

Abstract
PURPOSE
Platinum-based chemotherapy is the standard of care for platinum-sensitive ovarian cancer, but complications from repeated platinum therapy occur. We assessed the activity of two all-oral nonplatinum alternatives, olaparib or olaparib/cediranib, versus platinum-based chemotherapy.
PATIENTS AND METHODS
NRG-GY004 is an open-label, randomized, phase III trial conducted in the United States and Canada. Eligible patients had high-grade serous or endometrioid platinum-sensitive ovarian cancer. Patients were randomly assigned 1:1:1 to platinum-based chemotherapy, olaparib, or olaparib/cediranib. The primary end point was progression-free survival (PFS) in the intention-to-treat population. Secondary end points included activity within germline BRCA-mutated or wild-type subgroups and patient-reported outcomes (PROs).
RESULTS
Between February 04, 2016, and November 13, 2017, 565 eligible patients were randomly assigned. Median PFS was 10.3 (95% CI, 8.7 to 11.2), 8.2 (95% CI, 6.6 to 8.7), and 10.4 (95% CI, 8.5 to 12.5) months with chemotherapy, olaparib, and olaparib/cediranib, respectively. Olaparib/cediranib did not improve PFS versus chemotherapy (hazard ratio [HR] 0.86; 95% CI, 0.66 to 1.10; P = .077). In women with germline BRCA mutation, the PFS HR versus chemotherapy was 0.55 (95% CI, 0.32 to 0.94) for olaparib/cediranib and 0.63 (95% CI, 0.37 to 1.07) for olaparib. In women without a germline BRCA mutation, the PFS HR versus chemotherapy was 0.97 (95% CI, 0.73 to 1.30) for olaparib/cediranib and 1.41 (95% CI, 1.07 to 1.86) for olaparib. Hematologic adverse events occurred more commonly with chemotherapy; however, nonhematologic adverse events were higher with olaparib/cediranib. In 489 patients evaluable for PROs, patients receiving olaparib/cediranib scored on average 1.1 points worse on the NFOSI-DRS-P subscale (97.5% CI, –2.0 to –0.2, P = .0063) versus chemotherapy; no difference between olaparib and chemotherapy was observed.
CONCLUSION
Combination olaparib/cediranib did not improve PFS compared with chemotherapy and resulted in reduced PROs. Notably, in patients with a germline BRCA mutation, both olaparib and olaparib/cediranib had significant clinical activity.
INTRODUCTION
Ovarian cancer remains a leading cause of death for women worldwide, with an estimated 313,959 cases and 207,252 deaths occurring in 2020.1 Although response rates to initial treatment are high, most women experience disease recurrence. The cancer is considered platinum-sensitive when recurrence occurs 6 months or more after completing initial platinum-based therapy. The current standard of care in this clinical setting is a platinum-based doublet with or without bevacizumab. If bevacizumab is not used, poly-ADP-ribose polymerase (PARP) inhibitor switch maintenance therapy following completion of chemotherapy may be considered.
CONTEXT
Key Objective
This phase III trial examined whether all-oral nonplatinum regimens of combination olaparib and cediranib or olaparib monotherapy could improve progression-free survival compared with standard-of-care platinum-based chemotherapy in patients with relapsed platinum-sensitive ovarian cancer.
Knowledge Generated
Neither combination olaparib and cediranib nor olaparib monotherapy improved progression-free survival compared with standard-of-care chemotherapy. However, both olaparib/cediranib and olaparib monotherapy had evidence of clinical activity, most substantially in women with a germline BRCA mutation.
Relevance
Platinum-based chemotherapy remains the standard of care for women with relapsed platinum-sensitive ovarian cancer. The observed activity of olaparib/cediranib and olaparib, particularly in patients with BRCA-mutated tumors, warrants further exploration and development of non–platinum-based alternatives, especially in selected patient populations with platinum-sensitive ovarian cancer.
Platinum-based therapy is highly active in women with platinum-sensitive ovarian cancer. However, there are complications that can occur with repeated platinum therapy, including development of a platinum allergy, cumulative hematologic toxicity, and exacerbation of neuropathy.2 Because of these cumulative risks and the difficulty of extending chemotherapy beyond six cycles, a well-tolerated long-term nonplatinum alternative would be an attractive option in this population.
Studies have suggested that PARP inhibitors and antiangiogenic agents may act synergistically in platinum-sensitive ovarian cancer,3-6 potentially because of downregulation of genes associated with homologous recombination in the setting of hypoxic stress, resulting in increased homologous recombination deficiency.7,8 A preclinical study of cediranib, a small-molecule tyrosine kinase inhibitor with anti–angiogenic activity, together with olaparib suggested that the anti–platelet-derived growth factor receptor activity of cediranib could also lead to increased homologous recombination deficiency independent of hypoxic stress, with resulting increased sensitivity to PARP inhibition.9 A phase II study comparing olaparib monotherapy to combination olaparib/cediranib in platinum-sensitive relapsed ovarian cancer demonstrated superiority of the olaparib/cediranib combination, with a significantly increased progression-free survival (PFS) from 9.0 to 17.7 months (hazard ratio [HR] 0.42; 95% CI, 0.23 to 0.76).3,4 In the context of these emerging data, this phase III study was conducted to confirm whether combination olaparib/cediranib was superior to olaparib alone, and to examine whether it yielded a superior PFS compared with platinum-based chemotherapy in platinum-sensitive relapsed ovarian cancer.
PATIENTS AND METHODS
Patient Selection
Eligible patients had platinum-sensitive relapsed high-grade serous or high-grade endometrioid ovarian, primary peritoneal, or fallopian tube cancer (collectively referred to as ovarian cancer), who had received a first-line platinum-based chemotherapy and had recurred > 6 months after their most recent platinum-based chemotherapy. Key exclusion criteria included prior receipt of PARP inhibitor or receipt of antiangiogenic therapy in the recurrent setting. Full eligibility criteria are available in the Appendix 1 (online only).
Study Design
This was a phase III, open-label, randomized study conducted through the NRG Oncology research group at participating sites in the United States and Canada. Participants randomly assigned to receive platinum-doublet chemotherapy received carboplatin and paclitaxel, carboplatin and gemcitabine, or carboplatin and pegylated liposomal doxorubicin, per investigator choice. Chemotherapy could be continued for as long as deemed appropriate. After completion of chemotherapy, all patients were to be followed without further cancer-directed therapy, thus specifically precluding bevacizumab or PARP inhibitor switch maintenance therapy, until the time of disease progression.
Olaparib monotherapy included olaparib 300 mg tablets twice daily and olaparib/cediranib included olaparib 200 mg tablets twice daily with cediranib 30 mg tablet once daily; both regimens were continuously dosed. Participants continued treatment until disease progression, discontinuation because of adverse events (AEs), or withdrawal; participants could continue on just one study drug in select circumstances after discussion with the study chair.
Radiographic tumor assessments were performed every 9 weeks for the first year and then every 12 weeks until disease progression, independent of delays and/or changes in the treatment schedule.
Patient-reported outcomes (PRO) were captured through three instruments: NCCN/FACT-Ovarian Cancer Symptom Index-18 (NFOSI-18), FACT/GOG-Ntx-4 measure of sensory neuropathy, and the five-item EQ-5D measure of patient preference.10-12 PROs were collected every 12 weeks for 3 years including after stopping study treatment, unless the participant died or declined PRO completion.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines, and was approved by the appropriate institutional review board for each participating site. All participants provided written informed consent. The study was sponsored by the US National Cancer Institute (NCI) and registered on ClinicalTrials.gov identifier: NCT02446600.
