

Abstract

With advances in machine learning (ML) techniques, the quantitative structure–activity relationship (QSAR) approach is becoming popular for evaluating chemicals. However, the QSAR approach requires that the chemical structure of the target compound is known and that it should be convertible to molecular descriptors. These requirements lead to limitations in predicting the properties and toxicities of chemicals distributed in the environment as in the PubChem database; the structural information on only 14% of compounds is available. This study proposes a new ML-based QSAR approach that can predict the properties and toxicities of compounds using analytical descriptors of mass spectrum and retention index obtained via gas chromatography–mass spectrometry without requiring exact structural information. The model was developed based on the XGBoost ML method. The root-mean-square errors (RMSEs) for log Ko-w, log (molecular weight), melting point, boiling point, log (vapor pressure), log (water solubility), log (LD50) (rat, oral), and log (LD50) (mouse, oral) are 0.97, 0.052, 51, 23, 0.74, 1.1, 0.74, and 0.6, respectively. The model performed well on a chemical standard mixture measurement, with similar results to those of model validation. It also performed well on a measurement of contaminated oil with spectral deconvolution. These results indicate that the model is suitable for investigating unknown-structured chemicals detected in measurements. Any online user can execute the model through a web application named Detective-QSAR (http://www.mixture-platform.net/Detective_QSAR_Med_Open/). The analytical descriptor-based approach is expected to create new opportunities for the evaluation of unknown chemicals around us.
Chemicals greatly aid our daily lives; however, we are not acutely aware of which ones surround us. Chemical Abstracts Service has more than 250 million chemical species registered in its database.1 The publicly accessible database, PubChem, provides information on over 111 million unique compounds.2 Of these data, over 16 million chemical structures, which correspond to approximately 14% of the PubChem compounds, are provided with a link to the PATENTSCOPE patent database. Furthermore, approximately 32,000 compounds are linked to physical and physicochemical properties, and 11,000 are linked to toxicological information, including acute toxicity; these values account for only 0.3% and 0.1% of all PubChem compounds, respectively. The situation is similar or more dismal for other famous large databases such as ChemSpider and ChEMBL.3,4 The proportion is considerably smaller for the GDB-17 database, which contains 166.4 billion in silico chemical structures of up to 17 atoms of C, N, O, S, and halogens.5 This situation indicates that we can access information on the properties, hazards, and risks only for a small fraction of the chemicals.
The quantitative structure–activity relationship (QSAR) approach is popular for evaluating chemicals.6 QSAR is based on the similarity-property principle (SPP), wherein the chemical structure is strongly related to chemical activity, which includes pharmacological activity and toxicity to organisms. Thus, information on molecular features, such as chemical functional groups as substructures, properties that represent the structure, mathematical descriptors known as fingerprints,7 and other chemical descriptors from calculators including DRAGON (alvaDesc),8 RDKit,9 and OpenBabel,10 is used as explanatory variables for predicting the properties or biological effects, including the toxicity of the target compound. When the focus is on properties, this is referred to as the quantitative structure–property relationship (QSPR).
Recent advances in ML techniques allow QSAR to handle an expanded number of descriptors calculated from the chemical structure, resulting in a wider application range and enhanced predictive performance. The ML-based QSAR called istkNN, which employs k-nearest neighbors (kNN), demonstrated excellent performance with a mathematical descriptor of a structural key that represents a chemical structure with a binary code. This approach resulted in a root-mean-square error (RMSE) of 0.55 and an R2 of 0.63 for the external data of the logarithm of the median lethal dose (LD50) on a rat administered orally (mmol/kg).11,12 In another case, aiQSAR employed multiple methods, including an ML-based one that used a number of chemical descriptors calculated by DRAGON based on the target chemical structure. This approach resulted in an RMSE of 0.54 and an R2 of 0.65 for the external data of the log LD50 (rat, oral).11,13 These methods outperform traditional QSAR; for example, a univariant model that used in vitro cytotoxicity as a surrogate for log LD50 (rat, oral) achieved an R2 of 0.40 even for the training data set.14
QSAR approaches, which include ML-based methods, require the structure of the target compound to be available and convertible to any type of molecular descriptor. However, PubChem only provides structural information on 14% of its compounds, and the structures of the rest are undefined; thus, QSAR may face limitations in predicting the properties and toxicities of chemicals in the environment. For example, a study that explored environmental pollutants in 50 river water samples using a nontarget analytical technique of two-dimensional gas chromatography (GC × GC) interfaced with high-resolution time-of-flight mass spectrometry (MS) found 87,000 raw chromatographic peaks corresponding to single or multiple chemicals. However, only 0.2% of the total peaks were identified by a spectral database search with high reliability.15 Similarly, averages of 9550 and 9610 chromatographic peaks were found in the effluents of wastewater treatment plants using the positive and negative ion modes of liquid chromatography interfaced with high-resolution mass spectrometry (LC-HRMS), respectively. Only 1.7% and 0.6% of the peaks were identified and assigned based on measurement data of chemical standards in the positive and negative ion modes, respectively.16 Determination of the chemical structures for a vast number of peaks in measurements remains a challenge.
As indicated above, structure identification remains challenging for most chemicals in our surroundings detected with instrumental measurements, even using state-of-the-art instruments. Thus, approaches for predicting the properties and toxicities of unknown-structured chemicals detected in actual samples are required for hazard and risk assessment. However, QSAR requires the chemical structure as a first step, and there is currently a lack of ability to meet this requirement.
If measurement data such as those from MS are directly used for the prediction without the process of structure identification, it may be possible to expand the range of chemicals involved in hazard and risk assessments, even if exact information regarding the structure is not available. The mass spectrum obtained with electron impact ionization (EI), which is used for gas chromatography (GC) interfaced with MS, comprises instrumental signals of the fragment ions of a compound. Several studies have aimed to predict the GC–EI–MS mass spectrum from the chemical structure using deep neural networks (DNNs)17 and vice versa.18 The results of these studies suggest that the EI spectrum is strongly correlated with the chemical structure.
