. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537(7620):417–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, et al. 2016. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 166(6):1500–11.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, et al. 2019. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571(7764):270–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, et al. 2019. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571(7764):265–69 [DOI] [PubMed] [Google Scholar]
- 185.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, et al. 2019. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571(7764):211–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chen J, López-Moyado IF, Seo H, Lio CWJ, Hempleman LJ, et al. 2019. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567(7749):530–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, et al. 2019. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. PNAS 116(25):12410–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yao C, Lou G, Sun HW, Zhu Z, Sun Y, et al. 2021. BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8+ T cells. Nat. Immunol 22(3):370–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Merino A, Zhang B, Dougherty P, Luo X, Wang J, et al. 2019. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig 129(9):3770–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ebihara T, Taniuchi I. 2019. Exhausted-like group 2 innate lymphoid cells in chronic allergic inflammation. Trends Immunol. 40(12):1095–104 [DOI] [PubMed] [Google Scholar]
- 191.Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, et al. 2011. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 71(16):5412–22 [DOI] [PubMed] [Google Scholar]
- 192.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, et al. 2011. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity 35(3):400–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kiner E, Willie E, Vijaykumar B, Chowdhary K, Schmutz H, et al. 2021. Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol 22(2):216–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tibbitt CA, Stark JM, Martens L, Ma J, Mold JE, et al. 2019. Single-cell RNA sequencing of the T helper cell response to house dust mites defines a distinct gene expression signature in airway Th2 cells. Immunity 51(1):169–84.e5 [DOI] [PubMed] [Google Scholar]
- 195.Mimitou EP, Cheng A, Montalbano A, Hao S, Stoeckius M, et al. 2019. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16(5):409–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Efremova M, Vento-Tormo R, Park JE, Teichmann SA, James KR. 2020. Immunology in the era of single-cell technologies. Annu. Rev. Immunol 38:727–57 [DOI] [PubMed] [Google Scholar]
- 197.Mezger A, Klemm S, Mann I, Brower K, Mir A, et al. 2018. High-throughput chromatin accessibility profiling at single-cell resolution. Nat. Commun 9(1):3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, et al. 2019. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10(1):1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pierce SE, Granja JM, Greenleaf WJ. 2021. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat. Commun 12(1):2969. [DOI] [PMC free article] [PubMed] [Google Scholar]