Statistical Procedures and Analyses
The original design had power = 0.85 to detect an HR = 0.65 using a one-sided alpha level .025 test. Randomized treatment comparisons of the PFS and overall survival (OS) end points were grouped separately, with the PFS family tested first. Within each family, the order of testing was as follows: (1) olaparib/cediranib versus chemotherapy; (2) olaparib versus chemotherapy; and (3) olaparib/cediranib versus olaparib. Type I error was strongly controlled by a hierarchical gatekeeping procedure. The alpha was forwarded to each test in the sequence, stopping with the first null hypothesis that failed to reject. There was an interim PFS futility analysis at 50% information time; the protocol also allowed for an OS analysis when the PFS end point matured if the PFS null hypothesis for chemotherapy versus olaparib/cediranib was rejected. The original design included 450 participants, with PFS maturity when 204 events occurred for the first test in the PFS family. In July 2017, the sample size was increased to 549 patients to accommodate increased participation without changes to the statistical analysis plan. The protocol was amended in August 2018 to adjust for potential biases from participants initiating nonprotocol maintenance therapy, with the number of events for PFS maturity increased to 265 and power reduced to 80% for a diluted HR = 0.70. In August 2019, the protocol was amended to limit follow-up time to 24 months after last patient enrollment. Simulations suggested approximately 236 events would be available, with 90% power for an undiluted HR = 0.65 or 78% power for a diluted HR = 0.70. Additional details regarding the statistical design are provided in the Appendix 1.
Before random assignment, comprehensive analysis of germline BRCA status (BRACANalysis, Myriad Genetics, Salt Lake City, UT) was performed on patients without prior germline BRCA1 and BRCA2 (BRCA1/2) testing or with less than full sequencing of BRCA1/2 in a CLIA-certified setting. Participants were enrolled via a web-based registration system. Three protocol treatment regimens were assigned in a 1:1:1 fashion using random permuted blocks, stratified by germline BRCA1/2 mutation (yes v no), prior platinum-free interval (6-12 months v > 12 months), and prior receipt of antiangiogenic treatment (yes v no). Treatment assignment remained concealed until the registration process was completed.
The primary end point was investigator-assessed PFS by RECIST 1.1 criteria. For an intention-to-treat analysis, patients were grouped according to randomized treatment, regardless of compliance with the assigned treatment, and included all patients who received maintenance therapy in the control arm in violation of the protocol procedures. The primary analysis was supported by the 565 patients enrolled onto the phase III component of the study. PFS was defined as time in months from random assignment to either death or disease progression, whichever occurred first. Participants who were alive with no progression before the cutoff date were censored on the date of last tumor assessment, the date of the assessment before missing two consecutive assessments, or the date of consent withdrawal, whichever occurred first. Key secondary end points included OS and response. Responses were defined as best overall response per RECIST 1.1 of complete or partial response. Assessment of activity within subgroups by BRCA status was a key preplanned subanalysis.
The primary PRO end point was the NFOSI-DRS-P (a nine-item targeted symptom index from the NFOSI-18 designed for clinical research in advanced ovarian cancer).10-12 A mixed-effects model was used to estimate and compare the mean NFOSI-DRS-P scores during the 2 years following enrollment. The model was adjusted for assigned study treatment, age at enrollment, initial performance status, pretreatment PRO score, and assessment time.
RESULTS
Patients
Between February 4, 2016, and November 13, 2017, 565 eligible patients were enrolled and randomly assigned (Fig 1). Twenty-eight patients (20 chemotherapy, six olaparib/cediranib, and two olaparib) did not initiate their assigned study treatment. Patient baseline characteristics are shown in Table 1 and were balanced across the arms. Most (> 90%) of participants had high-grade serous cancer. Approximately 65% of patients had received only one prior therapy; fewer than 10% had received a prior antiangiogenic. Close to 25% of patients had a deleterious germline BRCA1/2 mutation. The distribution of chemotherapy choice for patients randomly assigned to platinum-based chemotherapy was carboplatin and pegylated liposomal doxorubicin in 89 patients (47.6%), carboplatin and gemcitabine in 51 patients (27.2%), and carboplatin and paclitaxel in 47 patients (25.1%).
FIG 1.

CONSORT diagram. Only patients in the chemotherapy arm completed planned treatment (completed chemotherapy course, followed by surveillance), as olaparib and olaparib + cediranib were continued until the time of disease progression. As of the data cutoff date (November 13, 2019). AE, adverse event; ITT, intention-to-treat.
TABLE 1.
Baseline Patient Characteristics

Efficacy
Olaparib/cediranib did not improve PFS compared with platinum-based chemotherapy, with an HR of 0.856 (95% CI, 0.66 to 1.10; P value 0.077; Fig 2). The observed median PFS was 10.4 (95% CI, 8.5 to 12.5) months for olaparib/cediranib and 10.3 (95% CI, 8.7 to 11.2) months for chemotherapy. Additional treatment comparisons were not formally conducted because the null hypothesis was not rejected for the comparison of olaparib/cediranib to chemotherapy. However, the HR of olaparib compared with chemotherapy was 1.2 (95% CI, 0.93 to 1.5), with a median PFS of 8.2 (95% CI, 6.6 to 8.7) months for olaparib. OS data are not mature; with 251 total events, there was no difference in median OS between the three arms, with point estimates of 31.3 (95% CI, 26.4 to 34.4) months for chemotherapy, 30.5 (95% CI, 27.0 to 36.5) months for olaparib/cediranib, and 29.2 (95% CI, 23.9 to 32.7) months for olaparib when the study data were frozen for these analyses. The objective response rate was 71.3% (95% CI, 63.4 to 78.1) to chemotherapy, 69.4% (95% CI, 61.8 to 76.1) to olaparib/cediranib, and 52.4% (95% CI, 44.8 to 60.1) to olaparib. There was no statistically significant difference in response rates between olaparib/cediranib and chemotherapy. However, the response rate to olaparib was statistically lower than chemotherapy (P < .001).
FIG 2.

Progression-free survival by randomized treatment. Distributions were estimated by means of the Kaplan-Meier method for progression-free survival by investigator assessment.
A prespecified subgroup analysis was performed based upon germline BRCA1/2 status. The median PFS was 10.5 (95% CI, 9.0 to 12.8) months with chemotherapy, 18.0 (95% CI, 12.6 to 22.1) months with olaparib/cediranib, and 12.7 (95% CI, 9.3 to 17.7) months with olaparib in the subgroup of women with deleterious germline BRCA1/2 mutation (Fig 3A). The PFS HR estimates compared with chemotherapy were 0.55 (95% CI, 0.32 to 0.94) for olaparib/cediranib and 0.63 (95% CI, 0.37 to 1.07) for olaparib. Response rates were 70.6% (95% CI, 53.8 to 83.2) with chemotherapy, 88.9% (95% CI, 74.7 to 95.6) with olaparib/cediranib, and 90.0% (95% CI, 76.9 to 96.0) with olaparib. Women without a deleterious germline BRCA1/2 mutation had a median PFS of 9.7 (95% CI, 8.4 to 11.2) months with chemotherapy, 8.9 (95% CI, 8.3 to 10.4) months with olaparib/cediranib, and 6.6 (95% CI, 6.2 to 8.1) months with olaparib (Fig 3B). The HR estimates compared with chemotherapy were 0.97 (95% CI, 0.73 to 1.30) for olaparib/cediranib and 1.41 (95% CI, 1.07 to 1.86) for olaparib. Response rates were 71.6% (95% CI, 62.5 to 79.2) with chemotherapy, 63.6% (95% CI, 54.8 to 71.7) with olaparib/cediranib, and 40.0% (95% CI, 31.7 to 48.9) with olaparib.
FIG 3.