Several homologues of certain compounds show the same mass spectral pattern in GC–MS (e.g., alkyl phthalates), even though their structures are different. In such cases, information on retention time differences in the GC is useful for distinguishing these homologues. Thus, the GC–MS output may contain sufficient information to infer chemical structures. This indicates that the instrumental output, i.e., analytical descriptor, has considerable potential to predict the properties and toxicities of chemicals.
The idea of an analytical descriptor has been partially introduced in some QSAR research fields. For example, QSAR-like approaches in genomics have used biological properties of gene expression profiles obtained by high-throughput screening as gene-based descriptors.19,20 Experimentally determined properties, such as elemental composition, zeta potential, size distribution, and shape, are used to reveal quantitative nanostructure–activity relationships, referred to as nano-QSAR.21,22 The characterization of material surfaces that interact with biological film formation was investigated using cell imaging techniques, such as time-of-flight secondary ion mass spectrometry (ToF-SIMS) and a QSAR-like approach.23−25 These studies have shown that high-content data obtained via ToF-SIMS have strong potential to elucidate the surface functions of materials against biofilm formation through self-organizing maps and partial least-squares regression.
However, the above-described studies with the idea of analytical descriptor were performed to reinforce the prediction performance of traditional QSAR models by combining them with other types of chemical descriptors. Such chemical descriptors eventually require the chemical structure to identify new or potentially useful chemical descriptors for predicting the target properties/toxicities or to investigate undefined phenomena by exploring meaningful correlations between analytical data and biological activity. QSAR methods that do not require an exact chemical structure and are fully based on analytical descriptors are currently scarce. However, they have strong potential for empowering research in various domains of chemistry, including chemical risk and safety.
This study proposes a new QSAR approach that predicts the properties and toxicities of compounds based solely on the analytical descriptors obtained by GC–MS using a combination of ML techniques. The predictive performance of the developed approach was evaluated, and metrics to capture the applicability domain (AD) of the method were implemented in a free web application called Detective-QSAR. This approach will be useful for evaluating various unknown-structured chemicals that exist in our surroundings and to prioritize them for detailed assessment.
Methods
An overview of the study is illustrated in Figure 1. This study predicted physicochemical properties and toxicities of compounds directly from the mass spectrum and retention index (RI) of GC–MS, equipped with EI unless specified otherwise. It does not require information on the exact chemical structure of interest. This requirement is mandatory for traditional and ML-based QSAR approaches that use chemical descriptors or mathematical descriptors, such as MACCS keys, PubChem fingerprints, and CDK-standard fingerprints obtained from chemical structures.
Figure 1.

Overview of direct prediction from analytical descriptors.
Data Set and Preparation
A data set of chemicals containing GC–MS mass spectra and RIs as analytical descriptors, properties, and toxicities as objective variables was prepared for modeling. The mass spectra and RIs were obtained from NIST17,26 MassBank,27,28 Fiehn laboratory,29 RIKEN,30,31 and in-house data. The RIs used in this study were obtained with semistandard nonpolar GC columns, such as DB-5, under ramped temperature conditions; if unavailable, RIs with standard nonpolar GC columns, such as DB-1, were used. There were only a few minor percentage differences between the RI values, unlike the differences in the polar GC columns. The property lists of log Ko-w, boiling point, melting point, vapor pressure, and water solubility were obtained from ChemIDplus32 and Comptox33,34 for over 110,000 candidate compounds that were GC amenable. The LD50 (rat, oral) and LD50 (mouse, oral) values were obtained from ChemIDplus. The molecular weights of the chemicals were obtained using OPERA.35 The data set of chemicals with GC–MS spectra and RI contained the molecular weight (g/mol), melting point (°C), boiling point (°C), log Ko-w (unitless), vapor pressure (Pa), water solubility (mmol/L), LD50 (rat, oral) (mmol/kg), and LD50 (mouse, oral) (mmol/kg) of 12810, 3836, 3385, 2674, 1299, 1383, 2080, and 1630 compounds, respectively (summarized in Table S-1).
The data on each objective list were split randomly into training, validation, and test data with a ratio of 0.8:0.1:0.1. The m/z range of the mass spectrum in the data set was 1–6420. The entire range of the mass spectrum was used for the modeling. Each m/z value was normalized such that the highest intensity was 1 so that data from different sources could be combined. The vector resulting from concatenation of the RI value and corresponding m/z values of the mass spectrum was used as input to the proposed model. The vector is converted to the format used for sparse model during the model execution.
In addition to the data set of the predictive and objective variables for modeling, molecular features of the chemicals in the data set were prepared to represent the chemical space of the chemicals used for modeling and to investigate the model performance. The well-known molecular features of Abraham parameters (E, S, A, B, L, and V) used for linear free energy relationship (LFER) or linear solvation energy relationship (LSER) in pharmacology and environmental chemistry were calculated by Absolv in the ACD/Laboratories software with the simplified molecular input line entry system (SMILES) as input.36−38 The number of mass spectral bins, which represents the number of centroided bars of the mass spectrum for each compound, was set based on the original mass spectral data. The number of elemental species constituting each molecule was obtained by converting its molecular formula.
Modeling and Model Comparison
Several regression and supervised ML techniques that suit the objective of the study, such as lasso,39 ridge regression,40 elastic net (Enet),41 DNN,42,43 random forest (RF),44,45 and eXtreme Gradient Boosting (XGBoost),46 were applied using the statistical programming software R.47 Methods from Keras42 and TensorFlow43 were used for the DNN, and methods from caret48 were used for the others. Lasso,39 ridge regression,40 and Enet41 are linear regression methods that use regularization terms to avoid overfitting. RF is an ensemble-learning method based on decision trees.44,45 A DNN comprises an unbounded number of layers that are unbounded in width with nonpolynomial activation functions, which permits practical application and optimized implementation.42,49 Further, a DNN requires model construction and parameter finetuning to achieve better predictive performance. The model construction and parameter finetuning are described in the Supporting Information (Section S-1).
