

ABSTRACT
Toxin-antitoxin (TA) systems are genetic modules that consist of a stable protein-toxin and an unstable antitoxin that neutralizes the toxic effect. In type II TA systems, the antitoxin is a protein that inhibits the toxin by direct binding. Type II TA systems, whose roles and functions are under intensive study, are highly distributed among bacterial chromosomes. Here, we identified and characterized a novel type II TA system PrrT/A encoded in the chromosome of the clinical isolate 39016 of the opportunistic pathogen Pseudomonas aeruginosa. We have shown that the PrrT/A system exhibits classical type II TA characteristics and novel regulatory properties. Following deletion of the prrA antitoxin, we discovered that the system is involved in a range of processes including (i) biofilm and motility, (ii) reduced prophage induction and bacteriophage production, and (iii) increased fitness for aminoglycosides. Taken together, these results highlight the importance of this toxin-antitoxin system to key physiological traits in P. aeruginosa.
IMPORTANCE The functions attributed to bacterial TA systems are controversial and remain largely unknown. Our study suggests new insights into the potential functions of bacterial TA systems. We reveal that a chromosome-encoded TA system can regulate biofilm and motility, antibiotic resistance, prophage gene expression, and phage production. The latter presents a thus far unreported function of bacterial TA systems. In addition, with the emergence of antimicrobial-resistant bacteria, especially with the rising of P. aeruginosa resistant strains, the investigation of TA systems is critical as it may account for potential new targets against the resistant strains.
KEYWORDS: Pseudomonas aeruginosa, toxin-antitoxin, biofilm, prophages, bacteriophages, biofilms
INTRODUCTION
Toxin-antitoxin (TA) systems are genetic modules that were initially identified as plasmid maintenance systems (postsegregational killing) (1, 2). Over the last few decades, thousands of TA loci were identified on plasmids, phages, and bacterial and Archael and chromosomes, harboring different functions (3, 4). The TA locus encodes for a toxin and a relatively unstable cognate antitoxin that neutralizes the toxic effect during normal bacterial growth (5). While the toxin gene typically encodes for a protein, the antitoxin gene product can be either an RNA or a low-molecular-weight protein, depending on the TA class (4). Type II TA system genes are commonly expressed under one bicistronic operon by a tightly autoregulated promoter (6), and the antitoxin is a protein that neutralizes the toxin by direct binding (7). The antitoxin usually negatively regulates the operon transcription by direct DNA binding, with relatively low affinity that can be enhanced by antitoxin-toxin complexes ratio (8). The activation of type II TA systems also undergoes posttranslational regulation, dependent on the antitoxin instability and degradation mediated by bacterial proteases (9). The proteolysis of the antitoxin promotes under several stress conditions, leading to the activation of the TA system (10).
The Type II TA class is considered the most abundant system in bacterial genomes and was identified across diverse bacterial species (6). As the original plasmid-stabilization function is not relevant for the diverse TA systems found in bacterial chromosomes, TA systems are hypothesized to be involved in other biological processes, for example, stabilizing chromosomal mobile elements (11, 12), bacteriophage inhibition by abortive infection (13), involvement in bacterial stress response, persisters formation (14, 15), and biofilm formation (16). The diverse functions of type II TA systems and their role in bacterial responses are extensively studied yet remain largely unknown (17).
Pseudomonas aeruginosa is a Gram-negative, rod-shaped bacterium with a single flagellum. It is an opportunistic pathogen of plants, nematodes, insects, animals, and humans (18). It can cause a wide range of acute and chronic infections, enhanced by the bacterium's low susceptibility to various antimicrobial substances, making most of the infections difficult to treat and life-threatening (19). Several type II TA systems were previously identified in the genome of P. aeruginosa; three were shown bioinformatically to be highly conserved between P. aeruginosa isolates; parE-parD (20), relE-relB (21), and higB-higA (22).
The ParE class of toxins acts through direct binding and inhibition of DNA gyrase, resulting in accumulation of DNA breaks, activation of SOS response, and bacterial death (17, 20). HigB toxin acts as an RNase, which rapidly degrades mRNAs and influences bacterial virulence by enhancing the type III secretion system (23), reducing pyochelin and pyocyanin production, biofilm formation, and swarming motility (22). HigA functions as an antitoxin that neutralizes the RNase activity of HigB (22). The higA gene has an independent promoter apart from the joint higB/A promoter, resulting in higher higA transcripts in the late stationary phase (24). Moreover, besides the auto-repression properties of HigA, it also binds and represses the mvfR promoter, a central virulence transcription regulator (24).
In the current study, we identified and characterized a novel type II TA system PrrT/A (named for Prophage Regulator Toxin/Antitoxin) encoded in the chromosome of the clinical isolate 39016 of P. aeruginosa. The toxin PrrT, carrying a ParE-like domain, is inhibited by PrrA, an antitoxin with a predicted HigA-like domain. We have found that the PrrT/A system affects bacterial growth, biofilm formation, swarming motility, prophage induction, bacteriophage production, and aminoglycosides fitness.
RESULTS
PrrT/A exhibits classical type II TA characteristics.
PA39016_100004 (prrA) gene product is a DNA binding protein with a predicted HigA-like domain. The sequence of the 39016 strain is poorly annotated, and many open reading frames (ORFs) are not properly detected in the sequence. To better understand the functionality of the prrA gene, the genomic region of prrA was scanned to detect unannotated genes that might influence the prrA gene function. The screening identified one such gene, 366 bp ORF adjacent to prrA, herein termed prrT. The protein sequence analysis revealed that the prrT product has a ParE-like domain, suggesting that PrrT and PrrA act together as a type II TA system.
To investigate whether the prrT/A gene pair indeed encodes for a TA system, deletion mutants of the prrA (ΔprrA), prrT (ΔprrT), and both genes (ΔprrTA) were created. The growth curve of the different mutants showed that the deletion of the prrA gene significantly decreased the bacterial growth rate. The double mutant strain did not show any significant change in its growth, indicating that the growth inhibition depends on PrrT toxin activity. Complementation by a genomic expressed copy of prrA (ΔprrA/prrA) restored the bacterial growth phenotype with no significant difference to the wild-type (WT) strain (Fig. 1A and Fig. S1A).
FIG 1.

The prrT/prrA gene pair act as a type II TA system. (A) Deletion of prrA in 39016 strain revealed a decrease in the growth rate; the growth curve of the mutants in comparison to the WT, and complementation by arabinose induced prrA expression in the mutant. (B) prrT expression is toxic only for the ΔprrA strain; the growth curve of prrT induced OE in the WT and mutant strains, and the inducer was added immediately after the dilution. (C) prrT/prrA gene pair expressed as a polycistron; the following sets of primers were used for the operon verification: (a) 500 bp upstream to prrT, (b) prrT_F, (c) prrT_R, (d) prrA_F, (e) prrA_R, and (f) 500 bp downstream to prrA. The cDNA results represent the transcripts, while gDNA results represent the bacterial genome as a positive control. (D) PrrT/PrrA form a protein complex together; BACTH assay on indicative LB plates containing IPTG and x-gal. The above (A) and (B) graphs are the averages of three independent experiments consisting of five replicates each. Error bars represent the standard deviations.
