

Abstract
EGFR oncogenic mutations predict sensitivity to EGFR inhibitors in NSCLC, but less is known about EGFR “variants of unknown significance.” Using preclinical models, 3D structure analyses, and patient response data, Robichaux et al. show in Nature that mutations in structural regions of EGFR predict responses to different EGFR inhibitors.
The response of EGFR mutant non-small cell lung cancer (NSCLC) to EGFR tyrosine kinase inhibitors (TKIs) is an early example of an acquired tumor mutation predicting response to an inhibitor targeting that mutant protein (Lynch et al., 2004). This has led to the clinical practice of testing NSCLC patients’ tumors for EGFR and other therapeutically actionable mutations using a CLIA-certified test at the time of diagnosis. If “classical mutations” in EGFR are found, which are known to be oncogenic and sensitive to EGFR inhibitors, EGFR TKI therapy is given. However, many different mutations may occur in EGFR, and not all mutations in EGFR, even those that appear very similar to classical mutations, are sensitive to EGFR TKIs (Russo et al., 2019). For these variants of uncertain significance (VUS) in EGFR, it is not known if the patient will derive any benefit from treatment with any EGFR TKI. Mutations in EGFR can also become a problem later. In patients that respond to EGFR TKIs, despite initial clinically beneficial responses (alleviating symptoms and promoting survival), essentially all patients ultimately relapse in 1–2 years (Cho et al., 2020). This relapse can be driven by a variety of mechanisms, some of which are mutations in EGFR that prevent the drug from inhibiting EGFR. A deeper understanding of how various EGFR mutations affect both EGFR signaling and sensitivity to EGFR TKIs would have a positive impact on clinical decisions and aid the development of new EGFR inhibitors, ultimately resulting in better outcomes for patients with EGFR mutant NSCLC.
A recent article published in Nature (Robichaux et al., 2021) has begun to address this issue. By analyzing multiple large clinical datasets, Robichaux et al. show that patients with classical mutations in EGFR had longer times to treatment failure than those with non-classical mutations when treated with an EGFR inhibitor, suggesting that the type of EGFR mutation is relevant for treatment outcomes. Preclinical models of mutant EGFR, including molecularly annotated NSCLC cell lines, patient-derived xenografts (PDXs), and genetically engineered mouse models (GEMMs), have been essential for progress and clinical translation of mutant EGFR-targeted therapy. Robichaux et al. developed a new preclinical model by establishing a panel of mouse BA/F3 cells transfected with 76 different mutant human EGFR cDNAs. The survival of these BA/F3 cells is dependent on the activity of the mutant EGFR protein; thus, the sensitivity (or resistance) of these mutant EGFR proteins to EGFR inhibitors can be established. Guided by the response of this cell line panel to 18 EGFR inhibitors, Robichaux et al. stratified “non-classical” EGFR mutations into four structural classes: “classical-like” mutations that were distant from the ATP-binding pocket, “T790M-like” mutations that lie in the hydrophobic core, insertions in the loop at the C-terminal end of the αC-helix in exon 20, and mutations predicted to be P loop and αC-helix compressing (“PACC” mutations). Compared to previously used proximity-based predictions, such as exon location, these structure-based classifications better predict response to an EGFR inhibitor in their BA/F3 preclinical model and could potentially inform inhibitor selection for more patients (Figure 1).
Figure 1. Structure-based classification of EGFR mutations matches more patients to efficacious EGFR inhibitors than prior schemes.