XGBoost was developed by Chen and Guestrin.46 Both RF and XGBoost are ensemble-learning methods that use a decision tree as the base learner. RF adapts bagging as the ensemble technique to improve predictive performance and control overfitting by averaging several decision trees generated in parallel on several subsamples of the data set. Alternatively, XGBoost adapts the boosting ensemble technique to improve the model based on a sequential learning process involving iterative calculation to update and adjust the parameters based on outliers in the previous model. It is based on the same idea as that employed for gradient boosting; the second-order method originates from Friedman et al.50 In addition to the second-order method, XGBoost has been improved for regularized objectives.
In brief, XGBoost—a tree ensemble model—uses K additive functions to predict the output
| 1 |
where 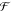 indicates
all possible trees, and the function fk at each step k maps the i-th sample of descriptor x to a certain
output
indicates
all possible trees, and the function fk at each step k maps the i-th sample of descriptor x to a certain
output  .
.
The following regularized objective is minimized to learn the set of functions used in the model
 |
2 |
where l represents a differentiable convex loss function, and the second term Ω penalizes the complexity of the model, which helps smooth the final learned weights to avoid overfitting.
A second-order approximation is used to optimize the objective using the following equation at the t-th iteration
| 3 |
where 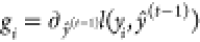 and
and 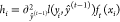 represent the first- and second-order gradient
statistics of the loss function, respectively. Two additional techniques,
shrinkage and column subsampling, were introduced in XGBoost to further
prevent overfitting. The shrinkage scales the newly added weights
by a factor η after each boosting step; this reduces the influence
of each tree and leaves space for future trees to improve the model.
Column (feature) subsampling considers only a random subset of descriptors
when building a given tree; this technique also accelerates the computation.
In addition, XGBoost uses a sparsity-aware split-finding approach
to efficiently train the model on sparse data, such as the mass spectral
data used in this study. This method requires parameter finetuning
to achieve higher predictive performance. The gbtree function was
selected as the booster, and a regression model with a squared loss
was applied as the learning objective. For parameter tuning, to avoid
model overfitting, the parameters eta (learning rate) = 0.02, alpha
(L1 regularization) = 1, lambda (L2 regularization) = 1, and minimum
child weight = 1 were determined via a parameter grid search. The
depth of the tree was chosen as 7 based on grid search optimization.
represent the first- and second-order gradient
statistics of the loss function, respectively. Two additional techniques,
shrinkage and column subsampling, were introduced in XGBoost to further
prevent overfitting. The shrinkage scales the newly added weights
by a factor η after each boosting step; this reduces the influence
of each tree and leaves space for future trees to improve the model.
Column (feature) subsampling considers only a random subset of descriptors
when building a given tree; this technique also accelerates the computation.
In addition, XGBoost uses a sparsity-aware split-finding approach
to efficiently train the model on sparse data, such as the mass spectral
data used in this study. This method requires parameter finetuning
to achieve higher predictive performance. The gbtree function was
selected as the booster, and a regression model with a squared loss
was applied as the learning objective. For parameter tuning, to avoid
model overfitting, the parameters eta (learning rate) = 0.02, alpha
(L1 regularization) = 1, lambda (L2 regularization) = 1, and minimum
child weight = 1 were determined via a parameter grid search. The
depth of the tree was chosen as 7 based on grid search optimization.
Model Validation and Evaluation
The developed prediction model based on XGBoost was validated with the validation data and further evaluated with the test data based on the RMSE, R2, and Q2 values on the predicted and measured data for each target list
| 4 |
| 5 |
| 6 |
where n and n′ represent the numbers of training and external data, respectively.
The ranges of all collected measurement data on all possible important molecular features of over 48,000 compounds out of the 110,000 candidates were compared with those of the data used for the modeling to check the chemical space of the data handled by the model. The total numbers of data on the properties and toxicities were 6341, 4635, 4416, 1467, 3535, 3559, and 2990 out of the 48,000 compounds for melting point, boiling point, log Ko-w, vapor pressure, water solubility, LD50 (rat, oral), and LD50 (mouse, oral), respectively (summarized in Table S-1). The performance of the model was evaluated using the actual measurement of a chemical standard mixture and sample of contaminated car engine oil running over 10,000 km. Details of the oil sample collection are presented elsewhere.51 The chemical lists for the evaluation are provided as Supporting Information. Prediction of the objective lists was performed online via Detective-QSAR (http://www.mixture-platform.net/Detective_QSAR_Med_Open/) using an input CSV file that includes the analytical descriptors of m/z values and RI of a target compound. Both the nominal mass and accurate mass spectra are available for the prediction. The mass spectrum is automatically normalized, and an intensity threshold truncates intensities of 0.5% against the highest intensity on the target spectrum in Detective-QSAR.
Calculation of Applicability Domain
The applicability domain (AD) of a model in QSAR is measured by an indicator to determine whether a chemical of interest is covered by the model.52−54 The similarity level between a target vector and model training data was applied as an indicator to determine whether the target is within the AD. The cosine distance between the vectors of input and training spectra combined with RI was used as the similarity index (SI)
| 7 |
where u represents an o-dimensional vector of RI and normalized m/z intensity for the target compound, and v represents a compound in the training data set.
The mean of SIs between the input and m most similar spectra in the training data was calculated as SI.t, which helped evaluate whether the model was suitable for predicting an objective list of interest in the input spectrum
| 8 |
where m represents the number of candidates and was set to 5.
A higher SI.t indicates that compounds expressed by input descriptors are included in the training data (the input is included in the AD of model). The RMSE for the validation data that exceed the specified SI threshold value (RMSE only for data within the AD) can be calculated using Detective-QSAR. Users can achieve a higher prediction accuracy for a target input that presents a higher SI.t (e.g., >0.7) by referring to the “RMSE with the SI threshold” calculated and provided by Detective-QSAR. The processing flow of the prediction by the software is illustrated in Figure S-1.