To examine the toxicity of PrrT, we inserted into an attTn7 site of the WT and mutant strains an intact copy of the prrT gene under arabinose inducible promoter. PrrT induction did not affect the WT strain, while in both the ΔprrA and ΔprrTA strains, the toxin induction resulted in significant growth inhibition (Fig. 1B and Fig. S1B). The recovery rate of the ΔprrTA strain was somewhat higher than ΔprrA, probably due to an unneutralized native toxin present in the ΔprrA strain.
Type II TA systems genes are commonly expressed as a bicistronic mRNA (25). To verify whether this is also the case for the prrTA, systemRNA was extracted from the WT 39016, reverse transcribed to cDNA, and was further amplified by PCR with specific primers for the pair of genes compared to genomic DNA (gDNA). The results showed that prrT and prrA are expressed by polycistronic mRNA, reinforcing our hypothesis that they act as a type II TA system (Fig. 1C).
As the type II TA system neutralization mechanism is mediated by specific interaction between the toxin and the antitoxin, we also examined the PrrT-PrrA binding. Both classical Co-IP and BACTH assays strongly showed that the proteins directly interact and form a complex (Fig. 1D, Fig. S2).
PrrA protein represses prrT/A operon expression.
The PrrA antitoxin is a predicted transcriptional regulator as it contains a DNA binding domain. To characterize its regulatory properties, self-regulation was evaluated by examining the prrT/A promoter activity in different mutant strains. For that, a transcription fusion was constructed. Briefly, 300 bp upstream to prrT were amplified, fused to mCherry reporter, and inserted into the attCtx site in the WT and mutant strains; the promoter activity was compared in the early stationary phase, 14 h postdilution. As expected, in the WT strain, the activity of the promoter was significantly decreased compared to the ΔprrAT operon mutant, indicating negative regulation of the proteins (Fig. 2A). To better characterize the contribution of each protein to the autorepression, single complementation strains, created by inserting an inducible copy of prrT or prrA into the attTn7 site of ΔprrTA/mCherry strain, were utilized. The fluorescent measurements of these strains clearly showed that PrrA expression caused significant promoter repression while the toxin induction resulted in promoter activity elevation (Fig. 2A). These results strongly suggest that the PrrA protein is sufficient for the operon autorepression, while in the absence of the antitoxin, PrrT can stimulate auto-expression.
FIG 2.

The prrT/A promoter activity is affected by PrrA, PrrT, and NOR stressor. (A) PrrA represses the promoter while PrrT expression elevates the activity; fluorescence measurement of the WT and the ΔprrTA strain with single complementation, and PrrA and PrrT were induced in the ΔprrTA strain at time zero with 10 mM arabinose. (B) NOR treatment elevated the promoter activity exclusively in strains with an intact prrA; fluorescence measurement of the WT and mutant strains with or without 0.1 mM NOR treatment at time zero. (C) PrrA directly binds to the prrT/A promoter; competition sample with biotinylated DNA and an unlabeled competitor DNA (lane 1), biotinylated DNA with the addition of decreasing amount of the PrrA protein (lanes 2–7). For the A and B graphs, m-Cherry fluorescence measurements were taken in the early stationary phase. The above A and B graphs are the average of three independent experiments consisting of five replicates each. Error bars represent the standard deviation. According to t test: *, P < 0.05; **, P < 0.01.
Type II TA systems are activated under different stress conditions, presumably due to the antitoxin cleavage (26). To confirm that the toxin effect on the promoter is not due to growth inhibition and/or stress conditions, we tested the ΔprrT strain exposed to different stressors. Out of all the tested stress conditions, only treatment with subinhibitory Norfloxacin (NOR) concentrations resulted in a significant increase of promoter activity in the WT strain (Fig. S3). We hypothesized that the increase in promoter activity depends on PrrA cleavage. Examination of the effect of NOR treatment on the different strains verified the antitoxin-dependent regulation. The treatment elevated the promoter activity only in strains harboring the prrA gene, while the treatment on the ΔprrTA strain showed no effect. The ΔprrA showed a decrease in fluorescence, perhaps due to the growth inhibition (Fig. 2B). The results reinforce the indication for the stress-independent positive regulation of PrrT, especially when considering that its effect was observed in the ΔprrTA strain.
Next, to confirm that the antitoxin represses the promoter by direct binding, electrophoretic mobility shift assay (EMSA) was carried out using a purified PrrA protein with 300 bp promoter region upstream to prrT start codon (prrT/A promoter). The results showed a clear shift in the sample containing both the tagged promoter and PrrA, indicating positive interaction between the antitoxin and the prrT/A promoter. The observed shift disappears with the addition of the competitor (an untagged DNA X120 excess) similar to the negative control, which does not contain the protein (Fig. 2C).
PrrT/A is involved in biofilm and motility regulation.
The impact of HigA antitoxin on biofilm regulation was previously published (22). To investigate whether PrrA influences biofilm formation and bacterial motility, the biofilm formation of the different mutant strains was quantified by crystal violet (CV) staining. Biofilm formation was significantly increased in the antitoxin mutant after 24 h, while the toxin mutant did not exhibit any difference compared to the WT strain (Fig. 3A). The double mutant strain showed nearly complete complementation with a significant decrease in comparison with the ΔprrA strain. Complementation by a genomic expressed copy of prrA completely restored the biofilm phenotype. Since some growth differences were detected after 24 h, we have also examined the biofilm formation with an extended incubation period (48 h). No planktonic growth differences were seen after 48 h, while the biofilm results were consistent with the shorter incubation time point (Fig. S4).
FIG 3.

prrA deletion influenced swarming motility and biofilm formation. (A) prrA deletion significantly increased biofilm formation; CV stained 24 h biofilm of the strains. (B) The deletion of prrA significantly impacts the amrZ gene; RT- PCR analysis comparing the expression levels of amrZ the IN prrA mutant strain compared to the WT and complementation strains. (C) prrA deletion reduced swarming motility; the WT, mutants, and complementation strains were grown for 48 h. The A graph is the average of three independent experiments with five replicates each. The B graph is the average of three independent experiments with three replicates each. The C pictures represent three independent experiments conducted with three replicates each. Error bar represents standard error. **, P < 0.01 with WT strain as a reference, according to t test.
To understand how the antitoxin deletion influences biofilm formation, we performed a transcriptomic analysis of the WT and the ΔprrA strains. Focus was given to biofilm-related genes, influencing the bacterial cyclic di-GMP (c-di-GMP) levels. In P. aeruginosa, E. coli, and other species, the c-di-GMP second messenger regulates the switch between planktonic and biofilm growth (27). Two protein families mainly regulate the bacterial c-di-GMP levels: the diguanylate cyclase genes (DGCs) that promote c-di-GMP synthesis (28), and the phosphodiesterase genes (PDEs) that are involved in the turnover of cyclic-di-GMP (29). The analysis revealed a significant decrease in the transcript levels of the amrZ gene, a master regulator of several PDEs and DGCs genes (30). The results were validated by RT-PCR (Fig. 3B). Several downstream DGCs and PDEs were also affected, and their transcripts levels were altered correspondingly with the observed phenotype (Fig. S5). The results indicate that the observed increase in biofilm formation in the antitoxin mutant may result from elevated bacterial c-di-GMP levels.