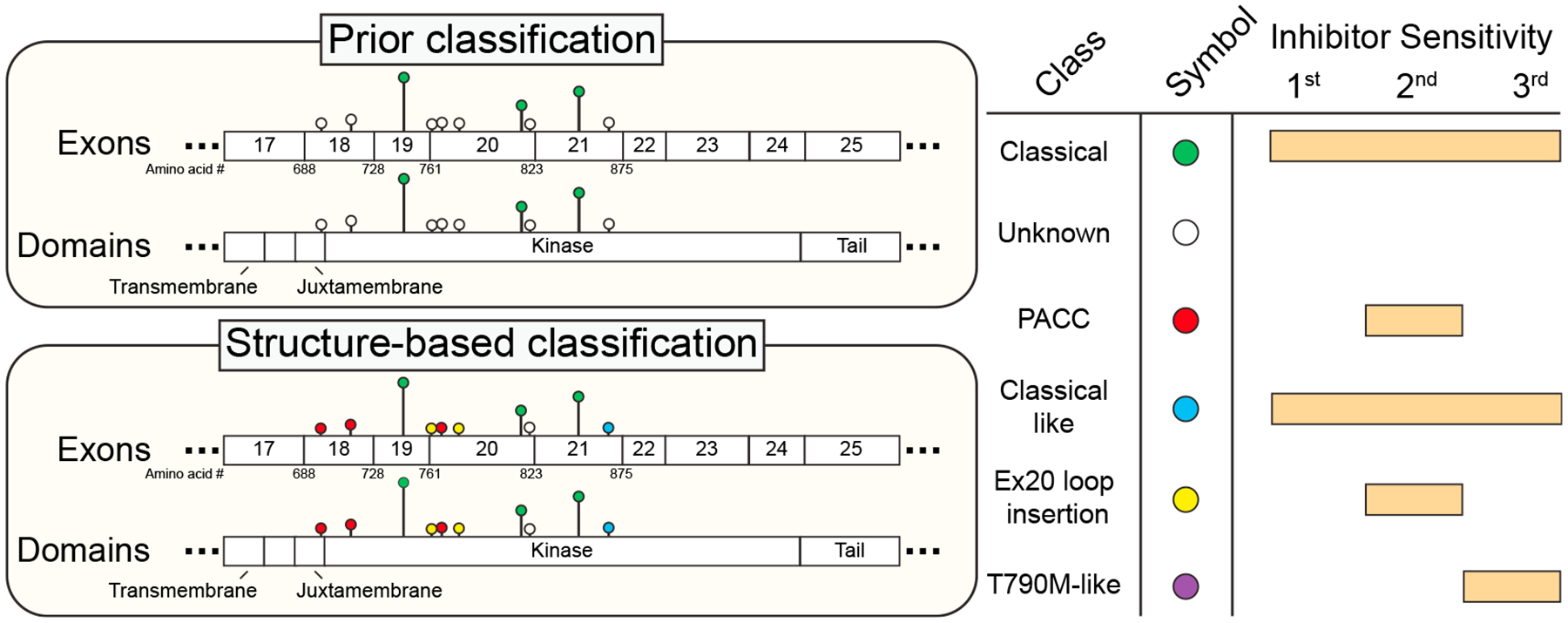
Left, cartoon lollipop diagram showing mutation location, frequency (height of lollipop; as identified by Robichaux et al., 2021) and mutation classes (colored circles) in the kinase domain of EGFR using the prior classification scheme (top) and the structure-based classification proposed by Robichaux et al. (bottom). Only mutations found at >1% frequency by Robichaux et al. are shown; T790M-like mutations are not displayed due to low frequency. Right, color key for circles in the lollipop diagram and predicted sensitivity of each mutation class to first-, second-, and third-generation EGFR inhibitors. Exon 20 (Ex20) loop insertions may be sensitive to second-generation or most EGFR inhibitors depending on their location (as discussed in the text) but are shown as only sensitive to second-generation inhibitors, for simplicity. First-generation EGFR inhibitors are erlotinib, gefitinib, AZD3759, and sapatinib; second-generation inhibitors are afatinib, dacomitinib, neratinib, and poziotinib; third-generation inhibitors are osimertinib, nazaratinib, olmutinib, rociletinib, naquotinib, and lazertinib.
Previous studies have shown that NSCLC tumors with exon 20 EGFR mutations have a heterogenous response to EGFR inhibitors (Kosaka et al., 2017; Robichaux et al., 2018). Using their BA/F3 model, Robichaux et al. found that most point mutations in exon 20 were PACC mutations and sensitive to second-generation EGFR inhibitors, while most exon 20 insertion mutations in the αC-helix behaved similarly to “classical-like” mutations and were sensitive to all EGFR inhibitors. The remainder of exon 20 insertions that occurred in the C-terminal loop of the αC-helix, referred to as Exon20ins-L mutations, were only sensitive to second-generation EGFR inhibitors. However, even within these Exon20ins-L mutations there was heterogeneity, with mutations near the C-terminal loop being more sensitive to EGFR inhibition than those farther from the C-terminal loop.
In another test of their structural classification, the authors tested the response of a NSCLC PDX with an EGFRG719A PACC mutation to various EGFR inhibitors and found that while osimertinib (a third-generation EGFR inhibitor) only resulted in moderate tumor growth inhibition, treatment with afatinib or poziotinib (both second-generation EGFR inhibitors) caused complete tumor growth inhibition or regression, respectively. Additionally, similar results were seen in a NSCLC patient with an EGFRE709K/G719S PACC mutant tumor that was treated with afatinib. Importantly, when a PACC mutation co-occurred with a classical EGFR mutation, the preclinical models were sensitive to second-generation but not third-generation EGFR inhibitors. This is clinically relevant because it is possible that patients with EGFR mutant NSCLCs that initially benefit from osimertinib treatment may co-develop PACC mutations and progress on osimertinib but still receive clinical benefit from treatment with a second-generation EGFR inhibitor. Finally, Robichaux et al. found that patients who receive an EGFR inhibitor as their first line of therapy and have a PACC mutation derive the most benefit from second-generation EGFR inhibitors such as afatinib, strongly suggesting that patients with PACC mutations should be treated with second-generation EGFR inhibitors.