Results and Discussion
Chemical Space of Data for Modeling
Clarity regarding the chemical space of the data (i.e., model coverage) is important for modeling the properties and toxicities of various chemicals.55 The data used in this study were curated from reliable sources considering duplication and outliers following the curation approach described in a previous study.56 The range of the molecular features of the training data alongside that of all available data on log Ko-w to capture the chemical space for the model is illustrated in Figure 2. The training data covered a wide range of molecular features, which were consistent with all available data over 48,000 GC amenable compounds in terms of the Abraham parameters (E, S, B, L, and V). The training data for parameter A were slightly skewed toward nonzero data. This means that there could have been slightly less low-acidity molecules regarding hydrogen bonds in the training data than in all data. The number of data on molecular weight in the training data set was slightly lower for compounds with low molecular weights. This indicates that the model developed with the training data performed well for compounds with low molecular weights but not for compounds with high molecular weights. Training data on the number of mass spectral bins that were possibly correlated with the molecular weight were concentrated around the lower side. Although the number of all available data was still not sufficient for the objective list of log Ko-w in the data-driven approach, the training data were chosen uniformly from all available data according to the data distributions. Distributions of other objective lists are shown in Figures S-2–S-8. Overall, severely biased data were not used for the modeling, and the wide ranges of chemical spaces within the GC amenable chemicals were covered by the training data.
Figure 2.

Chemical space of training data on log Ko-w with all available data.
Method Comparison for the Best Predictive Models
Figure S-9 shows the performance of models based on DNN, ridge regression, Enet, lasso, RF, and XGBoost. The distributions of absolute differences between the predicted and measured values on the test data set are shown as violin plots with their RMSEs. The RF and XGBoost-based models showed high accuracy for prediction by analytical descriptors of m/z values with RI as the input. The other methods fluctuated in performance between the test and validation data sets, as shown in Figure S-10. RF and XGBoost are ensemble-learning models for bagging and boosting, respectively, based on the decision tree model. Each spectral bin (or RI) variable is evaluated in the tree model, and its coefficient weight is assigned to the respective node of the tree. These models were superior to linear regression models with certain regularization(s), such as lasso, ridge regression, and Enet. The relatively poor quality by the DNN was not expected because generally DNN has high potential for superior prediction by iteratively calculating the coefficient weights of spectral bins through a fully connected multilayer at each depth. Other attempts at DNN optimization, including the application of various types of activation functions, regularization terms, and convolutional neural network approaches, did not provide high performance. DNNs are not always superior to other methods in predictive performance, as shown in previous studies.57,58 DNNs are susceptive of hyperparameters, architecture, and optimizers. It is difficult to achieve competitive performance with other methods in certain cases. Meanwhile, one study demonstrated comparative performance between DNN and XGBoost in terms of predictive accuracy.59 Therefore, the application of a more suitable algorithm to connect each layer and/or the usage of more training data may enhance the neural network’s performance. In this study, XGBoost, which showed the highest prediction accuracy, was applied and further evaluated.
Performance of the Developed Model
The performance of the XGBoost-based model was evaluated using the test data for the objective lists, as shown in Figure 3. The predictive performance was very close to that with the validation data, as shown in Figure S-11. Therefore, it is considered that the models were not biased by the validation process and were optimized based solely on the training data. The boiling and melting points were modeled using an antilog scale because the data range was not wider and normally distributed. These predictive accuracies were high considering RMSEs of 24 and 51. The other objective lists were modeled on a log scale because of the log-normal distribution of the data that span wide ranges. The partitioning property of Ko-w is known to be the log-transformed value of log Ko-w; therefore, the value of log Ko-w was considered without reconversion in this study. Good relationships in terms of y = x between the predicted and measured values were obtained for log (molecular weight), log (vapor pressure), and log Ko-w, followed by log (water solubility).
Figure 3.

Predictive performances of models for physicochemical properties and toxicities based on test data. The blue lines indicate direct proportions. The red lines represent the linear regression lines.
The developed model predicted the objective lists with a similar level of accuracy to other QSAR models that require molecular descriptors as inputs. Using the model T.E.S.T., which applies the consensus method of several methods including hierarchical clustering with a genetic algorithm-based technique, multilinear regressions, and nearest neighbor method with 797 molecular descriptors as input variables, the RMSEs of the melting point, boiling point, log (vapor pressure), log (water solubility), and log (LD50) (rat, oral) were 43.7, 20.5, 0.82, 0.87, and 0.60, respectively.60,61 The RMSEs of the melting point, boiling point, log Ko-w, log (vapor pressure), and log (water solubility) obtained using the OPERA model, which applies the kNN algorithm with 9–16 molecular descriptors as input variables, were 52.2, 22.1, 0.78, 1.00, and 0.86, respectively.35 There were differences in the coverage of the chemical space for each of the proposed model, which handles GC amenable chemicals, and these two above models, which handle wider ranges of compounds. Although such a difference exists, the predictive accuracies are comparable, except for log (water solubility), which was slightly inferior for the proposed model. The advantage of the proposed model is that it does not require an exact chemical structure to obtain molecular descriptors but instead requires measurement data. Detective-QSAR (Ver. Pred) is available (http://www.mixture-platform.net/Detective_QSAR_Pred_Open/) to emulate the predictions of T.E.S.T. and OPERA with the Detective-QSAR system.
Although the RMSEs of log (LD50) for rats and mice in the proposed model were as low as 0.70 and 0.73, respectively, the slopes of the linear regressions of the plots predicted against measured values were not adequate (0.37 and 0.32, respectively). Although the training data were chosen to be as comprehensive as possible, the ranges of values for log (LD50) were 4–5 orders of magnitude. These ranges were narrower than those of other objective lists, such as log Ko-w, log (vapor pressure), and log (water solubility), whose ranges were 10–15 orders of magnitude. Therefore, although the values of log (LD50) were predictable with an RMSE of approximately 0.7 when using this model to assess the toxicity of unknown compounds spread in AD, further refinement to capture the relationship between structural information and toxic mechanisms is necessary. Applicability domain judged by SI.t of the model is discussed in Section S-2.
Descriptor Importance in Model Prediction
The relative importance of variables for prediction results was calculated as the score of descriptor importance (or feature importance).62,63 The variable was provided with a relative value among the descriptors and showed a strong influence on the prediction results when the score of a certain variable was high, irrespective of the direction of influence. The results of the descriptor importance of the model inputs for all objective lists are shown in Figure S-16. The importance values were calculated for all descriptors (m/z values with RI) so that the importance value of the highest m/z value became 1, making the importance result visible as a mass spectrum. Clear trends were not captured from the patterns of m/z importance because several m/z values simultaneously affected the results in positive and negative directions.