Since AmrZ protein is a transcriptional factor that regulates motility and alginate synthesis (31), we further assessed the PrrA involvement in bacterial motility. The swarming of the different strains was examined (Fig. 3C). Consistently with the biofilm and the transcriptomic results, the ΔprrA strain exhibited significantly less swarming than the other strains. Unlike the biofilm phenotype, the double mutant strain showed complete restoration of the phenotype. Complementation by a genomic expressed prrA copy repaired the swarming phenotype.
We also examined the possible involvement of the PrrT/A system in bacterial persistence. The persistence assay showed that the WT and the prrT/A operon mutant showed similar persistence and the same biphasic death curve, indicating that the system is not involved in persistence formation (Fig. S6).
PrrT/A is involved in prophage regulation.
The prrA gene was first identified by PHAST (32) within a prophage region in 39016. However, after the prophage att sites were identified (data not shown), it appeared that the gene is not a part of the prophage and is actually located 3,504 bp upstream. PrrA protein contains a CRO/CI-type DNA binding domain, so although proven to be a bacterial gene, we hypothesized that prrA might also be involved in prophage regulation.
To examine this, we performed an RNA-seq analysis of the antitoxin mutant and the WT strains after induction for prophage excision (1 h postinduction) focusing on the genes encoded at the prophage regions of 39016, herein named PR1-5. The transcriptomic analysis revealed that the absolute majority of the “prophages” genes are significantly downregulated in the induced ΔprrA strain compared to the induced WT strain (Fig. 4A).
FIG 4.

The prrT/prrA system is involved in prophage regulation and impacts phage production. (A) The prrA deletion resulted in a significant downregulation for most prophage encoded genes; heat-map was constructed with the RNA-seq results of the WT and ΔprrA strain, 1 h post prophage induction. The following prophage region (coordinates predicted by PHASTER [60]) genes were analyzed; PR1, PR2, PR3, PR4, and PR5. (B) The deletion of prrA resulted in decreased PR5 phage production; PA14 strain was used as a host, and phages were induced and extracted from WT, mutants, and the complementation strain. (C) prrA gene confers partial defense against phage infection; phages extracted from 39016 were used to infect the strains PA14/pUCP18 (PA14_VEC) and PAO1/pUCP18 prrA (PA14_prrA). The plaque-forming units presented in the above B and C graphs are the average of three independent experiments consisting of three replicates each. Error bars represent the standard deviations. *, P < 0.05; **, P < 0.01; ***, P < 0.001 when the WT and VEC is the reference strain, according to t test.
To further investigate whether the prrA deletion influenced phage production and infectivity, we quantified the phages produced by the mutants by performing a plaque assay with PA14 strain as a host. The results revealed that the ΔprrA strain produced significantly fewer infective phages than the WT strain (Fig. 4B). The ΔprrT had an equal number of phages as the WT, and the ΔprrTA strain produced slightly less than the WT strain yet significantly more than the ΔprrA strain.
The CRO/CI DNA binding domain of PrrA might indicate its involvement in lysogenic conversion or serve as an immunity element upon infection. To test this, prrA was overexpressed in the PA14 strain, and the susceptibility was examined and compared to PA14 carrying an empty vector. The overexpression (OE) of prrA resulted in a significant susceptibility reduction for phages extracted from the 39016 strain, indicating that the PrrA protein provides partial immunity for the strain (Fig. 4C).
PrrT/A system affects bacterial fitness for subinhibitory concentrations of aminoglycosides.
Further mining of the RNA-seq results showed that members of the mexXY efflux pump system genes are significantly upregulated in the antitoxin mutant strain compared to the WT strain (Fig. 5). Since the mexXY system in P. aeruginosa is known to affect resistance against aminoglycoside antibiotics (33), we first checked the MIC of the different strains for Kanamycin (Kan) and Streptomycin (Strep) aminoglycosides. The results showed that although the MIC values were not affected by the deletion of prrA, in the subinhibitory concentrations, in both examined antibiotics, the mutant showed higher growth compared to the WT strain, while in normal conditions, its growth is significantly inhibited (Fig. 6A and B). To further examine the subinhibitory concentration effect, we performed a growth curve of the different strains with a particular subinhibitory concentration of either Kan or Strep antibiotics. The results showed that the prrA mutant strain began to recover earlier in both antibiotics than the WT and the complementation strains (Fig. 6C and D). We concluded that the PrrA role is presumably important to the recovery from antibiotic inhibition. To further examine the possible fitness advantage this might have, we performed a coculture competition assay between the antitoxin mutant and WT strain grown in the presence of subinhibitory concentrations of aminoglycosides. The results showed that, unlike the control culture in which the percentage of the prrA mutant remained roughly 50%, in the treated cocultures, the antitoxin mutant took over and became the primary strain during the growth, highlighting the increased fitness of this strain under these tested conditions (Fig. 6E).
FIG 5.
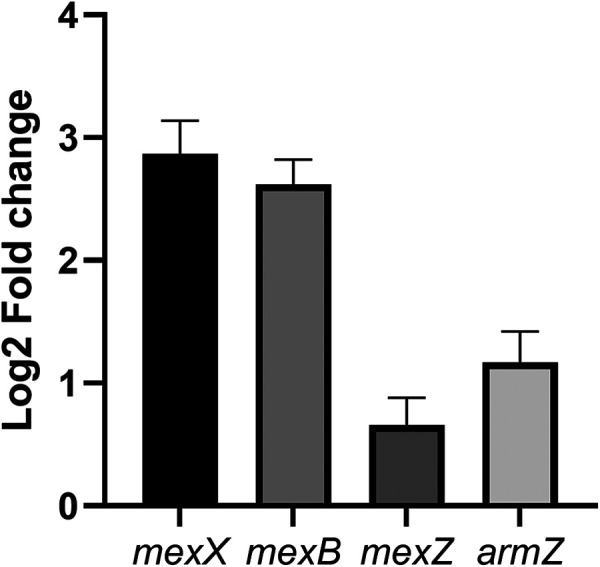
The MexXY system is upregulated in the prrA mutant. Log2 fold change of the mutant strain relative to the WT strain. Values were calculated with the transcriptomics levels. Error bars represent standard error.
FIG 6.

The prrA mutants show enhanced fitness in the subinhibitory treatment of aminoglycosides. (A) MIC experiment with Kan antibiotic, with concentrations ranging from 0 to 300 μg/mL. The MIC for Kan in both strains is 198 μg/mL, and the subinhibitory concentration is 131 μg/mL. (B) MIC experiment with Strep antibiotic, with concentrations ranging from 0 to 800 μg/mL. The MIC for Kan in both strains is 400 μg/mL, and the subinhibitory concentration is 200 μg/mL. (C) The growth curve of the different strains with the treatment of Kan in the concentration of 100 μg/mL. (D) The growth curve of the different strains with the treatment of Strep in the concentration of 400 μg/mL. (E) Competition assay for WT and prrA mutant coculture. The y axis represents the percentage of prrA strain in the different treatments. The above graphs are the average of three independent experiments with three replicates each. Error bars represent the standard deviations.
PrrT/A system is highly distributed among P. aeruginosa strains.