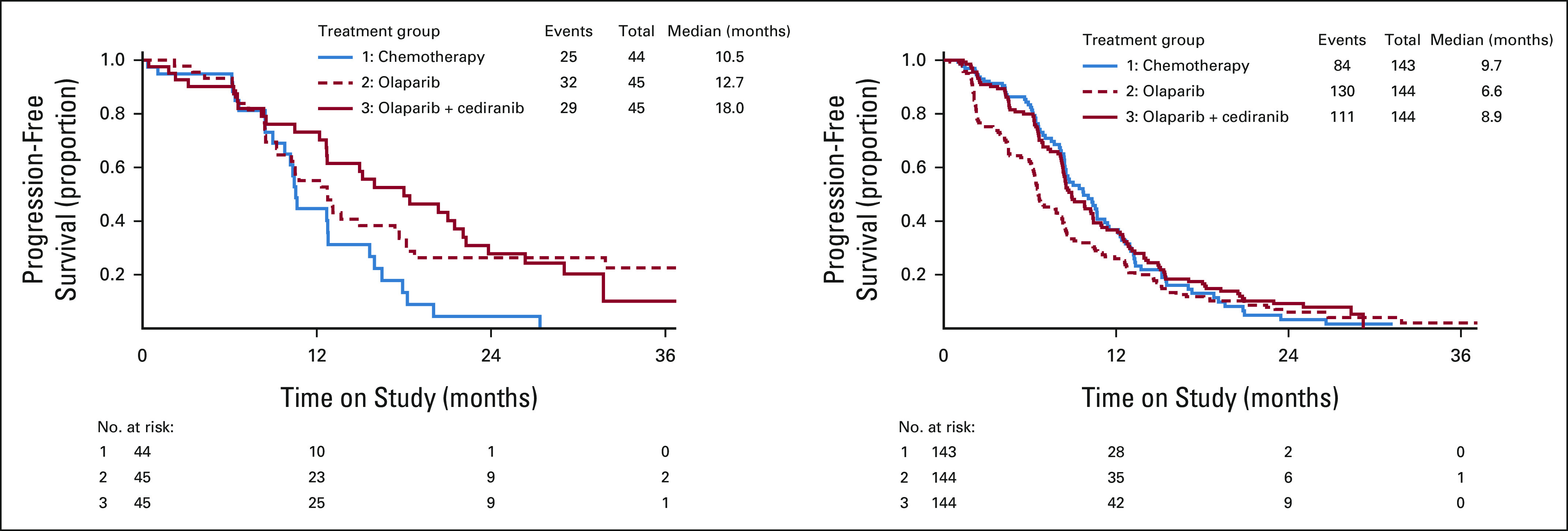
PFS in BRCA1/2-mutation subgroups. Progression-free survival estimates by Kaplan-Meier method are shown for (A) participants with a deleterious germline BRCA1/2 mutation and (B) participants without a deleterious germline BRCA1/2 mutation.
Twenty-eight percent of patients in the chemotherapy arm received non–protocol-directed therapy, predominantly PARP inhibitor maintenance, in the setting of approval of PARP inhibitors for maintenance therapy by the US Food and Drug Administration during the course of the study. In a prespecified sensitivity analysis added to censor patients at the time of initiating PARP inhibitor maintenance, the results were consistent with the primary analysis (median PFS 10.4 [95% CI, 8.5 to 12.5] months for olaparib/cediranib and 10.2 [95% CI, 8.6 to 11.2] months for chemotherapy, with an HR of 0.90 [95% CI, 0.69 to 1.18]; Appendix Fig A1, online only).
Safety
Treatment-emergent AEs occurring in at least 20% of patients in any of the three study arms are listed in Table 2. In general, patients receiving chemotherapy had more frequent total and grade 3 or higher hematologic AEs, although anemia was similar in occurrence and severity with olaparib. However, nonhematologic AEs were more common among patients receiving olaparib/cediranib, with the exception of peripheral sensory neuropathy, which occurred more frequently with chemotherapy. The most frequently observed nonhematologic AEs with olaparib/cediranib included diarrhea (82.6% of patients; 13.7% grade 3+), fatigue (80.9%; 17.5% grade 3+), nausea (73.8%; 4.4% grade 3+), and hypertension (69.9%; 31.7% grade 3+). Four patient deaths on study were deemed possibly related to study treatment: two patients were on chemotherapy (one sepsis and one aspiration), one on olaparib/cediranib (sepsis), and one on olaparib (worsening hydronephrosis). Four events of myelodysplastic syndrome or leukemia were reported: one on chemotherapy, two on olaparib/cediranib, and one on olaparib.
TABLE 2.
Treatment-Emergent Adverse Events

PROs
Quality-of-life data were evaluable for 489 participants; 76 participants were unevaluable because of lack of baseline PROs (22 chemotherapy, nine olaparib/cediranib, and five olaparib) or lack of follow-up PROs (14 chemotherapy, 13 olaparib/cediranib, and 13 olaparib). No statistically significant differences between the arms with regards to patient or disease characteristics were observed among patients evaluable for PROs. Patient-reported NFOSI-DRS-P scores over time are presented in Figure 4. The fitted linear mixed model showed no significant interaction effect between treatment groups and assessment time after adjustment for baseline age, performance status, and score. On average, patients on olaparib/cediranib scored 1.1 points worse on the NFOSI-DRS-P subscale score (97.5% CI, –2.0 to –0.2; P = .0063) than those on chemotherapy. The strongest decrement in the NFOSI-DRS-P subscale score was observed 12 weeks after starting treatment, when patients on olaparib/cediranib reported scores 1.9 points worse than those on chemotherapy (97.5% CI, –3.2 to –0.7; P = .0007). No significant difference between olaparib and chemotherapy in terms of the NFOSI-DRS-P subscale score was observed (estimated difference 0.2; 95% CI, –0.7 to 1.0; P = .7).
FIG 4.
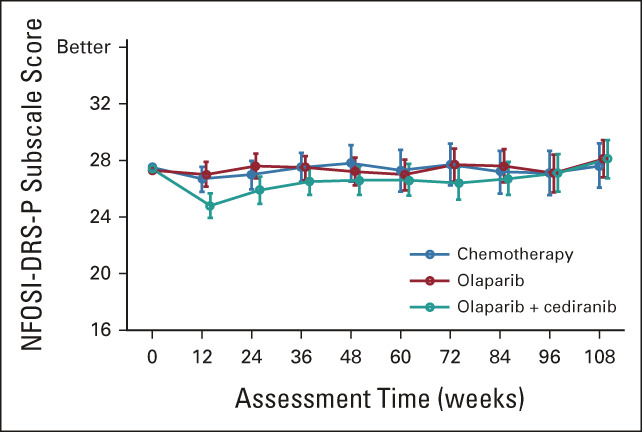
Mean NFOSI-DRS-P scores over time. Larger scores suggest a better or preferable state of health.
DISCUSSION
This is the first phase III trial to compare an all-oral nonplatinum regimen to platinum-based chemotherapy in women with platinum-sensitive recurrent ovarian cancer. Combination olaparib/cediranib did not meet the primary end point of improving PFS compared with platinum-based chemotherapy, but significant clinical activity in terms of both PFS and objective response rates was observed. In a prespecified subgroup analysis within patients with a known germline BRCA1/2 mutation, significant clinical activity was observed for both olaparib/cediranib and olaparib monotherapy.