Contrary to the trend of m/z values, differences in RI importance among objective lists were clearly observed. The boiling points followed by vapor pressure showed high scores of 460 and 300, respectively. The boiling point and vapor pressure showed a direct relationship with the GC retention times (i.e., RI). The RI importance values of the molecular weight and melting point were 36 and 19, respectively. Other objective lists showed a relatively low influence from RI; however, they were higher than all the m/z values.
Similarly, the effect of RI on the model was observed from the model performance results. The performance of the model that excluded RI from explanatory variables deteriorated with RMSEs of 0.060, 63, 45, 1.1, 1.5, 0.72, and 0.80 for molecular weight, melting point, boiling point, log Ko-w, vapor pressure, LD50 (rat, oral), and LD50 (mouse, oral), respectively. Owing to the requirement of RI data in addition to m/z values, 74, 40, 27, 39, 11, 28, 42, and 46% of data entries were removed from the training data set of the RI-excluding model, respectively. Only the RI-excluding model for water solubility did not deteriorate (RMSE decreased from 1.5 to 1.2). The RI-including model for water solubility was affected by the decrease in training samples (28%) because of the RI data requirement, which outweighed the benefit of RI information to the model performance. In addition, RI, which represents the extent of partitioning between the GC column phase and gas phase for the chemical of interest, was not simply correlated with water solubility, in contrast to vapor pressure. Comparison with the proposed RI-including model with a model based only on RI is described in the following section.
Practical Application of Detective-QSAR and Implications
The method developed through Detective-QSAR was applied to actual measurements. Prior to the application, it was a concern that raw measured data contain very little noise throughout the m/z range; the noise severely affected the prediction results for all the objective lists. The threshold cut of the intensity for the target input improved the noise issue, as indicated in Table S-2. A threshold cut of 0.5% intensity for the normalized mass spectrum had a positive effect on all the lists except for melting point and LD50. The negative effects on the melting point and LD50 were negligible. The varying effects of models are considered to be the results of a combination of noise conditions in the input and model descriptor importance which is different among the models. The noise conditions vary case-wise, and it may affect predictions of melting point and LD50 in some cases. Therefore, the process of the threshold cut with 0.5% intensity on the input was implemented for all the objectives in Detective-QSAR, as described in the Methods section.
Detective-QSAR was applied to 31 structure-identified chromatographic peaks in the chemical standard mixture and 14 peaks detected in the contaminated oil sample. Figure S-17 shows the results of all the objective lists on these peaks. Several properties with the chemical lists are provided in Tables S-3 and S-4. Detective-QSAR performed well on the peaks of the chemical standard mixture as similarly with the result of model validation. Comparison of predictive performance on structurally homologous compounds (nitrobenzenes, phthalate, PAHs) and nonhomologous compounds within the standard mixture is shown in Table S-5. Although deteriorated performance on log (LD50) (rat, oral) was observed for nitrobenzenes, the results did not show clear trends on other objective lists (Table S-5). As shown in Figure S-17, it also performed well on the peaks in contaminated oil after spectral deconvolution.64,66 The deconvolution improved spectral similarity with the reference spectrum (Figure S-18), resulting in improved predictive performance compared with the cases on the raw spectrum (Figure S-19). Without the deconvolution procedure, the model underperformed on peaks in contaminated oil compared with the results of model validation, especially for log Ko-w, boiling point, and log (water solubility). This was because these chromatographic peaks without deconvolution were influenced by signals of coeluted compounds and/or sample matrices. In addition to the instrumental noise discussed above, interference by coelution should be considered in the model application on the measurement of a raw spectrum. To obtain the spectrum stem from a sole compound convoluted in the coeluted peak, spectral deconvolution techniques are helpful.64−67 Once spectral deconvolution was performed for the detected peaks in the contaminated oil, the prediction results were improved (Figure S-19), and eventually, the results were comparative with those of the model validation. Performance of Detective-QSAR was comparable to that of a GC × GC property estimation model51,68 that uses only RI on the estimation of log Ko-w or superior to that on log (water solubility) as shown in Figure S-20. The applicability domain of the GC × GC model is currently limited to nonpolar compounds, but Detective-QSAR is not limited to them.
Further validation for various actual cases with different types of compounds is warranted for detailed model characterization and enhancing the model performance, and the results demonstrated the possibility of applying the method based on analytical descriptors for predictions. The approach will be useful for the hazard and risk assessment of unknowns found in measurements. In most cases of measurements of complex mixtures, such as environmental, food, beverage, waste material, biological, and manufactured product samples, GC–MS peaks of chemicals overlap with each other. As shown in this study, the developed method is applicable even to actual measured peaks comprising multiple chemicals with the help of spectral deconvolution techniques. To enhance the number of compounds in the mixture sample to be evaluated using the analytical descriptor-based method, further functionality in the deconvolution technique, such as judgment/estimation of spectral purity for unknowns in coeluted peak, is expected. The model has considerable potential for predicting properties and toxicities of unknown-structured compounds found in measurements as long as the target signal is not taken from mixtures (pure component) and is judged by SI.t to be within the model AD. The predicted values of all objective lists with SI.t can be calculated for approximately 10 min for 1000 spectra in the web application.
The objective lists considered throughout the study are only a fraction of what is required for the detailed assessment of chemicals. A further expansion of the lists, such as bioaccumulation factor, degradation rate, mutagenicity, ecotoxicity, cytotoxicity, and others, is expected in the future. In addition, the analytical descriptor-based approach has the potential to expand to other analytical methodologies that provide structure-specific signals, including GC interfaced with multistage mass spectrometry (MSn), LC-MSn, and LC interfaced with a diode array detector.