The abundance of the PrrT and PrrA homologs was analyzed in 233 P. aeruginosa genomes (all the existent complete genomes). Although the system was absent from the two laboratory strains PAO1 and PA14, the screen revealed that 132 genomes (56.6%) contained both PrrT and PrrA homologs. Moreover, in all PrrT/A-positive genomes, the PrrT and PrrA homologs were located adjacent to each other, mainly with a short overlap up to 10 bp apart (Table S3). We further investigated whether all of the homolog PrrT/A systems are located close to a prophage by calculating the genomic distance between the PrrT/A homologs and the next prophage. The analysis did not indicate any constant PrrT/A–prophage genomic distance, suggesting that the genomic proximity might not be essential for the PrrT/A function (data not shown).
DISCUSSION
We have identified and characterized a novel type II TA system of P. aeruginosa, which seems like a combined higA/parE-like system. We showed that prrA and prrT are cotranscribed and can interact to form a protein complex together. We examined the system's self-regulation and showed that PrrA antitoxin represses the promoter by direct binding and that PrrT toxin positively regulates the operon expression. We also found that toxin imbalance impacts bacterial growth, increases biofilm formation, reduces motility, and impacts the fitness at subinhibitory concentrations of aminoglycosides. Importantly, we showed that prrA deletion led to the repression of expression of prophages' genes over the entire genome, which correlated with reduced phage production. At the same time, prrA OE in the host bacteria resulted in decreased phage susceptibility (Fig. 7).
FIG 7.

Model for the PrrTA system and its involvement in bacterial processes.
The HigA antitoxin was shown to have an independent promoter apart from the higA/B promoter, which results in higher transcript levels of higA compared to higB toxin in the late stationary phase (24). In the current study, the toxin effect could only be detected by robust and artificial induction in the absence of prrA, indicating a significant stoichiometric advantage of PrrA antitoxin. Moreover, the toxin induction also influenced the ΔprrT strain, though only in the late stationary phase, suggesting that prrA, similarly to higA, may have an independent promoter located inside the prrT ORF.
In type II TA systems, the antitoxin alone or the TA complex act as a transcriptional auto-repressor (34). The experimental evidence showed that the PrrT-PrrA complex is not required for self-repression as both the prrT mutant strain and the prrT/A operon mutant with prrA induced expression exhibited complete repression with minimum promoter activity. Surprisingly, the PrrT toxin was found to stimulate and increase self-expression in the absence of the PrrA antitoxin. Toxin-driven transcriptional stimulation is mainly attributed to “conditional cooperativity,” a condition in which the repressors are destabilized due to a disruption of the antitoxin/toxin ratio, resulting from toxin excess, allowing resynthesis of both genes (35). Since the PrrT-dependent promoter stimulation was detected in the prrA mutant strain, conditional cooperativity poorly accounts for the observed phenomenon. It can only occur if some cross-regulation between other chromosomal TA systems has occurred. An interaction between noncognate complexes of toxins and antitoxins, which can bind to other TA promoter regions and regulate the expression, has been reported (36–38).
The PrrT effect can be attributed to a stress condition as it inhibits bacterial growth. Type II TA systems are influenced by stressors and are thought to influence bacterial survival and tolerance in stress conditions (26). A recently published paper showed that despite the transcriptional increase of the TA genes upon different stress conditions, the toxin is not activated in the examined conditions, and the upregulation is caused solely by antitoxin degradation (26). Consistently, we showed that stress caused by a subinhibitory concentration of NOR resulted in a significant increase in prrT/A promoter activity but only in strains carrying an intact prrA gene. Notably, NOR and PrrT act similarly; NOR is a fluoroquinolone antibiotic that acts through direct binding to the A subunit of the DNA gyrase (39), and PrrT toxin belongs to ParE family of toxins, which are also gyrase inhibitors that block DNA replication (40). Thus, despite the similar mechanism, the PrrT dependent stimulation effect cannot be attributed to the stress induction as it does not depend on PrrA cleavage.
The deletion of prrA antitoxin resulted in increased biofilm formation and reduced motility by elevating DGCs expression, leading to higher bacterial c-di-GMP levels. In contrast, it was shown that the higA mutant exhibits reversed phenotype with decreased biofilm formation, elevation in PDEs expression, and reduced c-di-GMP levels (22, 41). The contrasting results are probably attributable to the fact that prrT/A is a combined system in which the antitoxin mutant effect is mainly due to the upregulation of the toxin, as the ParE toxin was shown to enhance biofilm formation in E. coli (37) significantly.
Interactions between TA systems and prophages were found and characterized for different TA systems encoded by either the prophage itself (42, 43) or a residual plasmid (44). To our knowledge, no chromosomal type II TA system was described for prophage regulation properties. The deletion of the antitoxin resulted in significant global repression of the prophage's gene expression over the entire genome and decreased phage production. Nearly complete restoration of the phenotype was detected in the double mutant strain, indicating that the toxin upregulation influenced the observed prophage repression. Notably, considering the presumable mechanism of the PrrT toxin, it should have oppositely influenced the phages as it inhibits the gyrase and leads to SOS response activation, and thus the expected outcome would be prophage induction.
Interestingly, a link between phages and AmrZ levels was recently described (45). It has been shown that AmrZ represses CRISPR-Cas immunity genes upon surface attachment and that some phages of Pseudomonas carry amrZ homologs to avoid CRISPR defense. Here, we have demonstrated that upon antitoxin deletion, in addition to “prophages” genes’ downregulation and decreased phage induction, amrZ is also repressed, which correlates with the published anti-defense properties of amrZ, although the 39016 strain does not harbor a CRISPR system.
The OE of prrA-antitoxin in the PA14 strain resulted in lower susceptibility to PR5 phages produced by the 39016 strain. TA systems can compensate for each other by cross-reactivity. Therefore, single toxins or antitoxins can still influence the bacteria by reacting with other chromosomal or plasmid-encoded antitoxins or toxins (36, 37). Considering this cross-reactivity ability of TA systems, a reasonable assumption would be some toxin downregulation in the prrA OE strain due to an interaction with either a bacterial (PA14) or phage-encoded toxin.
Antibiotic tolerance driven by type II TA systems is mainly associated with the formation of persistence cells (46) or the maintenance of plasmids and genomic islands carrying antibiotic resistance genes (47). The activation of the mexXY efflux pump by toxin upregulation has not been described before for type II or other TA classes. As the mexXY pumps are not associated with biofilm resistance in P. aeruginosa (48), the enhanced fitness for aminoglycosides observed in the antitoxin mutant is presumably a direct effect of the PrrT toxin.
Although first identified and characterized in a single clinical isolate of P. aeruginosa, the PrrT/A system is highly distributed among the P. aeruginosa strains, indicating its high importance and the significance of the observed characteristics. The described functions of the PrrT/A system, especially the novel prophage regulation function, can significantly contribute to the developing research and knowledge about the chromosomal type II TA systems, their functions, and host contribution.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth media.