Resistance to EGFR inhibitors in NSCLC can occur via many mechanisms, with a major contributor being mutations in EGFR itself that alter inhibitor binding. The structural changes engendered by these mutations provide insight into inhibitor-EGFR interactions and essential information for creating the next generation of drugs that would work in the face of these drug resistance mutations. Thus, it is extremely important to identify and understand these drug resistance mutations. Recently two techniques have been developed to obtain this information. One is MITE-seq (Melnikov et al., 2014), where a library of synthetic oligos comprised of all possible mutations are cloned into an expression vector and then tested for drug sensitivity or resistance. The second is LentiMutate (Yenerall et al., 2021), which harnesses the error-prone property of lentiviral reverse transcriptase to randomly create mutations in a cDNA while the cDNA is delivered into cells (such as BA/F3), which requires the activity of the protein encoded by the cDNA. These cells are then treated with an inhibitor of the protein encoded by the cDNA, killing off cells lacking a resistance mutation, and the cDNAs in the surviving drug-resistant cells are sequenced to identify resistance mutations. Mutations identified by either MITE-seq or LentiMutate can then be modeled in 3D using algorithms designed to determine the impact of a mutation on the 3D structure of a protein (Jubb et al., 2018; Krebs et al., 2021).
Following up on these findings, an important next step would be to fully understand in 3D how the 76 EGFR mutant proteins tested by Robichaux et al. develop on-target EGFR mutation resistance to first-, second-, and third-generation EGFR inhibitors. While daunting, this study would provide both clinically and structurally important information. It is also important to consider how the findings by Robichaux et al. can be translated and validated in the clinic. Because there are several FDA-approved EGFR inhibitors, and many VUS can arise in EGFR, the design and execution of a prospective clinical trial would be monumental. However, as another approach for both EGFR and other frequently mutated onco-proteins, the development of national databases with paired mutation, treatment, and outcome data would enable better, “real-world” retrospective studies to understand which mutations predict response to which inhibitors in various cancer types.
ACKNOWLEDGMENTS
This article was supported by funding from the Simmons Comprehensive Cancer Center at UT Southwestern (developmental funds to R.K. from P30CA142543), CPRIT (RP120732-P3 to R.K., RP160652 to J.D.M.), the National Institutes of Health (NCI SPORE in lung cancer, CA07090,7 to J.D.M. and R01CA200787 to R.K.), and the Margot Johnson Foundation to J.D.M. R.K. is a John L. Roach Scholar in Biomedical Research and a CPRIT Scholar in Cancer Research.
Footnotes
DECLARATION OF INTERESTS
There is a patent pending for LentiMutate that lists P.Y., J.D.M., and R.K. as inventors. J.D.M. receives licensing royalties from the NCI and UT Southwestern for cell lines.
REFERENCES
- Cho JH, Lim SH, An HJ, Kim KH, Park KU, Kang EJ, Choi YH, Ahn MS, Lee MH, Sun JM, et al. (2020). Osimertinib for patients with non-small-cell lung cancer harboring uncommon EGFR mutations: a multicenter, openlabel, phase II trial (KCSG-LU15–09). J. Clin. Oncol 38, 488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb HC, Saini HK, Verdonk ML, and Forbes SA (2018). COSMIC-3D provides structural perspectives on cancer genetics for drug discovery. Nat. Genet 50, 1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Tanizaki J, Paranal RM, Endoh H, Lydon C, Capelletti M, Repellin CE, Choi J, Ogino A, Calles A, et al. (2017). Response het erogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 77, 2712–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs FS, Zoete V, Trottet M, Pouchon T, Bovigny C, and Michielin O (2021). Swiss-PO: a new tool to analyze the impact of mutations on protein three-dimensional structures for precision oncology. NPJ Precision Oncol. 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Melnikov A, Rogov P, Wang L, Gnirke A, and Mikkelsen TS (2014). Comprehensive mutational scanning of a kinase in vivo reveals substratedependent fitness landscapes. Nucleic Acids Res. 42, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, Li S, Chen T, Poteete A, Estrada-Bernal A, et al. (2018). Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat. Med 24, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux JP, Le X, Vijayan RSK, Hicks JK, Heeke S, Elamin YY, Lin HY, Udagawa H, Skoulidis F, Tran H, et al. (2021). Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 597, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A, Franchina T, Ricciardi G, Battaglia A, Picciotto M, and Adamo V (2019). Heterogeneous responses to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with uncommon EGFR mutations: new insights and future perspectives in this complex clinical scenario. Int. J. Mol. Sci 20, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenerall P, Kollipara RK, Avila K, Peyton M, Eide CA, Bottomly D, McWeeney SK, Liu Y, Westover KD, Druker BJ, et al. (2021). Lentiviral-driven discovery of cancer drug resistance mutations. Cancer Res. 81, 4685–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]