Conclusions
This study proposed a new QSAR approach that predicts the properties and toxicities of compounds based solely on the analytical descriptors obtained by GC–MS using a combination of ML techniques. The model based on XGBoost was developed to predict log Ko-w, log (molecular weight), melting point, boiling point, log (vapor pressure), log (water solubility), log (LD50) (rat, oral), and log (LD50) (mouse, oral). It performed well on a chemical standard mixture and contaminated oil with spectral deconvolution. This approach compensates for the limitation of traditional QSAR, i.e., the requirement of a chemical structure. Further, it will be useful to prioritize chemicals without structural information for detailed environmental hazard and risk assessment, safety checks of food and beverages, product and waste management, medical investigation, and further exploration in new fields. The analytical descriptor-based QSAR approach developed in this study will provide insights for evaluating unknown chemicals around us.
Data and Software Availability
Detective-QSAR is freely available at http://www.mixture-platform.net/Detective_QSAR_Med_Open/ (Ver. Med) and http://www.mixture-platform.net/Detective_QSAR_Pred_Open/ (Ver. Pred).
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research (B) from JSPS KAKENHI (Grant No. 19H04297). The author thanks the three reviewers and the Editor who provided valuable comments to improve the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c01667.
Subsections of Methods and Results and Discussion (Sections S-1 and S-2), data used for modeling (Table S-1 and Figures S-1–S-8), model performance (Figures S-9–S-11), information related to model AD (Figures S-12–S-15), descriptor importance (Figure S-16), predictive performance for intensity threshold cut of mass spectrum (Table S-2), and model performance on measurements of chemical standard mixture and contaminated oil (Figures S-17–S-20 and Tables S-3–S-5) (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- CAS. https://www.cas.org/ja/node/32521 (accessed 2022-05-27).
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49 (D1), D1388–D1395. 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ChemSpider. http://www.chemspider.com/ (accessed 2022-05-27).
- ChEMBL. https://www.ebi.ac.uk/chembl/ (accessed 2022-05-27).
- Reymond J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48 (3), 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Muratov E. N.; Bajorath J.; Sheridan R. P.; Tetko I. V.; Filimonov D.; Poroikov V.; Oprea T. I.; Baskin I. I.; Varnek A.; Roitberg A.; Isayev O.; Curtalolo S.; Fourches D.; Cohen Y.; Aspuru-Guzik A.; Winkler D. A.; Agrafiotis D.; Cherkasov A.; Tropsha A. QSAR Without Borders. Chem. Soc. Rev. 2020, 49 (11), 3525–3564. 10.1039/D0CS00098A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAYLIGHT. https://www.daylight.com/dayhtml/doc/theory/theory.finger.html (accessed 2022-05-27).
- alvaDesc. https://www.alvascience.com/alvadesc/ (accessed 2022-05-27).
- RDKit. https://www.rdkit.org/ (accessed 2022-05-27).
- OpenBabel. http://openbabel.org/wiki/Main_Page (accessed 2022-05-27).
- Gadaleta D.; Vuković K.; Toma C.; Lavado G. J.; Karmaus A. L.; Mansouri K.; Kleinstreuer N. C.; Benfenati E.; Roncaglioni A. SAR and QSAR Modeling of a Large Collection of LD50 Rat Acute Oral Toxicity Data. J. Cheminform 2019, 11 (1), 58. 10.1186/s13321-019-0383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganaro A.; Pizzo F.; Lombardo A.; Pogliaghi A.; Benfenati E. Predicting Persistence in the Sediment Compartment With a New Automatic Software Based on the k-Nearest Neighbor (k-NN) Algorithm. Chemosphere 2016, 144, 1624–1630. 10.1016/j.chemosphere.2015.10.054. [DOI] [PubMed] [Google Scholar]
- Vukovic K.; Gadaleta D.; Benfenati E. Methodology of aiQSAR: A Group-Specific Approach to QSAR Modelling. J. Cheminf 2019, 11 (1), 27. 10.1186/s13321-019-0350-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidig A. P.; Dekkers S.; Verwei M.; Zvinavashe E.; Bessems J. G. M.; van de Sandt J. J. M. Development of a QSAR for Worst Case Estimates of Acute Toxicity Of Chemically Reactive Compounds. Toxicol. Lett. 2007, 170 (3), 214–222. 10.1016/j.toxlet.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Zushi Y.; Hashimoto S.; Tanabe K. Nontarget Approach for Environmental Monitoring by GC × GC-HRTOFMS in the Tokyo Bay Basin. Chemosphere 2016, 156, 398–406. 10.1016/j.chemosphere.2016.04.131. [DOI] [PubMed] [Google Scholar]
- Schymanski E. L.; Singer H. P.; Longrée P.; Loos M.; Ruff M.; Stravs M. A.; Ripollés Vidal C.; Hollender J. Strategies to Characterize Polar Organic Contamination in Wastewater: Exploring the Capability of High-Resolution Mass Spectrometry. Environ. Sci. Technol. 2014, 48 (3), 1811–1818. 10.1021/es4044374. [DOI] [PubMed] [Google Scholar]
- Wei J. N.; Belanger D.; Adams R. P.; Sculley D. Rapid Prediction of Electron–Ionization Mass Spectrometry Using Neural Networks. ACS Cent. Sci. 2019, 5 (4), 700–708. 10.1021/acscentsci.9b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H.; Deng H.; Lu H.; Zhang Z. Predicting a Molecular Fingerprint from an Electron Ionization Mass Spectrum with Deep Neural Networks. Anal. Chem. 2020, 92 (13), 8649–8653. 10.1021/acs.analchem.0c01450. [DOI] [PubMed] [Google Scholar]
- Bologa C. G.; Ursu O.; Halip L.; Curpăn R.; Oprea T. I. Chemical and Biological Descriptor Integration Improves Computational Modeling of in Vivo Rat Toxicity. Rev. Roum. Chim 2015, 60 (2–3), 219–226. [PMC free article] [PubMed] [Google Scholar]