The bacterial strains and plasmids used in this study are listed in Table S1. Primers used in this study are listed in Table S2. All strains were grown in LB (Luria-Bertani broth, Difco) at 37°C unless otherwise specified. For the deletion mutants, the following media were used; Vogel Bonner Minimal Medium (VBMM) (49), Pseudomonas Isolation Agar (PIA, Difco), and No Salt Luria-Bertani (NSLB) + 10% sucrose. For DH5α heat shock, BHI (brain heart infusion broth, Difco) media was used. All strains were grown at 37°C unless otherwise specified. Antibiotic concentrations used in this study were 300 μg/mL Carbenicillin (Crb) and 50 μg/mL Gentamicin (Gm) for P. aeruginosa, and 100 μg/mL Ampicillin (Amp) and 30 μg/mL Gm for Escherichia coli.
DNA manipulation and plasmid construction.
The genomic extraction was performed using the DNeasy Blood & Cell Culture DNA Kit (Qiagen). For DNA fragment amplification, Phusion High-Fidelity DNA polymerase (Thermo) was used. For gene overexpression, primers were designed to complement the beginning and end of each gene, with the addition of either enzyme restriction sites for ligation or an overlap sequence for Gibson assembly. The amplified inserts were purified using NucleoSpin Gel and PCR Clean-Up (Macherey-Nagel). For the ligation assay, inserts and plasmids were digested using the appropriate fast digest restricted enzymes (Thermo). Ligation was conducted using Biogase Fast Ligation Kit (Bio-Lab Ltd.). For the Gibson assembly, inserts were incubated in the appropriate concentration with a linearized plasmid and 2× LigON mixture (EURx). For plasmid extraction, the QIAprep Spin Miniprep Kit (Qiagen) was used. For verification of successful plasmid transformations, the DNA polymerase ReddyMix PCR Kit and universal primers were used.
Strain construction.
PAO1 and PA14 strains overexpressing prrA and prrT were created as described previously (50). For 39016 OE strains, the mini-Tn7 vector was used for genomic expression under an arabinose-induced promoter. The creation of the OE 39016 strains using the mini-Tn7 vector and the mini-CTX vector was performed as previously described (51, 52). Gene deletions were performed by homologous recombination as previously described (49) with minor changes using the ampR cassette.
Growth curve.
LB (2 mL) was inoculated with bacterial strains from frozen stocks and incubated overnight at 37°C with shaking (250 rpm). For the OD measurements, the culture was diluted to 0.005 OD (595 nm) in fresh media and transferred to a 96-well plate, 200 μL in each well. Arabinose was added for gene induction (33.3Mm unless otherwise specified). The plates were incubated for 20 h at 37°C with agitation. Optical density measurements at 595 nm were taken every 30 min using the Synergy 2 Multi-Detection Microplate Reader (BioTek). For the plating efficiency measurements, the culture was diluted to 0.005 OD (595 nm) in fresh LB media to a final volume of 15 mL. Samples of 100 μL were taken at 2 h intervals, and serial dilutions were plated in 5 μL drops on top of an LB plate. The plates were incubated ON, and the appearing colonies were counted for CFU/mL calculation.
Bacterial two-hybrid (BACTH) assay.
The BACTH assay was conducted as described (53). The coding regions of prrA and prrT were cloned into pUT18C and pKT25, respectively. The recombinant plasmids were cotransformed into E. coli BTH101 competent cells with selection for kanamycin and ampicillin resistance. Eight different single colonies were then resuspended, and 5 μL were spotted on LB plates supplemented with kanamycin, ampicillin, IPTG (0.5 mM), and X‐gal (40 μg/mL). The colonies grew for 6 days at 30°C. Negative controls were included.
Protein extraction.
PAO1 strains were inoculated to 2 mL LB with antibiotic selection and grown overnight. Bacteria were then diluted 1:100 into M9+CA medium with L-(+)-Arabinose (33.3 μM) and grown to 0.6 OD (595 nm). From each strain, 1.5 OD (595 nm) of bacteria was taken and centrifuged at 14,000 g for 2 min, and the supernatant was removed. The cell pellet was then resuspended in lysis buffer (100 mM NaCl; 5% glycerol; 50 mM Tris PH 7.5) containing Benzonase Endonuclease (Millipore), cOmplete protease inhibitor cocktail (Roche), and Lysozyme (Sigma-Aldrich). Samples were then incubated for 15 min at 30°C with agitation followed by Sonication (90 sec, ON 5 sec, OFF 5 sec, 37% amplitude). The sonicated samples were centrifuged at 20,817 g for 10 min, and the upper liquid phase containing the proteins was collected.
Co-immunoprecipitation with ANTI-FLAG resin (co-IP).
Cell lysate (400 μL) was added to ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich), and lysis buffer was added to a final volume of 1 mL and incubated ON with gentle shaking at 4°C. The cell lysate and resin mix was centrifuged at 5,000 g for 30 sec, and the supernatant was removed. The mix was then washed three times with 500 μL of TBS (0.8% NaCl; 20 mM Tris 1M pH 7.4; water). One hundred μL of 3× FLAG peptide (Sigma-Aldrich) solution (150 ng/μL final concentration in TBS) was added to the mix and incubated with gentle shaking for 30 min at 4°C. The mix was then centrifuged at 5,000 g for 30 sec, and the supernatant was collected. For Western blot analysis, protein samples were diluted 3:1 with Sample BufferX3 (150 mM Tris-HCl pH = 6.8; 3% β-mercaptoethanol; 6% sodium dodecyl sulfate; 0.3% Bromophenol blue; 30% glycerol; water), incubated at 95°C for 10 min and then centrifuged at 14,000 g for 2 min. The samples were then separated on a 20% Tris-Glycine gel and transferred to a nitrocellulose membrane. After blocking with 1% alkali-soluble casein in TBS for His or 5% skim milk in TBS for Flag ON at 4°C, the membrane was incubated for 1 h with anti-His tag antibodies (1:1,000; Merck) and anti- FLAG antibodies (1:2,500; Sigma-Aldrich) separately. Following three washes with Tris-buffered saline with Tween 20 (TBST), the membrane was incubated with goat antimouse (HRP) antibodies (1:2,500; Santa Cruz Biotechnology) for an hour. After an additional three TBST washes, the membrane was developed with an ECL kit.
Electrophoretic mobility shift assay (EMSA).
EMSA was conducted using the LightShift Chemiluminescent EMSA kit (20148. Thermo Fisher Scientific, MA, USA) according to the manufacturer's protocol. The prrT/A promoter region was amplified with biotinylated primers (prom_biot_F and prom_biot_R). To validate the interaction between the PrrA protein and prrT/A promoter, decreasing concentrations of a purified PrrA protein (initial concentration of 10 ng/μL) were mixed with binding buffer (×10), NP-40 (1%), ultrapure water, and 50 fmol of a biotinylated prrT/A promoter sequence. To establish the position of an unshifted band in the gel, a mixture with the biotinylated prrT/A promoter sequence was prepared but without the PrrA protein. To demonstrate that the band shift observed results from a specific protein-DNA interaction, a competition experiment was performed by first incubating the PrrA protein for 10 min at room temperature with an excess of ×120 unlabeled prrT/A promoter sequence (6 pmol) (Lane 3 in Fig. 2D). Following an hour of incubation with the labeled prrT/A promoter sequence at room temperature, a loading buffer was added, and the samples were run on a 6% native polyacrylamide gel. The gel was then transferred to a Biodyne B nylon membrane (77016, Thermo Fisher Scientific, MA, USA) for 40 min, and the DNA was cross-linked to the membrane using a hand-held UV lamp with a 254 nm bulb for 10 min. The detection was done by chemiluminescence, according to the manufacturer's protocol.
mCherry reporter construction and fluorescent measurement.