An ongoing question in the ovarian cancer space has been whether PARP inhibitors could provide a nonplatinum alternative with comparable or superior activity to platinum-based therapy in women with BRCA1/2-mutated platinum-sensitive ovarian cancer. The SOLO3 study of olaparib monotherapy in recurrent platinum-sensitive ovarian cancer reported superior olaparib activity compared with non–platinum-based chemotherapy, but does not include the use of platinum-based chemotherapy.13 In NRG-GY004, neither olaparib/cediranib nor olaparib demonstrated superiority to platinum chemotherapy in this patient population; additionally, this trial was not powered for an analysis of noninferiority. However, the observed significant activity of both olaparib/cediranib and olaparib monotherapy in women with germline BRCA1/2 mutation supports further direct exploration of this question in this patient population. The results reported from the ARIEL4 study of rucaparib compared with chemotherapy in recurrent BRCA1/2-mutated ovarian cancer support the observations from NRG-GY004, although this study included women with both platinum-resistant and platinum-sensitive ovarian cancer, and women with platinum-sensitive disease whose recurrence was within 6-12 months of their last platinum-based chemotherapy were treated with weekly paclitaxel and not platinum-based chemotherapy.14
The median PFS observed with olaparib/cediranib in the prior phase II trial was 17.7 months. This compares favorably to historical activity reported for PFS to platinum-based chemotherapy in randomized phase III trials in this population, which ranges from 8.4 to 13.8 months.15-21 By contrast, in this trial, the observed median PFS with olaparib/cediranib was less at 10.4 months. This decreased PFS may be due to study population differences between trials; however, the median PFS for olaparib monotherapy between the two trials was similar (8.2 months in this trial and 9.0 months in the phase II trial), suggesting that the two study populations may not have been that dissimilar in terms of expected clinical outcome. It is possible that the decreased dosing intensity and increased rate of discontinuation also contributed to the decreased clinical activity of olaparib/cediranib observed in this trial. Numerous strategies were provided to participants to guide amelioration of cediranib-associated symptoms. Further strategies can include using a different dosing regimen such as olaparib tablets 300 mg twice daily with cediranib 20 mg once daily on a continuous basis, a regimen being examined in the ongoing ICON9 study. A bridging study examining use of olaparib tablets in combination with cediranib demonstrated similarity between the two regimens across activity and tolerance.22 Further strategies directed toward maximizing exposure while managing AEs and limiting detrimental effects on quality of life remain important in the further development of this combination.
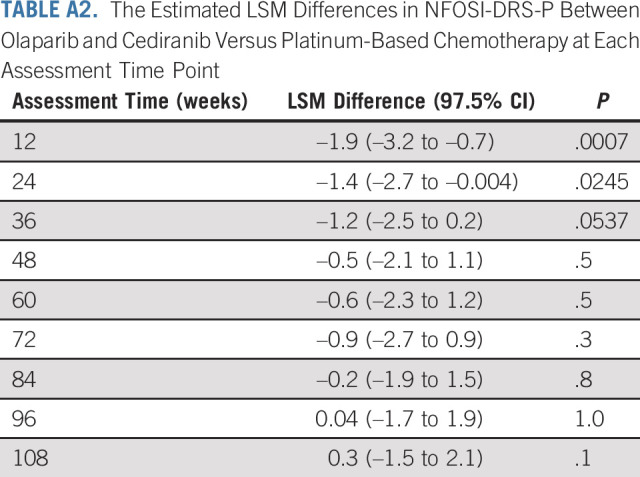
The AE profile for olaparib/cediranib was as expected and reported in prior trials.3,4,23 The most common AEs included diarrhea, fatigue, nausea, and hypertension. However, in this trial, 21.2% of participants withdrew from the olaparib/cediranib arm because of an AE, compared with 9.1% in a prior phase II trial.3 Additionally, dose modifications were common on olaparib/cediranib, with 71.6% of participants having at least one dose modification because of an AE within the first 12 cycles of therapy (Appendix Table A1, online only). The primary PRO outcome reported statistically worse disease-related physical symptoms, as measured with NFOSI-DRS-P, a finding in concordance with the observed AEs. As some measures included in the NFOSI-DRS-P (lack of energy, fatigue, cramps in the stomach area, and control of bowels) may overlap with common AEs reported with cediranib, the decrease in NFOSI-DRS-P observed in patients receiving olaparib/cediranib may reflect side effects of treatment instead of differences in disease-related symptoms. The difference in NFOSI-DRS-P scores was most pronounced at the 12-week assessment but subsequently decreased (Appendix Table A2, online only), which could reflect adjustments to the dosing regimen in response to AEs.
In this trial, of a nonchemotherapy containing combination therapy in platinum-sensitive relapsed ovarian cancer, olaparib/cediranib did not meet the primary end point of increasing PFS compared with platinum-doublet chemotherapy. Nonetheless, olaparib/cediranib demonstrated some evidence of clinical activity in patients with platinum-sensitive ovarian cancer. This observed activity opens the door for further investigation of nonchemotherapy approaches that may spare patients from cumulative chemotherapy-associated end-organ injuries. Nonchemotherapy alternatives to standard chemotherapy options remain of interest, and notably, 20 patients enrolled to this study withdrew from participation after being randomly assigned to receive chemotherapy, supporting the notion that patients are invested in finding nonchemotherapy alternatives. A small number of women remained on olaparib/cediranib (10 patients) or olaparib (15 patients) at the time of this analysis; ongoing biomarker studies may help identify patients most likely to benefit from these nonplatinum alternatives. Further studies of olaparib/cediranib in ovarian cancer are ongoing, including NRG-GY005 (ClinicalTrials.gov identifier: NCT02502266), which compares olaparib/cediranib to non–platinum-based chemotherapy in platinum-resistant ovarian cancer and ICON9 (ClinicalTrials.gov identifier: NCT03278717), which examines olaparib/cediranib as a maintenance therapy following platinum-based chemotherapy.
ACKNOWLEDGMENT
The following NRG Oncology/Gynecologic Oncology Group member institutions participated in this study: The US Oncology Network; Memorial Sloan-Kettering Cancer Center LAPS; New Mexico Minority Underserved NCORP; University of Iowa/Holden Comprehensive Cancer Center; University of Rochester; Cancer Research Consortium of West Michigan NCORP; CWRU Case Comprehensive Cancer Center LAPS; University of Minnesota/Masonic Cancer Center; Saitama Medical University International Medical Center; Sutter Cancer Research Consortium; Hawaii Minority Underserved NCORP; Duke University–Duke Cancer Institute LAPS; University of Oklahoma Health Sciences Center LAPS; Catholic Health Initiatives NCORP; University of Texas Southwestern Medical Center LAPS; Cancer Research for the Ozarks NCORP; Women and Infants Hospital; Washington University–Siteman Cancer Center LAPS; University of Pittsburgh Cancer Institute LAPS; Yale University–Yale Cancer Center LAPS; Dana-Farber/Partners Cancer Care LAPS; Fred Hutchinson Cancer Research Center LAPS; Metro Minnesota Community Oncology Research Consortium; NorthShore University Health System-Evanston Hospital; Kaiser Permanente NCI Community Oncology Research Program; University of Colorado Cancer Center LAPS; Wisconsin NCI Community Oncology Research Program; Georgia NCI Community Oncology Research Program; University of Cincinnati/Barrett Cancer Center; Pacific Cancer Research Consortium NCORP; Marin Cancer Care Inc; Northwestern University; Rush University Medical Center; AMITA Health Alexian Brothers Medical Center; University of Alabama at Birmingham/Deep South Research Consortium; LAPS; Roswell Park Cancer Institute LAPS; Ohio State University Comprehensive Cancer Center LAPS; Michigan Cancer Research Consortium NCORP; University of Pennsylvania/Abramson Cancer Center; Thomas Jefferson University Hospital; Southeast Clinical Oncology Research (SCOR) Consortium NCORP; Odette Cancer Centre–Sunnybrook Health Sciences Centre; UC San Diego Moores Cancer Center; University of Kansas Cancer Center; Greater Baltimore Medical Center; University of Virginia Cancer Center; Abington Memorial Hospital-Asplundh Cancer Pavilion; University Health Network-Princess Margaret Hospital; Delaware/Christiana Care NCI Community Oncology Research Program; Columbus NCI Community Oncology Research Program; Dartmouth College–Norris Cotton Cancer Center LAPS; University of Wisconsin Carbone Cancer Center LAPS; Wake Forest University Health Sciences; Avera Cancer Institute; Northwestern Medicine Cancer Center Warrenville; Cedars-Sinai Medical Center; Ottawa Hospital and Cancer Center-General Campus; Jewish General Hospital; Juravinski Cancer Centre at Hamilton Health Sciences; University of Arkansas for Medical Sciences; UCSF Medical Center-Mount Zion; University of Florida Health Science Center—Gainesville; Geisinger Cancer Institute NCI Community Oncology Research Program; Norton Hospital Pavilion and Medical Campus; University of Michigan Comprehensive Cancer Center LAPS; Wayne State University—Karmanos Cancer Institute LAPS; Mayo Clinic LAPS; UNC Lineberger Comprehensive Cancer Center LAPS; Henry Ford Hospital; Northwest NCI Community Oncology Research Program; WellSpan Health-York Hospital; Colorado Cancer Research Program NCORP; Hartford Hospital; University of Kentucky/Markey Cancer Center; JHU Sidney Kimmel Comprehensive Cancer Center LAPS; Maine Medical Center–Scarborough Campus; Montana Cancer Consortium NCORP; NHRMC Radiation Oncology—16th Street; Cleveland Clinic Akron General; Stroger Hospital of Cook County Minority Underserved NCORP; Thompson Cancer Survival Center; Wichita NCI Community Oncology Research Program; CHUM–Centre Hospitalier de l'Universite de Montreal; CHU de Quebec-L'Hotel-Dieu de Quebec (HDQ); Dayton NCI Community Oncology Research Program; Piedmont Hospital; Georgia Cares Minority Underserved NCORP; Heartland Cancer Research NCORP; Montefiore Minority Underserved NCORP; Trinity Cancer Care Center; Cooper Hospital University Medical Center; Ochsner NCI Community Oncology Research Program; Allegheny General Hospital; Sanford NCI Community Oncology Research Program of the North Central Plains; VCU Massey Cancer Center Minority Underserved NCORP; Froedtert and the Medical College of Wisconsin; West Virginia University Charleston Division; University of South Alabama Mitchell Cancer Institute; and Mercy Cancer Center—Sacramento.