- Sedykh A.; Zhu H.; Tang H.; Zhang L.; Richard A.; Rusyn I.; Tropsha A. Use of in Vitro HTS-Derived Concentration–Response Data as Biological Descriptors Improves the Accuracy of QSAR Models of in Vivo Toxicity. Environ. Health Perspect 2011, 119 (3), 364–370. 10.1289/ehp.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourches D.; Pu D.; Tassa C.; Weissleder R.; Shaw S. Y.; Mumper R. J.; Tropsha A. Quantitative Nanostructure–Activity Relationship Modeling. ACS Nano 2010, 4 (10), 5703–5712. 10.1021/nn1013484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourches D.; Pu D.; Tropsha A. Exploring Quantitative Nanostructure-Activity Relationships (QNAR) Modeling as a Tool for Predicting Biological Effects of Manufactured Nanoparticles. Comb. Chem. High Throughput Screen 2011, 14 (3), 217–225. 10.2174/138620711794728743. [DOI] [PubMed] [Google Scholar]
- Hook A. L.; Chang C.-Y.; Yang J.; Luckett J.; Cockayne A.; Atkinson S.; Mei Y.; Bayston R.; Irvine D. J.; Langer R.; et al. Combinatorial Discovery of Polymers Resistant to Bacterial Attachment. Nat. Biotechnol. 2012, 30 (9), 868–875. 10.1038/nbt.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiona R. M. T.; Welch N. G.; Muir B. W.; Winkler D. A.; Pigram P. J. Rapid Evaluation of Immobilized Immunoglobulins Using Automated Mass-Segmented ToF-SIMS. Biointerphases 2019, 14 (6), 061002. 10.1063/1.5121450. [DOI] [PubMed] [Google Scholar]
- Gardner W.; Hook A. L.; Alexander M. R.; Ballabio D.; Cutts S. M.; Muir B. W.; Pigram P. J. ToF-SIMS and Machine Learning for Single-Pixel Molecular Discrimination of an Acrylate Polymer Microarray. Anal. Chem. 2020, 92 (9), 6587–6597. 10.1021/acs.analchem.0c00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIST. http://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:amdis (accessed 2022-05-27).
- Horai H.; Arita M.; Kanaya S.; Nihei Y.; Ikeda T.; Suwa K.; Ojima Y.; Tanaka K.; Tanaka S.; Aoshima K.; et al. MassBank: A Public Repository for Sharing Mass Spectral Data for Life Sciences. J. Mass Spectrom 2010, 45 (7), 703–714. 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- MassBank. https://massbank.eu/MassBank/Search (accessed 2022-05-27).
- Mona. https://mona.fiehnlab.ucdavis.edu/ (accessed 2022-05-27).
- Tsugawa H.; Cajka T.; Kind T.; Ma Y.; Higgins B.; Ikeda K.; Kanazawa M.; VanderGheynst J.; Fiehn O.; Arita M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12 (6), 523–526. 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MS-DIAL. http://prime.psc.riken.jp/compms/msdial/main.html (accessed 2022-05-27).
- ChemID plus. https://chem.nlm.nih.gov/chemidplus/ (accessed 2022-05-27).
- CompTox. https://comptox.epa.gov/dashboard/ (accessed 2022-05-27).
- Williams A. J.; Grulke C. M.; Edwards J.; McEachran A. D.; Mansouri K.; Baker N. C.; Patlewicz G.; Shah I.; Wambaugh J. F.; Judson R. S.; Richard A. M. The CompTox Chemistry Dashboard: A Community Data Resource for Environmental Chemistry. J. Cheminf 2017, 9 (1), 61. 10.1186/s13321-017-0247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri K.; Grulke C. M.; Judson R. S.; Williams A. J. OPERA Models for Predicting Physicochemical Properties and Environmental Fate Endpoints. J. Cheminform 2018, 10 (1), 10. 10.1186/s13321-018-0263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. H. Scales of Solute Hydrogen-Bonding: Their Construction and Application to Physicochemical and Biochemical Processes. Chem. Soc. Rev. 1993, 22 (2), 73–83. 10.1039/cs9932200073. [DOI] [Google Scholar]
- Endo S.; Goss K.-U. Applications of Polyparameter Linear Free Energy Relationships in Environmental Chemistry. Environ. Sci. Technol. 2014, 48 (21), 12477–12491. 10.1021/es503369t. [DOI] [PubMed] [Google Scholar]
- ACD/Labs. https://www.acdlabs.com/products/percepta/ (accessed 2022-05-27).
- Tibshirani R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. B 1996, 58 (1), 267–288. 10.1111/j.2517-6161.1996.tb02080.x. [DOI] [Google Scholar]
- Hoerl A. E.; Kennard R. W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12 (1), 55–67. 10.1080/00401706.1970.10488634. [DOI] [Google Scholar]
- Zou H.; Hastie T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. B 2005, 67 (2), 301–320. 10.1111/j.1467-9868.2005.00503.x. [DOI] [Google Scholar]
- Chollet F.et al. ; Keras; 2017. https://github.com/fchollet/keras (accessed 2022-05-27).
- Abadi M.; Agarwal A.; Barham P.; Brevdo E.; Chen Z.; Citro C.; Corrado G. S.; Davis A.; Dean J.; Devin M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems; 2015. https://www.tensorflow.org/ (accessed 2022-05-27).
- Breiman L. Random Forests. Mach. Learn 2001, 45 (1), 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Wright M. N.; Ziegler A. ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Soft 2017, 77 (1), 1–17. 10.18637/jss.v077.i01. [DOI] [Google Scholar]
- Chen T.; Guestrin C.. XGBoost: A Scalable Tree Boosting System. In Proc. 22nd ACM SIGKDD Int. Conf. Know. Disc Data Mining; San Francisco, California, USA, 2016; 10.1145/2939672.2939785. [DOI]
- Ihaka R.; Gentleman R. R. A Language for Data Analysis and Graphics. J. Comput. Graph. Stat 1996, 5 (3), 299–314. 10.1080/10618600.1996.10474713. [DOI] [Google Scholar]
- Kuhn M. Building Predictive Models in R Using the caret Package. J. Stat. Soft 2008, 28 (5), 1–26. 10.18637/jss.v028.i05. [DOI] [Google Scholar]
- Krizhevsky A.; Sutskever I.; Hinton G. E.. ImageNet Classification with Deep Convolutional Neural Networks. In Proc. 25th Int. Conf. Neural Inf. Process. Syst., Lake Tahoe, Nevada; 2012.