LB (2 mL) was inoculated with bacterial strains carrying m-Cherry-fused promoter from frozen stocks and incubated overnight at 37°C with shaking (250 rpm). The cultures were diluted to 0.005 OD (595 nm) in fresh media and transferred to a 96-well plate, 200 μL in each well. Arabinose or NOR was added (10 mM and 0.1Mm, respectively). After 14 h of incubation at 37°C with shaking, optical density at 595 nm, fluorescent at an excitation wavelength of 580 nm, and emission wavelength of 610 nm were measured using the Synergy 2 Multi-Detection Microplate Reader (BioTek).
Static biofilm.
Bacteria were scraped from the LB plate and resuspended in 500 μL PBS, diluted to 0.05 OD (595 nm) in 1 mL M9+CA (with 33.3Mm arabinose if needed). Five replicates of 100 μL of each sample were then transferred to a 96-well plate followed by 24 h incubation at 37°C. The following day, OD was measured using the Synergy 2 Multi-Detection Microplate Reader (BioTek) (595 nm). Planktonic bacteria were washed twice with 200 μL deuterium-depleted water (DDW), followed by the addition of 150 μL crystal violet (CV, Sigma-Aldrich Israel Ltd.) and left for incubation at room temperature (RT) for 15 min. The plate was then rinsed with water to remove crystal violet residues, and 200 μL absolute ethanol was added; the plate was incubated at RT for 15 min. One hundred μL elution from each well was transferred into a new 96-well plate, and the new plate was read in the Synergy 2 Multi-Detection Microplate Reader (BioTek) (OD 595 nm).
Swarming motility.
For swarming motility assay, 2 mL medium (M9+CA) was inoculated with fresh colony and grown overnight at 37°C with shaking. On the following day, bacteria were diluted at 1:10 in the same medium and incubated for 3 h at 37°C with shaking. Bacteria (2.5 μL) were plated in the middle of a 0.5% agar M9 plate and incubated for 48 h.
Persistence assay (biphasic death curve).
Persistence assay was performed as previously described (54) with minor changes. Briefly, bacterial cultures were incubated for approximately 16 h, diluted in fresh LB media at 1:10 ratio, and further incubated until reaching the OD of 0.9 at 595 nm. The cultures were then treated with a 10-fold MIC of CIP (1.25 μg/mL); the control cultures for each of the strains were not treated. Samples of 100 μL were taken at the following time points: 1 h, 3 h, 5 h, 18 h, and 24 h. Serial dilutions were plated in 10 μL drops on top of an LB plate. The plates were incubated for 24 h, and the appearing colonies were counted for CFU/mL calculation.
Phage extraction.
LB (2 mL) was inoculated with bacterial strains incubated overnight. On the next day, the bacteria were diluted 1:50 with medium to a final volume of 4 mL and incubated until reaching OD 0.5 at 595 nm (~2 h). For phage induction, 0.4 μg/mL NOR (sigma) antibiotic was added, and the cultures were incubated for 1 h. Fresh LB was added (1.75 mL), and the cultures were incubated for an additional 1 h. Then, 1 mL of bacteria was centrifuged at 14,000 g for 2 min, and 900 μL of the supernatant was filtered using a 0.45 μm filter (Whatman).
Plaque assay.
LB (2 mL) was inoculated with recipient strain and incubated overnight. On the next day, bacteria were diluted at 1:50 with medium to a final volume of 4 mL and incubated for 1.5 h. Serial dilutions (1:10) of the induced phage stock were made. The diluted phage (100 μL) was added to the recipient bacteria (100 μL) and incubated for 15 min at 37°C. The mix of the phage and bacteria was then transferred into 5 mL heated (50°C) LB with 0.5% agar, then gently mixed and poured onto the surface of a 1.5% agar plate. The plate was incubated overnight until plaques were formed. The PFU/mL values are presented as the mean ± SEM.
RNA extraction.
The RNA extraction was performed as previously described (50). Bacterial strains were inoculated to LB and grown overnight. Bacteria were then diluted 1:100 into 15 mL M9 and grown to 0.5OD (595 nm). For the induced samples, 0.4 μg/mL NOR antibiotic was added, and all the cultures were incubated for an additional 1 h. Two mL of each sample was taken and incubated for 20 min with 4 mL RNAprotect Bacteria Reagent (Qiagen). After incubation, the bacteria were centrifuged at 3,220 g for 20 min and rinsed in Tris-EDTA buffer solution (pH 8; Sigma/fluka) to remove RNAprotect residues. Ninety μg/mL Lysozyme (Roche), 10 μL Proteinase K (Qiagen), and 1 mL warm Tri-reagent 37°C (Sigma) were added to pelleted cells. After 5 min of incubation at 65°C, 200 μL chloroform was added. The solution was centrifuged for 15 min at 20,817 g, and the upper liquid phase was transferred into 80% ethanol. The RNA was then extracted using an RNeasy minikit (Qiagen) according to the manufacturer's protocol.
RNA sequencing.
For RNA sequencing, 2 μg of total RNA was used for the RiboMinus Bacteria Transcriptome isolation kit (Invitrogen). The library was constructed with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB) according to the manufacturer’s instructions using 30 ng of depleted RNA. The final quality was evaluated by TapeStation High Sensitivity D1000 Assay (Agilent Technologies, CA, USA). Sequencing was performed based on Qubit values and loaded onto an Illumina MiSeq using the MiSeq V2 (50 cycles) Kit (Illumina, CA, USA). Single-end RNA-seq protocol was used, yielding about 1.34–1.74 million reads per sample. FastQC (v0.11.2) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc) was used to assess the quality of raw reads. Reads were aligned to P. aeruginosa 39016 strain (RefSeq sequence ID: NZ_CM001020) using the Bowtie2 (55) aligner software (version bowtie2-2.3.2) with default parameters. GTF annotation file for the 39016 strain wad downloaded from NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCF_000148745.1, downloaded March 2019), and the prrT_ORF coordinates were added manually to the annotation file. Raw read counts for 6469 gene-level features were determined using HTSeq-count (56) with the intersection-strict mode. Differentially expressed genes were determined with the R Bioconductor package DESeq2 (57). The P values were corrected with Benjamini-Hochberg FDR procedure. Genes with adjusted P values < 0.05 and |log fold change| > 1 were considered as differentially expressed.
Real-time PCR analysis (RT-PCR).
For cDNA production, the GoScript Reverse Transcription System (Promega) was used with 1 μg RNA according to the manufacturer's instructions. For the RT-PCR analysis, Fast SYBR Green Master Mix was used (Applied Biosystems, Thermo), according to the manufacturer's instructions. RT-PCR was conducted using CFX-96 Touch Real-Time PCR Detection System (Bio-Rad). Results were normalized using the PA3540 housekeeping gene.
MIC-MIC.
LB (2 mL) was inoculated with bacterial strains grown on LB plates and incubated overnight. Double dilutions of the antibiotics were performed. Bacteria were then diluted and transferred to a 96-well plate to a final concentration of 0.001 OD (595 nm). The plates were incubated overnight with shaking. Optical density measurements at 595 nm were taken using Synergy 2 Multi-Detection Microplate Reader (BioTek).
Competition assay.
LB (2 mL) was inoculated with bacterial strains grown on LB plates and incubated overnight. Initial amount of 106 cells were added from each strain into 15 mL of fresh LB media. The subinhibitory concentrations of the antibiotics were added immediately (except for the control coculture). Samples of 100 μL were taken at the following time points: 0 h, 3 h, 6 h, 24 h. Serial dilutions were plated in 5 μL drops on LB and LB-carbenicillin (CRB) plates. The plates were incubated for ON, and the appearing colonies were counted for CFU/mL calculation. Since the ΔprrA strain is the only CRB-resistant strain, its percentage was calculated as the ratio of plating efficiency observed on the LB-CRB to the plating efficiency on the LB plate, representing the overall efficiency.
PrrA and PrrT orthologs search.
Reciprocal best hit of BLAST (58) was used to find orthologs of prrA and prrT. The amino-acid sequences of PrrT and PrrA and complete genomes of 233 P. aeruginosa strains were downloaded from Pseudomonas Genome DB version 20.2 (59). First, tblastn (blast package, version 2.5.0 (was used to find matches (in protein level search) of PrrA or PrrT proteins in each of the 233 P. aeruginosa genomes. Then, for verification, for genomes with a match, tblastx was performed on the prrA and/or prrT orthologs (only matches with e-value < 0.05 were considered) against the 39016 genome to verify that it best matches to the PrrA or PrrT.
General bioinformatics.
Identification of prophage regions and genes was conducted using PHASTER (60) (PHAge Search Tool). Prophage gene identification was achieved using the Pseudomonas genome database (59). Gene comparison was conducted using BLAST (61). Gene annotation was performed using RAST (7).
Data availability.
The data that support the findings of this study are available in the GEO database (accession number GSE179116).
ACKNOWLEDGMENTS
This work is part of the PhD thesis of E.S. Partial funding for this work was through the Dyna and Fala Weinstock Foundation to E.B., the President's Scholarships, and the Merit-Based Scholarships at the Institute of Nanotechnology of Bar-Ilan University for E.S. We thank Gila Jacobi for editing the article.
Footnotes
Supplemental material is available online only.
Contributor Information
Ehud Banin, Email: Ehud.Banin@biu.ac.il.
Giordano Rampioni, University Roma Tre.
REFERENCES
- 1.Ogura T, Hiraga S. 1983. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci USA 80:4784–4788. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerdes K, Rasmussen PB, Molin S. 1986. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci USA 83:3116–3120. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall AM, Gollan B, Helaine S. 2017. Toxin–antitoxin systems: reversible toxicity. Curr Opin Microbiol 36:102–110. doi: 10.1016/j.mib.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Page R, Peti W. 2016. Toxin–antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol 12:208–214. doi: 10.1038/nchembio.2044. [DOI] [PubMed] [Google Scholar]
- 5.Christensen SK, Maenhaut-Michel G, Mine N, Gottesman S, Gerdes K, Van Melderen L. 2004. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol Microbiol 51:1705–1717. doi: 10.1046/j.1365-2958.2003.03941.x. [DOI] [PubMed] [Google Scholar]
- 6.Fraikin N, Goormaghtigh F, Van Melderen L. 2020. Type II toxin-antitoxin systems: evolution and revolutions. J Bacteriol 202:e00763-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aziz RK, Bartels D, Best A, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurėnas D, Van Melderen L. 2020. The variety in the common theme of translation inhibition by Type II toxin–antitoxin systems. Front Genet 11:262. doi: 10.3389/fgene.2020.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muthuramalingam M, White JC, Bourne CR. 2016. Toxin-antitoxin modules are pliable switches activated by multiple protease pathways. Toxins 8:214. doi: 10.3390/toxins8070214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeRoux M, Culviner PH, Liu YJ, Littlehale ML, Laub MT. 2020. Stress can induce transcription of toxin-antitoxin systems without activating toxin. Mol Cell 79:280–292.e8. doi: 10.1016/j.molcel.2020.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandey DP, Gerdes K. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res 33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christensen-Dalsgaard M, Gerdes K. 2006. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol Microbiol 62:397–411. doi: 10.1111/j.1365-2958.2006.05385.x. [DOI] [PubMed] [Google Scholar]
- 13.Pecota DC, Wood TK. 1996. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol 178:2044–2050. doi: 10.1128/jb.178.7.2044-2050.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. 2001. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci USA 98:14328–14333. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen SK, Pedersen K, Hansen FG, Gerdes K. 2003. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J Mol Biol 332:809–819. doi: 10.1016/S0022-2836(03)00922-7. [DOI] [PubMed] [Google Scholar]
- 16.Kim Y, Wang X, Ma Q, Zhang XS, Wood TK. 2009. Toxin-antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J Bacteriol 191:1258–1267. doi: 10.1128/JB.01465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen SB, Ghoul M, Griffin AS, Petersen B, Johansen HK, Molin S. 2017. Diversity, prevalence, and longitudinal occurrence of type II toxin-antitoxin systems of Pseudomonas aeruginosa infecting cystic fibrosis lungs. Front Microbiol 8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirnay JP, Bilocq F, Pot B, Cornelis P, Zizi M, Van Eldere J, Deschaght P, Vaneechoutte M, Jennes S, Pitt T, De Vos D. 2009. Pseudomonas aeruginosa population structure revisited. PLoS One 4:e7740. doi: 10.1371/journal.pone.0007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aloush V, Navon-Venezia S, Seigman-Igra Y, Cabili S, Carmeli Y. 2006. Multidrug-resistant Pseudomonas aeruginosa: risk factors and clinical impact. Antimicrob Agents Chemother 50:43–48. doi: 10.1128/AAC.50.1.43-48.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muthuramalingam M, White JC, Murphy T, Ames JR, Bourne CR. 2019. The toxin from a ParDE toxin-antitoxin system found in Pseudomonas aeruginosa offers protection to cells challenged with anti-gyrase antibiotics. Mol Microbiol 111:441–454. doi: 10.1111/mmi.14165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coskun USS, Cicek AC, Kilinc C, Guckan R, Dagcioglu Y, Demir O, Sandallı C. 2018. Effect of Mazef, Higba and Relbe toxin-antitoxin systems on antibiotic resistance in Pseudomonas aeruginosa and Staphylococcus isolates. Malawi Med J 30:67–72. doi: 10.4314/mmj.v30i2.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood TL, Wood TK. 2016. The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiologyopen 5:499–511. doi: 10.1002/mbo3.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Long Y, Liu Y, Liu Y, Chen R, Shi J, Zhang L, Jin Y, Yang L, Bai F, Jin S, Cheng Z, Wu W. 2016. HigB of Pseudomonas aeruginosa enhances killing of phagocytes by up-regulating the type III secretion system in ciprofloxacin induced persister cells. Front Cell Infect Microbiol 6:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Y, Sun C, Li Y, Tang K, Ni S, Wang X. 2019. Antitoxin HigA inhibits virulence gene mvfR expression in Pseudomonas aeruginosa. Environ Microbiol 21:2707–2723. doi: 10.1111/1462-2920.14595. [DOI] [PubMed] [Google Scholar]