APPENDIX 1. Supplemental Methods
Full Inclusion and Exclusion Criteria
Eligibility criteria (November 7, 2016) (July 31, 2017).
A patient cannot be considered eligible for this study unless ALL of the following conditions are met.
Patients must have platinum-sensitive recurrent high-grade serous or high-grade endometrioid ovarian, primary peritoneal, or fallopian tube cancers.
Patients with other (clear cell, mixed epithelial, undifferentiated carcinoma, or transitional cell carcinoma) high-grade histologies are also eligible, provided that the patient has a known deleterious germline BRCA1 or BRCA2 mutation identified through testing at a clinical laboratory.
Note: Because of the long acceptance of germline BRCA testing through Myriad, Myriad testing will be accepted. If testing for germline BRCA is done by other organizations, documentation from a qualified medical professional (eg, ovarian cancer specialty physician involved in the field, high-risk genetics physician, or genetics counselor) listing the mutation and confirming that the laboratory results showed a recognized germline deleterious BRCA1 or BRCA2 mutation or BRCA rearrangement is required. Please collect a copy of Myriad or other BRCA mutational analysis (positive or VUS or negative) reports.
○ Platinum-sensitive disease defined as no clinical or radiographic evidence of disease recurrence for > 6 months (or 182 days) after last receipt of platinum-based therapy.
○ Patients must have had a complete clinical response to their prior line of platinum therapy and cannot have had progression through prior platinum-based therapy.
• Patients must have signed an approved informed consent and authorization permitting release of personal health information.
• Patients must have evaluable disease—defined as one of the following:
○ RECIST 1.1 measurable disease OR
○ Evaluable disease (defined as solid and/or cystic abnormalities on radiographic imaging that do not meet RECIST 1.1 definitions for target lesions OR ascites and/or pleural effusion that has been pathologically demonstrated to be disease-related) AND a cancer antigen 125 (CA-125) that has doubled from the post-treatment nadir and is also > 2 times upper limit of normal (ULN).
• Prior therapy:
○ Prior chemotherapy must have included a first-line platinum-based regimen with or without intravenous consolidation chemotherapy.
○ Patients may have received an unlimited number of platinum-based therapies in the recurrent setting.
○ Patients may have received up to one non–platinum-based line of therapy in the recurrent setting. Prior hormonal therapy will not be considered to count as this non–platinum-based line.
○ Patients may not have had a prior antiangiogenic agent in the recurrent setting. Prior use of bevacizumab in the upfront or upfront maintenance setting is allowed.
○ Patients may not have previously received a poly (ADP-ribose) polymerase (PARP) inhibitor.
○ Prior hormonal-based therapy for ovarian, primary peritoneal, or fallopian tube cancer is acceptable.
• Patients must have an Eastern Cooperative Oncology Group performance status of 0, 1, or 2 (Karnofsky ≥ 60% [See Appendix I, Protocol])
• Patients must have adequate organ and marrow function, including
○ Absolute neutrophil count ≥ 1,500/mcL
○ Platelets > 100,000/mcL
○ Hemoglobin ≥ 10 g/dL
○ Creatinine ≤ the institutional ULN OR creatinine clearance ≥ 60 mL/min/1.73 m2 for patients with creatinine levels above institutional normal.
○ Urine protein: creatinine ratio (UPC) of ≤ 1 or ≤ 2+ proteinuria on two consecutive dipsticks taken no < 1 week apart. UPC is the preferred test. Patients with ≥ 2+ proteinuria on dipstick must also have a 24-hour urine collection demonstrating ≤ 500 mg over 24 hours.
○ Total bilirubin ≤ 1.5× the institutional ULN
○ AST and ALT ≤ 3 times institutional ULN.
• Toxicities of prior therapy (excepting alopecia) should be resolved to less than or equal to grade 1 as per NCI-CTCAE (located on the CTEP website at National Cancer Institute).24 Patients with long-standing stable grade 2 neuropathy may be considered after discussion with the overall PI, but may not receive carboplatin and paclitaxel as the reference regimen, if randomly assigned to that arm.
• Patients must be able to swallow and retain oral medications and without gastrointestinal illnesses that would preclude absorption of cediranib or olaparib.
• Patients must have adequately controlled blood pressure (BP), with a BP of > 140 mmHg (systolic) and 90 mmHg (diastolic) for eligibility. Patients must have a BP of ≤ 140/90 mmHg taken in the clinic setting by a medical professional within 2 weeks before starting study. Patients with hypertension may be managed with up to a maximum of three antihypertensive medications. It is strongly recommended that patients who are on three antihypertensive medications be followed by a cardiologist or blood pressure specialist for management of blood pressure while on protocol.
• Patients must be willing and able to check and record daily blood pressure readings. Blood pressure cuffs will be provided to patients randomly assigned to Arm III. Please refer to sections 9.7 and Appendix IX (Protocol).
• Cediranib has been shown to terminate fetal development in the rat, as expected for a process dependent on vascular endothelial growth factor signaling. For this reason, women of child-bearing potential must have a negative pregnancy test before study entry. Women of child-bearing potential must agree to use two reliable forms of contraception (hormonal or barrier method of birth control; abstinence) before study entry, for the duration of study participation, and for 6 weeks after cediranib discontinuation. Should a woman become pregnant or suspect she is pregnant while participating in this study, she should inform her treating physician immediately.
• Adequately controlled thyroid function, with no symptoms of thyroid dysfunction and thyroid stimulating hormone within normal limits.
• Age > 18 years.
Ineligibility criteria (November 7, 2016).
Patients with one or more of the following conditions are NOT eligible for this study.
• Patients who have had chemotherapy or radiotherapy within 4 weeks (6 weeks for nitrosoureas or mitomycin C) of starting treatment or those who have not recovered from adverse events (AEs) because of agents administered more than 4 weeks earlier. Patients may not have had hormonal therapy within 2 weeks before entering the study. Patients receiving raloxifene for bone health as per US Food and Drug Administration (FDA) indication may remain on raloxifene absent other drug interactions.
• Patients may not be receiving any other investigational agents nor have participated in an investigational trial within the past 4 weeks.
• Patients may not be receiving any medication that may markedly affect renal function (eg, vancomycin, amphotericin, and pentamidine).