- Friedman J.; Hastie T.; Tibshirani R. Additive logistic regression: A statistical view of boosting (With discussion and a rejoinder by the authors). Ann. Stat 2000, 28 (2), 337–407. 10.1214/aos/1016218223. [DOI] [Google Scholar]
- Zushi Y.; Yamatori Y.; Nagata J.; Nabi D. Comprehensive Two-Dimensional Gas-Chromatography-Based Property Estimation to Assess the Fate and Behavior of Complex Mixtures: A Case Study of Vehicle Engine Oil. Sci. Total Environ. 2019, 669, 739–745. 10.1016/j.scitotenv.2019.03.157. [DOI] [PubMed] [Google Scholar]
- Eriksson L.; Jaworska J.; Worth A. P.; Cronin M. T. D.; McDowell R. M.; Gramatica P. Methods for Reliability and Uncertainty Assessment and for Applicability Evaluations of Classification- and Regression-Based QSARs. Environ. Health Perspect 2003, 111 (10), 1361–1375. 10.1289/ehp.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazonovas A.; Japertas P.; Didziapetris R. Estimation of Reliability of Predictions and Model Applicability Domain Evaluation in the Analysis of Acute Toxicity (LD 50). SAR QSAR Environ. Res. 2010, 21 (1–2), 127–148. 10.1080/10629360903568671. [DOI] [PubMed] [Google Scholar]
- OECD. OECD principles for the Varidation, for Regulatory Purpose, of (Q)SAR Models. https://www.oecd.org/chemicalsafety/risk-assessment/validationofqsarmodels.htm (accessed 2022-05-27).
- Dobson C. M. Chemical Space and Biology. Nature 2004, 432 (7019), 824–828. 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- Fourches D.; Muratov E.; Tropsha A. Curation of Chemogenomics Data. Nat. Chem. Biol. 2015, 11 (8), 535–535. 10.1038/nchembio.1881. [DOI] [PubMed] [Google Scholar]
- Liu X.; Taylor M. P.; Aelion C. M.; Dong C. Novel Application of Machine Learning Algorithms and Model-Agnostic Methods to Identify Factors Influencing Childhood Blood Lead Levels. Environ. Sci. Technol. 2021, 55 (19), 13387–13399. 10.1021/acs.est.1c01097. [DOI] [PubMed] [Google Scholar]
- Zorn K. M.; Foil D. H.; Lane T. R.; Hillwalker W.; Feifarek D. J.; Jones F.; Klaren W. D.; Brinkman A. M.; Ekins S. Comparison of Machine Learning Models for the Androgen Receptor. Environ. Sci. Technol. 2020, 54 (21), 13690–13700. 10.1021/acs.est.0c03984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan R. P.; Wang W. M.; Liaw A.; Ma J.; Gifford E. M. Extreme Gradient Boosting as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model 2016, 56 (12), 2353–2360. 10.1021/acs.jcim.6b00591. [DOI] [PubMed] [Google Scholar]
- Martin T. M.; Young D. M. Prediction of the Acute Toxicity (96-h LC50) of Organic Compounds to the Fathead Minnow (Pimephales promelas) Using a Group Contribution Method. Chem. Res. Toxicol. 2001, 14 (10), 1378–1385. 10.1021/tx0155045. [DOI] [PubMed] [Google Scholar]
- USEPA. User’s Guide for T.E.S.T. (version 5.1) (Toxicity Estimation Software Tool): A Program to Estimate Toxicity from Molecular Structure; 2020. https://www.epa.gov/sites/default/files/2016-05/documents/600r16058.pdf (accessed 2022-05-27).
- Polishchuk P. Interpretation of Quantitative Structure–Activity Relationship Models: Past, Present, and Future. J. Chem. Inf. Model 2017, 57 (11), 2618–2639. 10.1021/acs.jcim.7b00274. [DOI] [PubMed] [Google Scholar]
- Zhong S.; Zhang K.; Bagheri M.; Burken J. G.; Gu A.; Li B.; Ma X.; Marrone B. L.; Ren Z. J.; Schrier J.; et al. Machine Learning: New Ideas and Tools in Environmental Science and Engineering. Environ. Sci. Technol. 2021, 55 (19), 12741–12754. 10.1021/acs.est.1c01339. [DOI] [PubMed] [Google Scholar]
- Zushi Y. NMF-Based Spectral Deconvolution with a Web Platform GC Mixture Touch. ACS Omega 2021, 6 (4), 2742–2748. 10.1021/acsomega.0c04982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S. E. An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. J. Am. Soc. Mass Spectrom. 1999, 10 (8), 770–781. 10.1016/S1044-0305(99)00047-1. [DOI] [Google Scholar]
- Zushi Y.; Hashimoto S.; Tanabe K. Global Spectral Deconvolution Based on Non-Negative Matrix Factorization in GC × GC–HRTOFMS. Anal. Chem. 2015, 87 (3), 1829–1838. 10.1021/ac5038544. [DOI] [PubMed] [Google Scholar]
- Smirnov A.; Qiu Y.; Jia W.; Walker D. I.; Jones D. P.; Du X. ADAP-GC 4.0: Application of Clustering-Assisted Multivariate Curve Resolution to Spectral Deconvolution of Gas Chromatography–Mass Spectrometry Metabolomics Data. Anal. Chem. 2019, 91 (14), 9069–9077. 10.1021/acs.analchem.9b01424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi D.; Gros J.; Dimitriou-Christidis P.; Arey J. S. Mapping Environmental Partitioning Properties of Nonpolar Complex Mixtures by Use of GC × GC. Environ. Sci. Technol. 2014, 48 (12), 6814–6826. 10.1021/es501674p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detective-QSAR is freely available at http://www.mixture-platform.net/Detective_QSAR_Med_Open/ (Ver. Med) and http://www.mixture-platform.net/Detective_QSAR_Pred_Open/ (Ver. Pred).