- 25.Guglielmini J, Van Melderen L. 2011. Bacterial toxin-antitoxin systems. Mob Genet Elements 1:283–306. doi: 10.4161/mge.18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeRoux M, Culviner PH, Liu YJ, Littlehale ML, Laub MT. 2020. Stress can induce transcription of toxin-antitoxin systems without activating toxin. Mol Cell 79:280–292.e8. doi: 10.1016/j.molcel.2020.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valentini M, Filloux A. 2016. Biofilms and cyclic di-GMP (c-di-GMP) signaling: lessons from Pseudomonas aeruginosa and other bacteria. J Biol Chem 291:12547–12555. doi: 10.1074/jbc.R115.711507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol 187:1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simm R, Morr M, Kader A, Nimtz M, Römling U. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessibility to motility. Mol Microbiol 53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- 30.Jones CJ, Newsom D, Kelly B, Irie Y, Jennings LK, Xu B, Limoli DH, Harrison JJ, Parsek MR, White P, Wozniak DJ. 2014. ChIP-seq and RNA-seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by pseudomonas aeruginosa. PLoS Pathog 10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones CJ, Ryder CR, Mann EE, Wozniak DJ. 2013. AmrZ modulates Pseudomonas aeruginosa biofilm architecture by directly repressing transcription of the psl operon. J Bacteriol 195:1637–1644. doi: 10.1128/JB.02190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morita Y, Tomida J, Kawamura Y. 2012. Mexxy multidrug efflux system of Pseudomonas aeruginosa. Front Microbiol 3:408. doi: 10.3389/fmicb.2012.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goeders N, Van Melderen L. 2014. Toxin-antitoxin systems as multilevel interaction systems. Toxins (Basel) 6:304–324. doi: 10.3390/toxins6010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Unterholzner S, Poppenberger B, Rozhon W. 2013. Toxin–antitoxin systems: biology, identification, and application. Mob Genet Elements 3:e26219. doi: 10.4161/mge.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kasari V, Mets T, Tenson T, Kaldalu N. 2013. Transcriptional cross-activation between toxin-antitoxin systems of Escherichia coli. BMC Microbiol 13:45. doi: 10.1186/1471-2180-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu C, Holt M, Ruan S, Bourne C. 2020. Evaluating the potential for cross-interactions of antitoxins in Type II TA systems. Toxins 12:422. doi: 10.3390/toxins12060422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang M, Gao C, Wang Y, Zhang H, He ZG. 2010. Characterization of the interaction and cross-regulation of three Mycobacterium tuberculosis RelBE modules. PLoS One 5:e10672. doi: 10.1371/journal.pone.0010672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein EJC. 1987. Norfloxacin, a fluoroquinolone antibacterial agent: classification, mechanism of action, and in vitro activity. Am J Med (82):3–7. doi: 10.1016/0002-9343(87)90612-7. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y, Pogliano J, Helinski DR, Konieczny I. 2002. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44:971–979. doi: 10.1046/j.1365-2958.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Xia B, Li M, Shi J, Long Y, Jin Y, Bai F, Cheng Z, Jin S, Wu W. 2018. HigB reciprocally controls biofilm formation and the expression of type III secretion system genes through influencing the intracellular c-di-GMP level in Pseudomonas aeruginosa. Toxins (Basel) 10:424. doi: 10.3390/toxins10110424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zander I, Shmidov E, Roth S, Ben‐David Y, Shoval I, Shoshani S, Danielli A, Banin E. 2020. Characterization of PfiT/PfiA toxin‐antitoxin system of Pseudomonas aeruginosa that affects cell elongation and prophage induction. Environ Microbiol 22:5048–5057. doi: 10.1111/1462-2920.15102. [DOI] [PubMed] [Google Scholar]
- 43.Yao J, Guo Y, Wang P, Zeng Z, Li B, Tang K, Liu X, Wang X. 2018. Type II toxin/antitoxin system ParESO/CopASO stabilizes prophage CP4So in Shewanella oneidensis. Environ Microbiol 20:1224–1239. doi: 10.1111/1462-2920.14068. [DOI] [PubMed] [Google Scholar]
- 44.Karoui H, Bex F, Drèze P, Couturier M. 1983. Ham22, a mini-F mutation which is lethal to host cell and promotes recA-dependent induction of lambdoid prophage. EMBO J 2:1863–1868. doi: 10.1002/j.1460-2075.1983.tb01672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borges AL, Castro B, Govindarajan S, Solvik T, Escalante V, Bondy-Denomy J. 2020. Bacterial alginate regulators and phage homologs repress CRISPR–Cas immunity. Nat Microbiol 5:679–687. doi: 10.1038/s41564-020-0691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goormaghtigh F, Fraikin N, Putrinš M, Hallaert T, Hauryliuk V, Garcia-Pino A, Sjödin A, Kasvandik S, Udekwu K, Tenson T, Kaldalu N, Van Melderen L. 2018. Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. mBio 9:e00640-18. doi: 10.1128/mBio.00640-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamruzzaman M, Iredell J. 2019. A ParDE-family toxin antitoxin system in major resistance plasmids of Enterobacteriaceae confers antibiotic and heat tolerance. Sci Rep 9:9872. doi: 10.1038/s41598-019-46318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulet X, Moyá B, Juan C, Macià MD, Pérez JL, Blázquez J, Oliver A. 2011. Antagonistic interactions of Pseudomonas aeruginosa antibiotic resistance mechanisms in planktonic but not biofilm growth. Antimicrob Agents Chemother 55:4560–4568. doi: 10.1128/AAC.00519-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hmelo LR, Borlee RB, Almblad H, Love ME, Randall TE, Tseng TS, Lin C, Irie Y, Storek KM, Yang JJ, Siehnel RJ, Howell PL, Singh PK, Nielsen-Tolker T, Parsek MR, Schweizer PH, Harrison JJ. 2015. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc 10:1820–1841. doi: 10.1038/nprot.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ben-David Y, Zlotnik E, Zander I, Yerushalmi G, Shoshani S, Banin E. 2018. SawR a new regulator controlling pyomelanin synthesis in Pseudomonas aeruginosa. Microbiol Res 206:91–98. doi: 10.1016/j.micres.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Choi KH, Schweizer HP. 2006. Mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 52.Becher A, Schweizer HP. 2000. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29:948–952. doi: 10.2144/00295bm04. [DOI] [PubMed] [Google Scholar]
- 53.Battesti A, Bouveret E. 2012. The bacterial two-hybrid system based on adenylate cyclase reconstitution in Escherichia coli. Methods 58:325–334. doi: 10.1016/j.ymeth.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 54.Chowdhury N, Kwan BW, Wood TK. 2016. Persistence increases in the absence of the alarmone guanosine tetraphosphate by reducing cell growth. Sci Rep 6:20519. doi: 10.1038/srep20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anders S, Pyl PT, Huber W. 2015. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FSL. 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ. 1990. Basic Local Alignment Search Tool. J Mol Biol 215:8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.01182-22-s001.pdf, PDF file, 1 MB (998.8KB, pdf)
Data Availability Statement
The data that support the findings of this study are available in the GEO database (accession number GSE179116).