• Patients may not have received prior treatment affecting the vascular endothelial growth factor pathway (including but not limited to thalidomide, sunitinib, pazopanib, sorafenib, and nintedanib). Bevacizumab used in the upfront setting in conjunction with chemotherapy and/or as maintenance to treat newly diagnosed disease will be allowed.
• Patients may not have previously received a PARP inhibitor.
• CA-125 only disease without RECIST 1.1 measurable or otherwise evaluable disease as per section 3.1.3 (Protocol).
• Patients with untreated brain metastases, spinal cord compression, or evidence of symptomatic brain metastases or leptomeningeal disease as noted on computed tomography or magnetic resonance imaging scans should not be included on this study, since neurologic dysfunction may confound the evaluation of neurologic and other AEs. Screening imaging to rule out brain metastases is not required for screening, but should be performed before study enrollment if clinically indicated. Patients with treated brain metastases and resolution of any associated symptoms must demonstrate stable post-therapeutic imaging for at least 6 months following therapy before starting study drug.
• History of allergic reactions attributed to compounds of similar chemical or biologic composition to cediranib or olaparib.
• Participants receiving any medications or substances that are strong inhibitors or inducers of CYP3A4 are ineligible. Refer to a frequently updated drug information reference for a list of strong inducers and inhibitors. See Appendix II (Protocol). Strong inhibitors and inducers of UGT/PgP should be used with caution.
• History of gastrointestinal perforation. Patients with a history of abdominal fistula will be considered eligible if the fistula was surgically repaired or has healed, there has been no evidence of fistula for at least 6 months, and the patient is deemed to be at low risk of recurrent fistula.
• History of intraabdominal abscess within the past 3 months.
• Current signs and/or symptoms of bowel obstruction or signs and/or symptoms of bowel obstruction within 3 months before starting study drugs.
• Dependency on IV hydration or total parenteral nutrition.
-
• Any concomitant or prior invasive malignancies with the following curatively treated exceptions:
○ Treated limited-stage basal cell or squamous cell carcinoma of the skin.
○ Carcinoma in situ of the breast or cervix.
○ Primary endometrial cancer meeting the following conditions: Stage not greater than IA, grade 1 or 2, no more than superficial myometrial invasion, without vascular or lymphatic invasion; no poorly differentiated subtypes, including papillary serous/serous, clear cell, or other International Federation of Gynecology and Obstetrics (FIGO) grade 3 lesions.
○ Prior cancer treated with a curative intent with no evidence of recurrent disease 3 years following diagnosis and judged by the investigator to be at low risk of recurrence.
-
• Patients with any of the following:
○ History of myocardial infarction within 6 months
○ Unstable angina
○ Resting ECG with clinically significant abnormal findings.
○ NYHA classification of III or IV
• If cardiac function assessment is clinically indicated or performed: left ventricular ejection fraction less than normal per institutional guidelines, or < 55%, if threshold for normal not otherwise specified by institutional guidelines.
Patients with the following risk factors should have a baseline cardiac function assessment:
○ Prior treatment with anthracyclines
○ Prior treatment with trastuzumab
○ Prior central thoracic radiation therapy (RT), including RT to the heart
○ History of myocardial infarction within 6-12 months (patients with history of myocardial infarction within 6 months are excluded from the study)
○ Prior history of impaired cardiac function
• History of stroke or transient ischemic attack within 6 months.
• Any prior history of hypertensive crisis or hypertensive encephalopathy.
• Clinically significant peripheral vascular disease or vascular disease (including aortic aneurysm or aortic dissection).
• Major surgical procedure, open biopsy, or significant traumatic injury within 28 days before starting cediranib.
• Uncontrolled intercurrent illness including, but not limited to, ongoing or active infection, symptomatic congestive heart failure, unstable angina pectoris, cardiac arrhythmia (other than atrial fibrillation with controlled ventricular rate), or psychiatric illness/social situations that would limit compliance with study requirements.
• Pregnant women are excluded from this study because cediranib and olaparib are agents with the potential for teratogenic or abortifacient effects. Because there is an unknown but potential risk of AEs in nursing infants secondary to treatment of the mother with cediranib and olaparib, breastfeeding should be discontinued if the mother is treated with cediranib or olaparib. These potential risks may also apply to other agents used in this study.
• Known HIV-positive individuals are ineligible because of the potential for pharmacokinetic interactions with cediranib or olaparib. In addition, these individuals are at increased risk of lethal infections when treated with marrow-suppressive therapy.
• Patients may not use any complementary or alternative medicines including natural herbal products or folk remedies as they may interfere with the effectiveness of the study treatments.
• No features suggestive of myelodysplastic syndrome or acute myelogenous leukemia on peripheral blood smear or bone marrow biopsy, if clinically indicated.
• No prior allogeneic bone marrow transplant or double umbilical cord blood transplantation.
Additional Statistical Characteristics and Protocol History
Hypothesis within the testing hierarchy were assessed using a stratified log-rank test. HRs and corresponding CIs were derived from proportional hazards models with covariate adjustment for the stratification factors reported at random assignment. A prespecified sensitivity analysis was also conducted where chemotherapy patients who switched to a nonprotocol anticancer treatment before disease progression were censored on the date of the last tumor assessment preceding the start of the nonprotocol therapy. Secondary end points included OS, PFS within stratification subgroups by germline BRCA1/2 status, and objective response rate.
In July 2017, the sample size was increased to 549 patients to increase participation from international sites, provide more power for translational studies, and shorten the study duration by about 5 months. No changes to the statistical design were required. Following FDA approval of niraparib (in March 2017) and later olaparib (August 2017) and rucaparib (April 2018) for switch maintenance therapy following platinum-based chemotherapy for platinum-sensitive relapsed ovarian cancer, a number of patients receiving the chemotherapy regimen were found to have received nonprotocol maintenance therapy before documentation of disease progression. Ultimately, a total of 53 (28%) of the 187 patients receiving the chemotherapy regimen started nonprotocol anticancer therapy before documentation of disease progression. It was anticipated that in the intention-to-treat analysis, the unplanned maintenance therapy would reduce the observed risk for disease progression in the standard chemotherapy arm, artificially understating the HR estimate in the olaparib versus chemotherapy and olaparib/cediranib versus chemotherapy comparisons.
The protocol was amended in August 2018 to adjust for these potential biases. The number of events for progression-free survival maturity was increased to 265 (from 204) in the olaparib/cediranib and chemotherapy arms, and the power was reduced to 80% (from 85%) for a diluted hazard ratio (HR) of 0.70. In August 2019, the protocol was amended again to limit follow-up time to 24 months after the last patient enrolled. Simulations suggested approximately 236 events in the olaparib/cediranib and chemotherapy arms would be available, providing 90% power for an undiluted HR = 0.65, or 78% power for a diluted HR = 0.70. At the final data cutoff data, 249 progression-free survival events supported the olaparib/cediranib versus chemotherapy comparison.
FIG A1.

Sensitivity analysis for PFS censoring patients in the chemotherapy arm at the time of receipt of PARP inhibitor maintenance. The results were consistent with the primary analysis, with a median PFS on the chemotherapy arm of 10.2 months. The HR for PFS for combination cediranib/olaparib compared with chemotherapy was 0.90 (95% CI, 0.69 to 1.18), with a P value of .124. HR, hazard ratio; PARP, poly (ADP-ribose) polymerase; PFS, progression-free survival.
TABLE A1.
Number of Patients With a Dose Modification Because of a Treatment-Emergent AE During the First 12 Cycles

TABLE A2.
The Estimated LSM Differences in NFOSI-DRS-P Between Olaparib and Cediranib Versus Platinum-Based Chemotherapy at Each Assessment Time Point
Joyce F. Liu
Consulting or Advisory Role: Tesaro, Mersana, Clovis Oncology, Genentech/Roche, GlaxoSmithKline, Regeneron, AstraZeneca
Research Funding: Genentech/Roche (Inst), AstraZeneca (Inst), Boston Biomedical (Inst), Acetylon Pharmaceuticals (Inst), Bristol Myers Squibb (Inst), Agenus (Inst), CytomX Therapeutics (Inst), Regeneron (Inst), Tesaro (Inst), Clovis Oncology (Inst), Surface Oncology (Inst), 2X Oncology (Inst), Vigeo Therapeutics (Inst), Aravive (Inst), Arch Oncology (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Merck
Uncompensated Relationships: Merck
Mark F. Brady
Consulting or Advisory Role: Cel-Sci
Ursula A. Matulonis
Honoraria: Advaxis, Alkermes, Symphogen
Consulting or Advisory Role: Merck, Novartis, NextCure, AstraZeneca, Blueprint Medicines, Trillium Therapeutics, GlaxoSmithKline, Agenus
Research Funding: Merck, Novartis, Tesaro, Syndax, Immunogen, Mersana, Leap Therapeutics, Fujifilm, SQZ Biotech
Travel, Accommodations, Expenses: AstraZeneca
Austin Miller
Consulting or Advisory Role: AstraZeneca/Merck, Regeneron (Inst)
Uncompensated Relationships: Genetect
Elizabeth M. Swisher
Leadership: IDEAYA Biosciences
David Cella
Stock and Other Ownership Interests: FACIT.org
Consulting or Advisory Role: AbbVie, GlaxoSmithKline, Pfizer, Astellas Pharma, Novartis, Bristol Myers Squibb, Asahi Kasei, Ipsen, Mei Pharma
Research Funding: Novartis (Inst), Ipsen (Inst), Pfizer (Inst), PledPharma (Inst), Bristol Myers Squibb (Inst), AbbVie (Inst), Regeneron (Inst), Clovis Oncology (Inst)
Noelle G. Cloven
Consulting or Advisory Role: Toray Industries
Carolyn Y. Muller
Research Funding: AstraZeneca (Inst), Genmab (Inst), VBL Therapeutics (Inst), Roche/Genentech (Inst), TapImmune Inc (Inst), Linnaeus Therapeutics (Inst), agenus (Inst), Incyte (Inst), Merck (Inst)
Patents, Royalties, Other Intellectual Property: Have a pending patent on the cancer use for R-ketorolac - not yet its own new drug (Inst)
Other Relationship: NCI, NCI, Department of Defense
Richard G. Moore
Honoraria: Fujirebio Diagnostics
Consulting or Advisory Role: Abcodia, Fujirebio Diagnostics
Research Funding: Angle
Steven E. Waggoner
Consulting or Advisory Role: Regeneron
Melissa A. Geller
Research Funding: Tesaro, Genentech, FATE Therapeutics, Morphotek, Bayer
Keiichi Fujiwara
Honoraria: Kyowa Hakko Kirin, Zeria Pharmaceutical, Nippon Kayaku, Chugai Pharma, Eisai, Taiho Pharmaceutical, Daiichi Sankyo, Ono Pharmaceutical, Takeda
Consulting or Advisory Role: MSD, Taiho Pharmaceutical, Eisai, Takeda, Genmab, NanoCarrier
Research Funding: Eisai (Inst), Kaken Pharmaceutical (Inst), Chugai Pharma (Inst), Immunogen (Inst), Oncotherapeutics (Inst), AstraZeneca (Inst), Zeria Pharmaceutical (Inst), Ono Pharmaceutical (Inst), MSD (Inst), Regeneron (Inst), Merck KGaA (Inst), Ono Pharmaceutical (Inst), Genmab (Inst), Seattle Genetics (Inst)
Travel, Accommodations, Expenses: MSD
Angeles Alvarez Secord
Honoraria: Myriad Genetics
Research Funding: Tesaro (Inst), AstraZeneca (Inst), Genentech (Inst), Boehringer Ingelheim (Inst), AbbVie (Inst), Merck (Inst), PharmaMar (Inst), Clovis Oncology (Inst), Eisai (Inst), Seattle Genetics (Inst), Immutep (Inst), GlaxoSmithKline (Inst), VBL Therapeutics (Inst), OncoQuest Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: GlaxoSmithKline
Uncompensated Relationships: Roche/Genentech, VBL Therapeutics, GOG Foundation, OncoQuest Pharmaceuticals, Regeneron, Aravive
Katherine M. Moxley
Consulting or Advisory Role: Tessa Therapeutics (Inst), Clovis Oncology (Inst), GlaxoSmithKline
Michael A. Bookman
Employment: The Permanente Medical Group
Consulting or Advisory Role: AstraZeneca, AbbVie, Immunogen, Merck Sharp & Dohme, Genentech/Roche, Seattle Genetics, Aravive
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Previously presented in the Oral Abstract Session for Gynecologic Cancers at the 2020 ASCO Virtual Scientific Program, May 29-31, 2020.
SUPPORT
Supported by National Cancer Institute grants to NRG Oncology SDMC (1U10 CA180822), NRG Operations (U10CA180868), and U24CA180803 (IROC). Additionally, Canadian Cancer Trials Group (CCTG) participation in this trial is supported through its grant from the US National Cancer Institute of the National Institutes of Health under the award CA180863. Additional programmatic funding support for the CCTG is provided by the Canadian Cancer Society (#704970) and the Canada Foundation for Innovation. Funding was also received from AstraZeneca.
CLINICAL TRIAL INFORMATION
DATA SHARING STATEMENT
Within approximately 1 year of publication, deidentified data from this article will be available for data sharing proposals at the National Cancer Institute NCTN/NCORP data archive: https://nctn-data-archive.nci.nih.gov.
AUTHOR CONTRIBUTIONS
Conception and design: Joyce F. Liu, Mark F. Brady, Elise C. Kohn, Elizabeth M. Swisher, David Cella, Carolyn Y. Muller, David P. Bender, Keiichi Fujiwara
Provision of study materials or patients: Joyce F. Liu, Ursula A. Matulonis, Elizabeth M. Swisher, Carolyn Y. Muller, David P. Bender, Richard G. Moore, David P. Michelin, Steven E. Waggoner, Michael Carney, Angeles Alvarez Secord
Collection and assembly of data: Joyce F. Liu, Mark F. Brady, Elizabeth M. Swisher, Carolyn Y. Muller, Richard G. Moore, David P. Michelin, Steven E. Waggoner, Keiichi Fujiwara, Angeles Alvarez Secord, Katherine M. Moxley
Data analysis and interpretation: Joyce F. Liu, Mark F. Brady, Austin Miller, Elise C. Kohn, Elizabeth M. Swisher, David Cella, David P. Bender, Richard G. Moore, Steven E. Waggoner, Melissa A. Geller, Angeles Alvarez Secord, Katherine M. Moxley
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Olaparib With or Without Cediranib Versus Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer (NRG-GY004): A Randomized, Open-Label, Phase III Trial
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Joyce F. Liu
Consulting or Advisory Role: Tesaro, Mersana, Clovis Oncology, Genentech/Roche, GlaxoSmithKline, Regeneron, AstraZeneca
Research Funding: Genentech/Roche (Inst), AstraZeneca (Inst), Boston Biomedical (Inst), Acetylon Pharmaceuticals (Inst), Bristol Myers Squibb (Inst), Agenus (Inst), CytomX Therapeutics (Inst), Regeneron (Inst), Tesaro (Inst), Clovis Oncology (Inst), Surface Oncology (Inst), 2X Oncology (Inst), Vigeo Therapeutics (Inst), Aravive (Inst), Arch Oncology (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Merck
Uncompensated Relationships: Merck
Mark F. Brady
Consulting or Advisory Role: Cel-Sci
Ursula A. Matulonis
Honoraria: Advaxis, Alkermes, Symphogen
Consulting or Advisory Role: Merck, Novartis, NextCure, AstraZeneca, Blueprint Medicines, Trillium Therapeutics, GlaxoSmithKline, Agenus
Research Funding: Merck, Novartis, Tesaro, Syndax, Immunogen, Mersana, Leap Therapeutics, Fujifilm, SQZ Biotech
Travel, Accommodations, Expenses: AstraZeneca
Austin Miller
Consulting or Advisory Role: AstraZeneca/Merck, Regeneron (Inst)
Uncompensated Relationships: Genetect
Elizabeth M. Swisher
Leadership: IDEAYA Biosciences
David Cella
Stock and Other Ownership Interests: FACIT.org
Consulting or Advisory Role: AbbVie, GlaxoSmithKline, Pfizer, Astellas Pharma, Novartis, Bristol Myers Squibb, Asahi Kasei, Ipsen, Mei Pharma
Research Funding: Novartis (Inst), Ipsen (Inst), Pfizer (Inst), PledPharma (Inst), Bristol Myers Squibb (Inst), AbbVie (Inst), Regeneron (Inst), Clovis Oncology (Inst)
Noelle G. Cloven
Consulting or Advisory Role: Toray Industries
Carolyn Y. Muller
Research Funding: AstraZeneca (Inst), Genmab (Inst), VBL Therapeutics (Inst), Roche/Genentech (Inst), TapImmune Inc (Inst), Linnaeus Therapeutics (Inst), agenus (Inst), Incyte (Inst), Merck (Inst)
Patents, Royalties, Other Intellectual Property: Have a pending patent on the cancer use for R-ketorolac - not yet its own new drug (Inst)
Other Relationship: NCI, NCI, Department of Defense
Richard G. Moore
Honoraria: Fujirebio Diagnostics
Consulting or Advisory Role: Abcodia, Fujirebio Diagnostics
Research Funding: Angle
Steven E. Waggoner
Consulting or Advisory Role: Regeneron
Melissa A. Geller
Research Funding: Tesaro, Genentech, FATE Therapeutics, Morphotek, Bayer
Keiichi Fujiwara
Honoraria: Kyowa Hakko Kirin, Zeria Pharmaceutical, Nippon Kayaku, Chugai Pharma, Eisai, Taiho Pharmaceutical, Daiichi Sankyo, Ono Pharmaceutical, Takeda
Consulting or Advisory Role: MSD, Taiho Pharmaceutical, Eisai, Takeda, Genmab, NanoCarrier
Research Funding: Eisai (Inst), Kaken Pharmaceutical (Inst), Chugai Pharma (Inst), Immunogen (Inst), Oncotherapeutics (Inst), AstraZeneca (Inst), Zeria Pharmaceutical (Inst), Ono Pharmaceutical (Inst), MSD (Inst), Regeneron (Inst), Merck KGaA (Inst), Ono Pharmaceutical (Inst), Genmab (Inst), Seattle Genetics (Inst)
Travel, Accommodations, Expenses: MSD
Angeles Alvarez Secord
Honoraria: Myriad Genetics
Research Funding: Tesaro (Inst), AstraZeneca (Inst), Genentech (Inst), Boehringer Ingelheim (Inst), AbbVie (Inst), Merck (Inst), PharmaMar (Inst), Clovis Oncology (Inst), Eisai (Inst), Seattle Genetics (Inst), Immutep (Inst), GlaxoSmithKline (Inst), VBL Therapeutics (Inst), OncoQuest Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: GlaxoSmithKline
Uncompensated Relationships: Roche/Genentech, VBL Therapeutics, GOG Foundation, OncoQuest Pharmaceuticals, Regeneron, Aravive
Katherine M. Moxley
Consulting or Advisory Role: Tessa Therapeutics (Inst), Clovis Oncology (Inst), GlaxoSmithKline
Michael A. Bookman
Employment: The Permanente Medical Group
Consulting or Advisory Role: AstraZeneca, AbbVie, Immunogen, Merck Sharp & Dohme, Genentech/Roche, Seattle Genetics, Aravive
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2021 CA Cancer J Clin 717–332021 [DOI] [PubMed] [Google Scholar]
- 2. Caiado J, Castells M. Presentation and diagnosis of hypersensitivity to platinum drugs. Curr Allergy Asthma Rep. 2015;15:15. doi: 10.1007/s11882-015-0515-3. [DOI] [PubMed] [Google Scholar]
- 3.Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study Lancet Oncol 151207–12142014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu JF, Barry WT, Birrer M, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer Ann Oncol 30551–5572019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirza MR, Avall Lundqvist E, Birrer MJ, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial Lancet Oncol 201409–14192019 [DOI] [PubMed] [Google Scholar]
- 6.Chiou VL, Kohn EC, Davarpanah N, et al. Novel therapeutic strategies for angiogenis inhibition in recurrent ovarian cancer Curr Angiogenesis 3179–1922015 [Google Scholar]
- 7.Bindra RS, Gibson SL, Meng A, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs Cancer Res 6511597–116042005 [DOI] [PubMed] [Google Scholar]
- 8.Bindra RS, Schaffer PJ, Meng A, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells Mol Cell Biol 248504–85182004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaplan AR, Gueble SE, Liu Y, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med. 2019;11:eaav4508. doi: 10.1126/scitranslmed.aav4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jensen SE, Kaiser K, Lacson L, et al. Content validity of the NCCN-FACT ovarian symptom index-18 (NFOSI-18) Gynecol Oncol 136317–3222015 [DOI] [PubMed] [Google Scholar]
- 11.Jensen SE, Rosenbloom SK, Beaumont JL, et al. A new index of priority symptoms in advanced ovarian cancer Gynecol Oncol 120214–2192011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaunfield S, Jensen S, Fisher AP, et al. Further content validation of the 18-item NCCN/FACT Ovarian Symptom Index and its Disease Related Symptom-Physical (DRS-P) subscale for use in advanced ovarian cancer clinical trials. Health Qual Life Outcomes. 2019;17:185. doi: 10.1186/s12955-019-1253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penson RT, Valencia RV, Cibula D, et al. Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): A randomized phase III trial J Clin Oncol 381164–11742020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kristeleit R, Lisyanskaya A, Fedenko A, et al. 1 Rucaparib vs chemotherapy in patients with advanced, relapsed ovarian cancer and a deleterious BRCA mutation: Efficacy and safety from ARIEL4, a randomized phase III study. Gynecol Oncol 162:S3-S4, 2021 (abstr)
- 15.Aghajanian C, Blank SV, Goff BA, et al. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer J Clin Oncol 302039–20452012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coleman RL, Brady MF, Herzog TJ, et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial Lancet Oncol 18779–7912017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ledermann JA, Embleton AC, Raja F, et al. Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): A randomised, double-blind, placebo-controlled phase 3 trial Lancet 3871066–10742016 [DOI] [PubMed] [Google Scholar]
- 18.Parmar MK, Ledermann JA, Colombo N, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: The ICON4/AGO-OVAR-2.2 trial Lancet 3612099–21062003 [DOI] [PubMed] [Google Scholar]
- 19.Pfisterer J, Plante M, Vergote I, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: An intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG J Clin Oncol 244699–47072006 [DOI] [PubMed] [Google Scholar]
- 20.Pfisterer J, Shannon CM, Baumann K, et al. Bevacizumab and platinum-based combinations for recurrent ovarian cancer: A randomised, open-label, phase 3 trial Lancet Oncol 21699–7092020 [DOI] [PubMed] [Google Scholar]
- 21.Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, et al. Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse J Clin Oncol 283323–33292010 [DOI] [PubMed] [Google Scholar]
- 22. Liu JF, Lee JM, Luo W, et al. A phase 1 study optimizing the dosing of olaparib tablet formulation combined with cediranib in recurrent ovarian cancer. J Clin Oncol. 2015;33 suppl; abstr 5559. [Google Scholar]
- 23.Liu JF, Tolaney SM, Birrer M, et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer Eur J Cancer 492972–29782013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.National Cancer Institute: Division of Cancer Treatment & Diagnosis. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm