

Abstract
Neurovascular unit (NVU) is organized multi-cellular and multi-component networks that are essential for brain health and brain homeostasis maintaining. Neurovascular unit dysfunction is the central pathogenesis process of ischemic stroke. Thus integrated protection of NVU holds great therapeutic potential for ischemic stroke. Catalpol, classified into the iridoid monosaccharide glycoside, is the main active ingredient of the radix from traditional Chinese medicine, Rehmannia glutinosa Libosch, that exhibits protective effects in several brain-related diseases. In the present study, we investigated whether catalpol exerted protective effects for NVU in ischemic stroke and the underlying mechanisms. MCAO rats were administered catalpol (2.5, 5.0, 10.0 mg·kg−1·d−1, i.v.) for 14 days. We showed that catalpol treatment dose-dependently reduced the infarction volume and significantly attenuated neurological deficits score in MCAO rats. Furthermore, catalpol treatment significantly ameliorated impaired NVU in ischemic region by protecting vessel-neuron-astrocyte structures and morphology, and promoting angiogenesis and neurogenesis to replenish lost vessels and neurons. Moreover, catalpol treatment significantly increased the expression of vascular endothelial growth factor (VEGF) through up-regulating PI3K/AKT signaling, followed by increasing FAK and Paxillin and activating PI3K/AKT and MEK1/2/ERK1/2 pathways. The protective mechanisms of catalpol were confirmed in an in vitro three-dimensional NVU model subjected to oxygen-glucose deprivation. In conclusion, catalpol protects NVU in ischemic region via activation of PI3K/AKT signaling and increased VEGF production; VEGF further enhances PI3K/AKT and MEK1/2/ERK1/2 signaling, which may trigger a partly feed-forward loop to protect NVU from ischemic stroke.
Keywords: ischemic stroke, catalpol, neurovascular unit (NVU), VEGF, PI3K/AKT, MEK1/2/ERK1/2
Introduction
Neurovascular unit (NVU), proposed at the 2001 Stroke Progress Review Group meeting of the National Institute of Neurological Disorders and Stroke, is an organized multi-cellular and multi-component networks that are essential for brain health and brain homeostasis maintaining [1, 2]. This brain-localized NVU is emerging as an important player during multiple acute brain injuries and chronic neurological disorders, including ischemic stroke, traumatic brain injury and Alzheimer’s disease [3]. NVU is systematacially divided into neural and vascular components, and represented fundamental unit of brain structure and function. Generally, the neural component/neuron is critical for the cognitive ability in a variety of neurodegeneration diseases as they are susceptible to various pathological factors, such as hypoxia and oxidative stress. The vascular component/endothelial cells form the important multi-cellular structure, blood–brain barrier (BBB), by interacting with neural component [4, 5] to separate the brain from systemic blood circulation [6]. Vascular component of NVU is sensitive to ischemia and is typically damaged in the super acute stage of ischemia [7]. Cerebral blood flow is rapidly decreased in ischemic region after stroke, which will result in various pathological events, including but not limited to oxidative stress, apoptosis, and inflammation [8–10]. Undoubtedly, neurovascular dysfunction is a major process in the pathophysiological process of ischemic stroke and various neurodegenerative diseases. The NVU plays its dominant roles in the pathogenesis process of ischemic stroke, with profound effects on the BBB integrity and neurovascular coupling maintenance. Damaged NVU can also lead to toxic compounds accumulation in the brain and lose its barrier ability for harmful molecules outside of the brain [11], which may result in both the onset and progression of neurodegeneration. Therefore, no matter what the therapies targeting the NVU, cell-based or pharmacological, may provide remarkable strategies for disastrous hallmarks induced by ischemic stroke. Recently, the holistic neurovascular unit as a target in ischemic stroke therapeutic philosophy, not the neuronal or vascular, is increasingly recognized. In order to further tap the therapeutic potential of targeting NVU in ischemic stroke, in clinical or non-clinical settings, considering the entire framework of the NVU and multiple interactions between cells are urgently needed [12].
Catalpol is isolated from the radix of Rehmannia glutinosa Libosch, which is classed into iridoid monosaccharide glycoside. As the main active component of Rehmannia glutinosa Libosch, evidence supports that catalpol exhibits potential protective effects in several brain-related diseases [13–16]. The difficulty in recovering from the brain-related diseases is mainly related to the low intrinsic activity of neurons and the difficulty in axonal regeneration. Recent report indicated that catalpol promotes the axonal growth of cortical neurons and may improve the microenvironment for the neurons, which is conducive to the neurons survival and regeneration [17]. Catalpol has also been reported to attenuate neurons apoptosis [18], and repress oigodendrocytes death in various ischemia animal models [19]. Furthermore, our previous study has demonstrated that repeated treatment of ischemic stroke rats with catalpol improved neurological deficits and reduced infarction volume [20, 21]. However, whether catalpol has global protection for entire framework of the NVU in stroke remains to be determined; also, dissecting the its underlying mechanisms could pave the way for exploring novel therapeutic drugs and targets toward NVU dysfunction in ischemic stroke.
Vascular endothelial growth factor (VEGF) is secreted by most types of cells, including neurons, astrocytes, and endothelial cells, and is deemed to contribute to angiogenesis and vascular permeability [22]. Both VEGF-binding receptors, VEGFR1 (Flt-1) and VEGFR2 (KDR/Flk-1), are tyrosine kinases. There are much evidences that VEGFR2 delivers key mitogenic, angiogenic and permeability-enhancing signals of VEGF, and is the major mediator of endothelial cells mitogenesis and survival, as well as angiogenesis and microvascular permeability [23, 24]. VEGF was once considered to be the most specific growth factor in endothelial cells, but there are increasing evidences supporting that VEGF also exhibits neuroprotective effects [22, 25–30]. The fact that VEGF protected NVU model from oxygen glucose deprivation (OGD) was also reported [31]. In the super acute stage of cerebral ischemia, VEGF is strongly induced, which not only plays its vital roles in vascular permeability improvement and angiogenesis promotion, but also in neuroprotection. The promotion of VEGF production and binding to VEGFR2 recovers collateral circulation, improves microcirculation, and strengthens neuroprotection. Nevertheless, this feedback self-protection is lost in recovery phase of ischemic stroke [22]. Enhancing the production of endogenous VEGF during recovery phase may be considered as a potential and promising therapeutic strategy for ischemic stroke.
In the present study, we aimed at investigating the potential NVU protective effect of catalpol in ischemic stroke rats from the aspects of vascular and neuronal protection, angiogenesis and neurogenesis promotion. In addition, we generated an in vitro three-dimensional NVU (3D NVU) with primary neural stem cells (NSCs) and brain microvascular endothelial cells (BMECs) to model ischemic stroke-induced pathological NVU by employing widely recognized oxygen-glucose deprivation (OGD) pattern, to dissect the underlying pharmacological mechanisms of NVU protection by catalpol.
Materials and methods
Rats care and usage
Neonatal rats (1-day-old, 10-day-old), and male Sprague-Dawley (SD) rats (two-month-old, 180–220 g) were all purchased from the Experimental Animal Center (Chongqing Medical University, SCXK(Yu) 2019-0014, Chongqing, China). The animal protocols were approved by the Southwest University Animal Use and Care Committee (SYXK (Yu) 2017-0003). The National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978) and Southwest University animal guidelines were obeyed in all animal experiments.
Middle cerebral artery occlusion (MCAO) models, modified neurological severity score (mNSS), and drug administration
MCAO models
Male SD rats (180–220 g) were deeply anesthetized with isoflurane and used to generate the MCAO-induced ischemic stroke models by electrocoagulation according to reported protocols [32, 33]. In brief, anesthetized rats were placed into a stereotaxic frame, followed by isolating temporal muscle from harnpan. A small hole was made by using an electric drill to expose the middle cerebral artery (MCA). Then bipolar coagulation forceps were used to coagulate the exposed MCA below the intersection of MCA and inferior vena cava. The rats with exposed but uncoagulated MCA were assigned to sham group.
mNSS
The waking rats were tested by mNSS for successful ischemic stroke rats screening (Supplementary Table S1), as previously reported [34–36]. All rats were tested mNSS again on day 7 and 14 in treatment process.
Drug administration
Catalpol groups were treated with catalpol (98.04%, HY-N0820, MedChemExpress, USA) for 14 days (i.v, 2.5, 5.0, 10.0 mg·kg−1·d−1, diluted in saline solution), and the same volume of saline solution was given to MCAO and Sham groups.
2, 3, 5-Triphenyl tetrazolium chloride (TTC) staining for brain slices
On day 14, partial rats were sacrificed for TTC staining. The sliced brain coronal sections were stained with 1% TTC (T8877, Sigma–Aldrich, USA) at 37 °C for 20–30 min and the stained sections were transferred into 4% paraformaldehyde for fixing and images capturing.
Generation of 3D NVU, OGD, and treatment for 3D NVU
We cultured primary NSCs and BMECs and generated 3D NVU according to our previous report [31]. For 3D NVU generation, primary NSCs and BMECs were co-cultured in Matrigel (356234, BD Biosciences, USA) at a density ratio of 1:10 (final density of BMECs and NSCs was 2.0 × 106/mL and 2.0 × 105/mL). After 7 days, the 3D NVU was used for OGD and treatment. For OGD, 3D NVU was incubated in anaerobic chamber (BINGDER150, Germany) filled with a gas mixture of 5% CO2, 95% N2. Cell culture medium was replaced with deoxygenated, glucose-free Earle’s balanced salt solution (EBSS) containing (mg/L) 6800 NaCl, 400 KCl, 264 CaCl2·2H2O, 200 MgCl2·7H2O, 2200 NaHCO3, 140 NaH2PO4·H2O, pH 7.2, and the 3D NVU was incubated in anaerobic chamber at 37 °C for 8 h [31]. Control 3D NVU was incubated in normal EBSS containing 25 mM glucose for 8 h at 37 °C, 5% CO2. 3D NVU was pre-treated with catalpol (25, 50, 100 μM) or VEGF (10 ng/mL, as a positive control, 78073, StemCell, Canada) for 16 h, SU1498 (10 μM, HY-19326, MedChemExpress, USA), LY294002 (10 μM, HY-10108, MedChemExpress, USA), or PD98059 (10 μM, HY-12028, MedChemExpress, USA) for 2 h and then continuouly treated with catalpol or inhibitors and placed in OGD conditions for 8 h.
Immunostaining
Immunostaining for brain slices
Part of rats was sacrificed and the brain was removed and fixed for paraffin sections slicing, after treatment with catalpol for 14 days. The paraffin blocks were then cut into 5 μm sections (Leica SM2010R sliding microtome, Leica Microsystems Inc., Buffalo Grove, IL, USA), mounted on glass slides, followed by performing deparaffinization and antigen retrieval (heating for 30 min in Citrate-EDTA Buffer containing 10 mM citric acid (pH 6.2), 2 mM EDTA and 0.05% Tween-20) according to previous report [21]. For immunofluorescence, the brain sections were washed with PBS (3 × 10 min) and blocked with blocking solution (0.1% Triton X-100 and 10% serum, which was generated from the species of the secondary antibodies) at room temperature for 1 h. The sections were then incubated with primary antibody in blocking solution at 4 °C overnight. After washing in PBS and incubating with Alexa Fluor secondary antibodies for 2 h at room temperature, the brain sections were finally mounted with Prolong Diamond Anti-fade mountant (P36982, Life Technologies, USA) for image capturing. Horseradish Peroxidase was also used in immunohistochemistry.
Immunostaining for 3D NVU
According to previous report [37] and our modified protocol, the 3D NVU was washed with Tris buffered saline supplemented with 0.1% (v/v) Tween-20 (TBST), fixed with 4% paraformaldehyde, and then permeabilized in TBST containing 0.5% (v/v) Triton X-100 and 4% donkey serum at room temperature for 2–3 h. The 3D NVU was then blocked in blocking solution (50 mM Tris (pH 7.4), 0.1% Tween-20, 4% donkey serum, 1% BSA, 0.1% gelatin and 0.3 M glycine) at room temperature for 5 h with gentle rocking. The 3D NVU was washed with TBST and incubated with primary antibodies at 4 °C overnight. After being washed 5 times and placed in TBST at 4 °C overnight, the 3D NVU was then incubated with matched secondary antibodies for 3–5 h at room temperature. After washing 5 times with TBST, Prolong Glass Anti-fade mountant was added to stain 3D NVU before image capturing. All the useful primary and secondary antibodies can be found in Supplementary Table S2.
Western blot assay
The proteins from the infarcted brain tissues or 3D NVU were collected with cell lysis solution for performing Western blot on SDS-PAGE gels. Cell lysis buffer with protease/phosphatase inhibitor cocktails (HY-K0010, HY-K0022, HY-K0023, MedChemExpress, USA) was used to avoid proteins degradation. All the used antibodies were listed in Supplementary Table S2.
5-ethynyl-2’-deoxyuridine (EdU) labeling of dividing cells in brain and 3D NVU
EdU (ST067, Beyotime, China, i.p, 50 mg·kg−1·d−1) was used from day 7 to day14 to label the proliferating cells in rats. 3D NVU was incubated with medium containing 10 mM EdU for 2 h (for BMECs) or 24 h (for neurons) to label the proliferating cells in 3D NVU. After immunostaining for brain sections or 3D NVU, EdU labeling for S-phase cells was detected using the BeyoClick™ EdU-594 Cell Proliferation Assay Kit (C0078, Beyotime, China).
Cytokines assay in 3D NVU
The levels of VEGF and brain-derived neurotrophic factor (BDNF) in the culture medium were detected by an enzyme-linked immunosorbent assay (ELISA) kit (Sino Best Biological Technology, China). A microplate ELISA reader (BioTek, Winooski, VT, USA) was used to measure absorbance.
Measurement of transendothelial electrical resistance (TEER) values
TEER values of 3D NVU in culture inserts (662640, Greiner, Germany) were measured using epithelial-volt-ohm resistance meter (ERS-2, Millipore, USA) according to the previous report [38]. TEER was measured by placing micro-electrode probes in three different regions of NVU models. The background TEER values were measured in the same well under the same condition with no 3D NVU seeded. The final result was calculated as the TEER values of different groups subtracted of the corresponding background TEER values and then multiplied by the area of insert membrane. The background TEER values were taken from the same condition with no cell seeded.
Permeability measurement of 3D NVU
For sodium fluorescein assay, the 3D NVU in the inserts was incubated with 0.5 mL culture medium containing 100 μg/mL sodium fluorescein (F6377, Sigma, USA) for 2 h. Absorbance of samples taken from the basolateral chamber was determined by using plate reader. Absorbance of samples was analyzed for evaluating the permeability of vascular network.
Lactate dehydrogenase (LDH) measurement
CyQUANT™ LDH cytotoxicity assay kit (C20300, ThermoFisher Scientific, USA) was used to measure LDH. After 3D NVU was exposed to OGD, the medium containing released LDH (extracellular LDH) was transferred to a 96-well plate and mixed with reaction mixture. To measure intracellular LDH, the 3D NVU was then lysed with lysis buffer and transferred to another 96-well plate, followed by adding reaction mixture. Stop solution was added into well after 30 min incubation at room temperature. Absorbance at 490 nm and 680 nm was measured on plate reader to calculate LDH activity. LDH activity = LDH490 nm – LDH680 nm. LDH leakage rate (%) = extracellular LDH activity / (extracellular LDH activity + intracellular LDH activity) × 100%.
Image acquisition and analysis
The images of the 3D NVU and the sections from brain were captured by using confocal microscope (NIKON, A1 + R10802, Japan) and fluorescence microscope (Leica DFC310, Germany). Images were analyzed by ImageJ (NIH, USA). To calculate a mean value of cell counts/intensity, 5 images were taken from different areas of each sample for each technical replicate and particles analysis and mean gray value analysis in ImageJ was used. All parameters of each image capturing, such as gain, exposure time, and contrast, were maintained the same for each fluorescence channel. Neuron J and Angiogenesis Analyzer plugins for ImageJ were used respectively to calculate the axon length and vascular length.
Statistical analysis
All in vivo experiments consisted of 8 biological replicates and in vitro experiments consisted of 3 biological replicates. All data were analyzed by using GraphPad Prism 8 and unpaired two-tailed t test or one-way ANOVA was used. Graphs were produced by using GraphPad Prism 8 with bars reflecting the value range. Data are presented as the mean ± S.D. and P value inferior to 0.05 was taken as statistically significant.
Results
Catalpol protected NVU structure of ischemic stroke rats
In this study, the rats were tested with mNSS on days 0, 7, and 14 and then were sacrificed for other tests (Fig. 1a). With continuous catalpol treatment, the neurological deficits score of rats were significantly reduced by catalpol, in contrast to that from MCAO group, suggesting the improved neurological function of ischemic stroke rats (Supplementary Fig. S1a, b). Furthermore, TTC staining of brain slices showed deep red staining from sham group but large infarcted areas in MCA-blocked hemisphere from MCAO group (Supplementary Fig. S1c). And as expected, compared with that from the MCAO group, the infarction volume in the catalpol groups was significantly decreased (Supplementary Fig. S1d), suggesting that the catalpol has anti-ischemic stroke activity. On day 14, ischemic necrosis, liquefaction, porous-mesh structure and blurred cell border were observed in brain tissues surrounding the blocking point or the cortex near the infarcted area. In contrast, alleviated brain tissue liquefaction and reduced necrosis and disorganized structure were observed in catalpol groups (2.5, 5, 10 mg/kg) (Fig. 1b, c).
Fig. 1. NVU was protected by catalpol in ischemic stroke.

a Sketch of the procedure for in vivo experiments. b Brain morphology of the ischemic stroke rats with or without catalpol treatment. The alleviated tissue liquefaction near the blocking point was observed. c Histological morphology suggested that ischemic necrosis and mesh-like structure were reduced with catalpol treatment. d NVU structures of the ischemic stroke rats were improved by catalpol. The rats with catalpol treatment exhibited more clearly cortical vascular morphology, more neurons, and more close astrocytes-vessels spatial arrangement (marked with white line). Scale bar, 100 μm (c), 20 μm (d).
Herein, we focused on exploring the potential protection for global NVU by catalpol. We then observed the integral structure of the NVU by multiple immunostaining. In the sham group, the NVU border was clear, vessel (CD31+)-neuron (MAP-2+)-astrocyte (GFAP+) unit near the infarcted area was ordered and had obvious relative configuration (white). Unlike rats from sham groups, MCAO rats exhibited disorganized unit, including disordered vascular structure, sparse neurons, and loose arrange position of astrocytes relative to vessels. The NVU structure near the infarct foci from catalpol-treated rats recovered significantly, in contrast to those in the MCAO rats (Fig. 1d). The above results indicated that catalpol alleviated NVU damage effectively in ischemic stroke rats.
Vascular and neuronal component damage was improved by catalpol
To count the number of vessels and neurons and observe vessel-astrocyte arrangement in the vicinity of infarcted area, CD31, NeuN and GFAP was used to label vessels, neurons, and astrocytes, respectively. Vessel and neuron density was decreased and vessel-astrocyte configuration was disordered in the MCAO group on day 14. When the rats were treated with catalpol for 14 days, the number of vessels and neurons was significantly increased and vessel-astrocyte configuration was improved in cortex near the infarcted area (Fig. 2a, b, f, g). The vascular and neuronal morphology was then observed by using immunochemistry and Nissl staining. Compared with those in sham group, vascular morphology in the vicinity of the infarcted area of MCAO group was disordered and exhibited basement membrane degradation (Fig. 2c). The cortical neurons were disorganized, the number of neuron was decreased, the Nissl staining became shallower, the Nissl bodies and nucleus were blurred, and the axonal length was reduced near the infarcted area of the ischemic stroke rats from the MCAO group, in contrast to that in the sham group (Fig. 2d, e, h). Expectedly, the rats of the catalpol groups (2.5, 5, 10 mg/kg) exhibited restored vascular structure (Fig. 2c). Moreover, compared with that in the MCAO group, the cortical neurons of catalpol-treated rats (2.5, 5.0, 10.0 mg/kg) were neatly arranged, with increased neuron number, obvious Nissl bodies and nucleus, and increased axonal length (Fig. 2d, e, h). Collectively, all the above results demonstrated that catalpol protected the NVU from ischemic stroke by protecting vascular and neuronal morphology and up-regulating vessel and neuron density.
Fig. 2. Catalpol protected vascular and neural component of NVU in the cortex near the infarcted area.

a, f, g The numbers of vessels and neurons in the cortex near the infarcted area were up-regulated in catalpol groups. b Vessel-astrocyte configuration near the infarcted area was improved by catalpol. The catalpol groups showed a significant improvement in vessel-astrocyte spatial arrangement on day 14. c Vascular morphology was protected by catalpol. Clearer vascular morphology and degradation recovery of basement membrane were shown in catalpol groups. d, e, h The catalpol groups showed a significant recovery of neurons layout and increased axonal length on day 14. Scale bar, 50 μm (b), 100 μm (a, c, d, e). The data are presented as the mean ± SD, n = 8, *P < 0.05, **P < 0.01.
Neurogenesis and angiogenesis was promoted by catalpol in the brain of ischemic stroke rats
Generally, the neurogenesis in adult mammalian brain is mainly generated in two areas: subgranular zone of hippocampal dentate gyrus (DG-SGZ) and subventricular zone of lateral ventricle (LV-SVZ) [39]. Therefore, we performed in vivo EdU assay to observe whether catalpol promotes brain neurogenesis and angiogenesis in DG-SGZ and LV-SVZ after stroke. Unlike sham group, the DCX+/EdU+ double positive cell number was decreased significantly in DG-SGZ and LV-SVZ of MCAO group rats. Whereas, after treatment with catalpol for 14 days, the DCX+/EdU+ double positive cell number was significantly up-regulated in catalpol groupthan that in the MCAO rats (Fig. 3a, c, d). We then tested angiogenesis in cortex of ischemic stroke rats with or without treatment with catalpol. The results suggested that the CD31+/EdU+ double positive cell number was decreased significantly in cortex of MCAO group compared with that in sham group. In contrast to those in MCAO group, the number of CD31+/EdU+ positive cells was increased significantly when treated with catalpol (Fig. 3b, e). All the above results indicated that catalpol promoted neurogenesis and angiogenesis in the brain of ischemic stroke rats, which provides robust evidence that catalpol protected NVU against ischemia stroke.
Fig. 3. Both neurogenesis and angiogenesis were promoted by catalpol.

a, c, d Neurogenesis in DG-SGZ and LV-SVZ was increased in catalpol groups. The number of DCX+/EdU+ double positive cells was significantly increased in ischemia brain after catalpol treatment for 14 days. b, e Angiogenesis was promoted by catalpol in the cortex. CD31+/EdU+ positive endothelial cells were significantly increased on day 14. Scale Bar = 100 μm. The data are presented as the mean ± S.D., n = 8, *P < 0.05, **P < 0.01.
VEGF-PI3K/AKT and VEGF-MEK1/2/ERK1/2 pathways were activated by catalpol
Subsequently, we studied the mechanisms of catalpol in protecting NVU from ischemic stroke. VEGF, which is secreted by endothelial cells, has been proved that it has the prominent role in angiogenesis and neurogenesis motivation [22]. The immunostaining results indicated that the production of VEGF, Paxillin, p-AKT, and p-ERK1/2 were significantly inhibited in the MCAO group, compared to that in the sham group. However, the VEGF, Paxillin, p-AKT, and p-ERK1/2 were all increased significantly in the catalpol groups (2.5, 5.0, and 10.0 mg/kg) (Fig. 4a–h), in contrast to that in MCAO rats. We further detected VEGF-related downstream protein expression changes under catalpol stimulation by Western blotting. The results suggested that compared to that in the sham group, the protein expression levels of VEGF, p-VEGFR2/VEGFR2, PI3K, p-AKT/AKT, FAK, p-Paxillin/Paxillin, MEK1/2, and p-ERK1/2/ERK1/2 were all reduced in the MCAO group. Nevertheless, the expression levels of above proteins were rescued by catalpol (Fig. 4i–m). Collectively, these results indicated that NVU protection by catalpol is dependent on the activation of VEGF-PI3K/AKT and VEGF-MEK1/2/ERK1/2 pathways followed by protecting vascular and neuronal morphology and promoting neurogenesis and angiogenesis.
Fig. 4. VEGF-PI3K/AKT and VEGF-MEK1/2-ERK1/2 pathways were activated by catalpol in the brain of cerebral ischemic rats.

a–h The fluorescence intensity of VEGF, Paxillin, p-AKT, and p-ERK1/2 was significantly increased in catalpol groups. i–m The production of VEGF downstream proteins was significantly up-regulated by catalpol on day 14. Scale bar, 100 μm (a, c, d) and 50 μm (b). The data are presented as the mean ± S.D., n = 8, *P < 0.05, **P < 0.01.
In vitro NVU structure was protected by catalpol under OGD exposure
To investigate the mechanisms of NVU protection of catalpol in detail, next, in vitro assays were performed by employing our previous reported 3D NVU model, which is generated by primary BMECs and NSCs co-culture [31] (Fig. 5a, b). When 3D NVU was exposed to OGD, the vessel-neuron-astrocyte spatial arrangement was destroyed, compared with that in the control (CTRL) group. The disordered NVU was restored after treatment with sustained catalpol (25, 50, 100 μM), with the more complete vascular-like structure, and the restored vessel-neuron-astrocyte configuration, consistent with VEGF (as a positive control) group (Fig. 5c). Furthermore, we noticed that oxidative stress-induced damage of 3D NVU was also alleviated by catalpol. After OGD-exposed 3D NVU was treated with catalpol, the dead cells, the superoxide anion levels, and the LDH leakage in 3D NVU were significantly decreased, in contrast to that in OGD group 3D NVU (Supplementary Fig. S2).
Fig. 5. Catalpol improved damaged vessel-neuron-astrocyte configuration of OGD-exposed 3D NVU.

a Schematic of the procedure of in vitro experiments. b Primary BMECs exhibited CD31-positive immunostaining and primary NSCs exhibited SOX-2/NESTIN-positive immunostaining. c The vessel-neuron-astrocyte configuration of OGD-exposed 3D NVU was improved by catalpol. Scale bar, 50 μm or 20 μm (b), 100 μm (c).
Catalpol promoted angiogenesis and neurogenesis and improved barrier function of OGD-exposed 3D NVU
We asked whether catalpol promotes angiogenesis and neurogenesis in OGD-exposed 3D NVU. Unlike 3D NVU in CTRL group, the CD31+/EdU+ double positive cell number was significantly decreased, vascular-like structure was destroyed and vascular length was significantly decreased of OGD-exposed 3D NVU. However, when treated with catalpol (25, 50, 100 μM) or VEGF for 24 h, the CD31+/EdU+ double positive cell number was increased significantly in catalpol or VEGF group (Fig. 6a, b, e, f). We then tested neurogenesis in OGD-exposed 3D NVU after treatment with catalpol. The results indicated that the DCX+/EdU+ double positive cell number and the axonal length were decreased significantly in OGD-exposed 3D NVU compared with those in CTRL 3D NVU. However, compared with those in the OGD group, DCX+/EdU+ double positive cell and axonal length in the catalpol groups or VEGF group were increased significantly (Fig. 6c, d, g, h). In addition, we also found the increased expression levels of phosphorylated ribosomal protein S6 (p-S6) and BDNF in catalpol groups (Supplementary Fig. S3). The expression of p-S6 is significantly up-regulated in newborn neurons [40]. Therefore, p-S6 is often deemed as indication of the neurons activity. BDNF is also an important factor affecting neurological function. When 3D NVU was exposed to the OGD environment, the expression levels of p-S6 and BDNF were significantly decreased in 3D NVU; however, compared with that in the OGD group, the up-regulated expression levels of p-S6 and BDNF were detected in 3D NVU of catalpol groups (Supplementary Fig. S3), which indicated the multi-targets effects of catalpol in NVU protection.
Fig. 6. Catalpol promoted angiogenesis and neurogenesis and improved barrier function of OGD-exposed 3D NVU.

a, e CD31+/EdU+ double positive cells were detected by immunostaining. The number of CD31+/EdU+ positive cells under OGD exposition was increased in the catalpol groups. b, f Integrity of vascular-like structure in OGD-exposed 3D NVU was maintained by catalpol. c, g The number of DCX+/EdU+ positive cells in OGD-exposed 3D NVU was increased in the catalpol groups. d, h Length of axon in OGD-exposed 3D NVU was maintained in the catalpol groups. i, k Tight junction integrity of 3D NVU was observed by immunostaining. The expression of ZO-1 in OGD-exposed 3D NVU was increased in the catalpol groups. j, l Astrocyte of 3D NVU was observed by immunostaining. The expression of GFAP in OGD-exposed 3D NVU was increased in the catalpol groups. m Leakage of sodium fluorescein in OGD-exposed 3D NVU was alleviated in the catalpol groups. n TEER values in OGD-exposed 3D NVU were increased by catalpol. Scale bar = 100 μm. The data are presented as the mean ± S.D., n = 3, *P < 0.05, **P < 0.01.
We then performed permeability assays since the barrier permeability has critical roles in homeostasis of NVU. After the 3D NVU was exposed to OGD, the expression of tight junction protein ZO-1 in OGD 3D NVU was significantly reduced compared with those in CTRL 3D NVU; but the expression level of ZO-1 was significantly increased in both VEGF and catalpol (25, 50, 100 μM) treated 3D NVU (Fig. 6i, k). In ischemic stroke, the neurovascular coupling microenvironment constructed by astrocytes will be destroyed. Compared with that in the CTRL 3D NVU, the positive area of astrocytes in the OGD-exposed 3D NVU was significantly reduced. However, the positive area of astrocytes was enlarged by catalpol (50, 100 μM) (Fig. 6j, l), but not VEGF, compared with the 3D NVU in OGD group. Furthermore, the absorbance of sodium fluorescein was significantly increased and the TEER values were significantly decreased in the OGD-exposed 3D NVU, revealing the impaired barrier function. As expected, the leakage rate of sodium fluorescein was decreased and the TEER values were increased significantly in VEGF and catalpol groups, respectively (Fig. 6m, n), which both indicated that the barrier function of 3D NVU was improved by catalpol.
VEGF-PI3K/AKT and VEGF-MEK1/2/ERK1/2 signals were enhanced by catalpol in the 3D NVU
We then asked whether VEGF signals were enhanced by catalpol in 3D NVU, as same as in vivo. Similarly, the decreased production of VEGF in the OGD-exposed 3D NVU was rescued by catalpol (Fig. 7a–c). Immunostaining results also showed that the positive area of Paxillin was decreased in the OGD group and was increased in catalpol groups (Fig. 7d, e). We further tested the expression of the VEGF downstream pathways related proteins. The results showed that the expression levels of VEGF, p-VEGFR2/VEGFR2, FAK, p-Paxillin/Paxillin, or the PI3K/AKT and MEK1/2/ERK1/2 in the OGD-exposed 3D NVU were significantly decreased or inhibited than that in the CTRL group. However, the expression levels of the above proteins in 3D NVU were increased after treatment with catalpol (Fig. 7f–j). These results demonstrated that both VEGF-PI3K/AKT and -MEK1/2/ERK1/2 pathways were activated in 3D NVU of catalpol groups.
Fig. 7. The VEGF-PI3K/AKT and -MEK1/2/ERK1/2 pathways were activated by catalpol.

a, b VEGF in OGD-exposed 3D NVU was significantly increased by catalpol, compared with those in OGD-exposed 3D NVU. c, VEGF released into medium was significantly increased by catalpol. d, e The fluorescence intensity of Paxillin in catalpol-treated 3D NVU was significantly increased. f–j The VEGF pathways in 3D NVU were activated by catalpol. Scale bar = 100 μm. Data are presented as the mean ± S.D., n = 3, *P < 0.05, **P < 0.01.
Catalpol protected vascular component of NVU by activating VEGF-PI3K/AKT pathway
To further confirm that VEGF mediates the protective effects of catalpol on vascular component through VEGF-PI3K/AKT activation, 3D NVU was incubated in the presence or absence of the VEGFR2 tyrosine kinase inhibitor, SU1498, or a broad-spectrum inhibitor of PI3K, LY294002. Vascular-like structures protection of catalpol was significantly inhibited by SU1498 and LY294002 (Fig. 8b, d), whereas the expression of p-AKT was slightly reduced but not significantly, between catalpol (50 μM) group and catalpol (50 μM)+SU1498 group (Fig. 8a, c, e, g). Moreover, the expression levels of the p-VEGFR2/VEGFR2, FAK, and p-Paxillin/Paxillin were significantly decreased in the presence of SU1498. Nevertheless, the expression level of PI3K had also no significant changes between catalpol (50 μM) group and catalpol (50 μM)+SU1498 group (Fig. 8e–h). We then used PI3K/AKT pathway inhibitor LY294002 to observe whether catalpol activates PI3K/AKT pathway directly. The results showed that the PI3K/AKT pathway was significantly inhibited in catalpol (50 μM)+LY294002 group. Intriguingly, the expression levels of VEGF and p-VEGFR2/VEGFR2 in catalpol (50 μM)+LY294002 group were also significantly decreased compared with those in the catalpol group (Fig. 8i–k). Collectively, these findings suggested that catalpol directly activated PI3K/AKT pathway to promote VEGF production and then protected vascular structures of NVU by increasing FAK and Paxillin and activating PI3K/AKT pathway.
Fig. 8. Activation of VEGF-PI3K/AKT pathway in OGD-exposed 3D NVU was inhibited by SU1498 or LY294002.

a, c Intensity of p-AKT was decreased in catalpol (50 μM)+LY294002 group. b, d Vascular-like structure length was decreased in the presence of SU1498 or LY294002 groups. e–h Effects of SU1498 on the production of VEGF downstream proteins. i–k Activation effects of catalpol on the production of VEGF, p-VEGFR2/VEGFR2, PI3K, and p-AKT/AKT were inhibited by LY294002. Scale bar = 50 μm. The data are presented as the mean ± S.D., n = 3, *P < 0.05, **P < 0.01.
Catalpol protected neural component of NVU by activating VEGF-MEK1/2/ERK1/2 pathway
In order to clarify whether the diffusible VEGF regulates the neuroprotection of catalpol via MEK1/2/ERK1/2 pathways, SU1498, or a selective MEK inhibitor, PD98059, was used. The expression of p-ERK1/2 was significantly reduced, followed by the decreased axonal length in 3D NVU after treatment with SU1498 or PD98059 (Fig. 9a–d), compared with those in single catalpol-treated 3D NVU. In addition, the exression levels of VEGF downstream proteins p-VEGFR2/VEGFR2, MEK1/2, and p-ERK1/2/ERK1/2 in the catalpol (50 μM)+SU1498 or catalpol (50 μM)+PD98059 group were reduced significantly, compared with those in single catalpol (50 μM) treatment group (Fig. 9e–g). Taken together, these results demonstrated that VEGF-MEK1/2/ERK1/2 pathway mediates the neuroprotection of catalpol.
Fig. 9. Activation of VEGF-MEK1/2/ERK1/2 pathway in OGD-exposed 3D NVU was inhibited by SU1498 or PD98059.

a, c Fluorescence intensity of p-ERK1/2 was reduced by SU1498 or PD98059. b, d Axonal length was decreased in catalpol (50 μM)+SU1498 or catalpol (50 μM)+PD98059 groups. e–g Promotion effects of catalpol on the production of p-VEGFR2/VEGFR2, MEK1/2, and p-ERK1/2/ERK1/2 were inhibited by SU1498 or PD98059. Scale bar = 50 μm. The data are presented as the mean ± S.D., n = 3, *P < 0.05, **P < 0.01.
Discussion
Neurovascular dysfunction is the central pathogenesis process of most neurodegenerative diseases, including stroke and Alzheimer’s disease [1]. NVU dysfunction leads to restriction of the transmission of oxygen and nutrients, an impaired ability to remove toxic compounds, and induces harmful molecules and immune cell infiltration, or through the barrier, promotes the neurovascular dysfunction and occurrence and development of neurodegeneration. Both vascular damages and neuron dysfunction are main pathological hallmarks of ischemic stroke, including reduced vessel density and neuron apoptosis or loss in ischemic tissue, followed by hypo-oxygenation and neurological function deficits [41]. For therapies of ischemic stroke, vascular component protection and angiogenesis promotion is necessary for repairing ischemic tissue and improving neurological function deficits; neuroprotection and the recovery of neural component structure and function are also critical for improving the prognosis of ischemia. In summary, NVU plays its vital role in neurovascular function repair during or after ischemic stroke, as the cell preservation was forcefully supported by the homeostasis of the NVU via an accurate regulation of neurotrophic factors and subsequent signaling pathways. NVU also secrets a various proteins to protect BBB from breakdown and to support its functional recovery [12]. Therefore, it is a promising strategy to combine vascular protection and neuroprotection to strengthen the overall protection for NVU in ischemic stroke.
In the present study, the global protective effects of catalpol on NVU were investigated from the perspectives of vascular and neural component protection. We found that vascular structure was more complete, vessels density and the dividing endothelial cells (CD31+/EdU+) were increased in the infarction cortex of the ischemic stroke rats after catalpol treatment for 14 days, compared with that in MCAO rats. In addition, catalpol improved the neuron morphology, promoted axon growth, and increased DCX/EdU double positive cells. These results all indicated that catalpol exhibits robust NVU protection for ischemic stroke rats: catalpol alleviates the damages of the vascular and neural component of NVU induced by the insufficient nutrients supply, and maintains the vascular structure and neuronal morphology; moreover, catalpol promotes angiogenesis and neurogenesis, partly replenishes necrotic vessels and lost neurons, improves the neurological function deficits, and reduces the infarction volume.
Hypoxic environment induces the dynamic expression of VEGF. It is generally believed that VEGF expression is strongly induced in the acute phase 1–3 h, peaked at 24–48 h, and significantly decreased in recovery phase after ischemic stroke [42]. Consistent with that, the VEGF expression in the brain of ischemic stroke rats was decreased on day 14. With aspect to the detailed mechanisms of catalpol in the process of NVU protection, both the results from rats and in vitro 3D NVU indicated that catalpol protects NVU by promoting the production of VEGF, which is dependent on PI3K/AKT pathways. FAK and Paxillin play their important roles in cell migration and angiogenesis, and PI3K/AKT signaling is also critical for endothelial cell survival [24, 43, 44]. We provided evidences that VEGF was increased by catalpol, then the binding of VEGF to VEGFR2 activated the PI3K/AKT pathway and increased the production of FAK and Paxillin to maintain endothelial cells activity, accelerate endothelial cells migration, and promote angiogenesis. In addition, MEK1/2/ERK1/2 signals exhibit vital regulation effects in neuroprotection [22, 45]. Our data showed that the binding of VEGF to VEGFR2 also activated the MEK1/2/ERK1/2 pathway, followed by increasing the axonal length, maintaining neuronal activity, and promoting neurogenesis. In addition, various doses of catalpol were set in this study to find the optimal dose of catalpol. The results suggested that catalpol at 5 and 10 mg/kg in vivo and 50 and 100 μΜ in vitro exhibited better therapeutic effects. The results also indicated that the different doses we set were not the most suitable gradients as the dose-dependent effects were not observed in all assays. Indeed, we should make more precise gradient dosages and more catalpol groups to find the optimal dose of catalpol in the treatment of stroke rats in the following study. Also, we noticed that the same dose of catalpol exhibited different effects on different indicators. For example, catalpol (25 μM) had little effect on axon length but maintain total vascular length of OGD-exposed 3D NVU (Fig. 6). In this study, we found that PI3K/AKT and MEK1/2/ERK1/2 pathways were activated by VEGF to mediate vascular protection and neuroprotection, separately. Catalpol mediated the cross-talk of PI3K/AKT and MEK1/2/ERK1/2 pathways through targeting VEGF to protect NVU, nevertheless, catalpol also activated PI3K/AKT pathway directly in 3D NVU (Fig. 8). Therefore, catalpol may play stronger activation on PI3K/AKT pathway to protect vascular component of NVU than its effects on MEK1/2/ERK1/2 pathway, even at the same dose. On this account, it is explicable that catalpol (25 μM) plays a role in total vascular length promotion of OGD-exposed 3D NVU but not on the axonal length.
Conclusions
Here, we provided robust evidences that catalpol protects NVU from ischemic stroke by improving vascular and neuronal morphology, maintaining barrier function, and promoting angiogenesis and neurogenesis, then alleviates neurological deficits and reduces infarction volume of ischemic rats. We proposed that the primary and detailed mechanisms of NVU protection by catalpol is dependent on PI3K/AKT to promote VEGF production, which mediated NVU protection by increasing FAK and Paxillin expression levels, and activating VEGF-PI3K/AKT and VEGF-MEK1/2/ERK1/2 pathways, in a partly feedforward loop manner (Fig. 10). Our findings also provide rationales for the clinic application of catalpol as a promising agent for ischemic stroke.
Fig. 10. The diagram of catalpol enhances VEGF-PI3K/AKT and VEGF-MEK1/2/ERK1/2 signals to protect neurovascular unit from ischemic stroke.
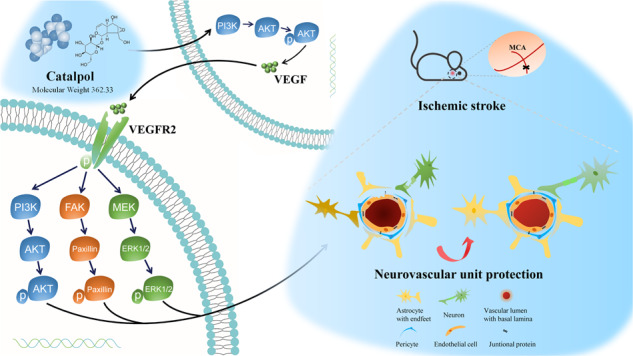
VEGF was up-regulated by catalpol mediated by PI3K/AKT to protect NVU, both in ischemic hemisphere of rats and in vitro NVU model, followed by the increase of FAK and Paxillin and the activation of PI3K/AKT and MEK1/2/ERK1/2 pathways.
Supplementary information
Acknowledgements
This work was supported by National Natural Science Foundation of China (81473549 to XYX and 81773984 to YC), the Fundamental Research Funds for the Central Universities (XDJK2020D037 to HJW), and the Chongqing Postgraduate Research Innovation Project (CYS18098 to HJW). In addition, we thank all sacrificial animals for this study, and other Xiao-yu Xu’ lab members for helpful discussions.
Author contributions
HJW designed and performed the experiments, and wrote and revised the paper. HFR and YY performed in vitro experiments. XGX, BXJ, SQY, YJC, and HJR performed in vivo experiment. SF and JFZ reviewed and revised the paper. YC, QX, and XYX designed the study and revised the paper. All authors contributed to analyze the results and approved the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Qiang Xue, Email: ygzxq3@sina.com.
Xiao-yu Xu, Email: xuxiaoyu@swu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00803-4.
References
- 1.Potjewyd G, Moxon S, Wang T, Domingos M, Hooper NM. Tissue engineering 3D neurovascular units: a biomaterials and bioprinting perspective. Trends Biotechnol. 2018;36:457–72. doi: 10.1016/j.tibtech.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96:17–42. doi: 10.1016/j.neuron.2017.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C, Li P. Neurovascular unit protection—novel therapeutic targets and strategies. CNS Neurosci Ther. 2021;27:5–6. doi: 10.1111/cns.13588. [DOI] [Google Scholar]
- 4.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 5.Muoio V, Persson PB, Sendeski MM. The neurovascular unit–concept review. Acta Physiol. 2014;210:790–8. doi: 10.1111/apha.12250. [DOI] [PubMed] [Google Scholar]
- 6.Birgit O, Richard D, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–96. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu YC, Chang YC, Lin YC, Sze CI, Huang CC, Ho CJ. Cerebral microvascular damage occurs early after hypoxia–ischemia via nNOS activation in the neonatal brain. J Cereb Blood Flow Metab. 2014;34:668–76. doi: 10.1038/jcbfm.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyle KP, Simon RP, Stenzelpoore MP. Neuropharmacology – special issue on cerebral ischemia mechanisms of ischemic brain damage – review article. Neuropharmacology. 2008;55:310–8. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267:156–71. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6:11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862:887–900. doi: 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Xiong X, Zhang L, Shen J. Neurovascular Unit: a critical role in ischemic stroke. CNS Neurosci Ther. 2021;27:7–16. doi: 10.1111/cns.13561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, Liu Q, Zhang R, Liu S, Xia Z, Hu Y. Catalpol ameliorates beta amyloid-induced degeneration of cholinergic neurons by elevating brain-derived neurotrophic factors. Neuroscience. 2009;163:1363–72. doi: 10.1016/j.neuroscience.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 14.Xia Z, Zhang R, Wu P, Xia Z, Hu Y. Memory defect induced by β-amyloid plus glutamate receptor agonist is alleviated by catalpol and donepezil through different mechanisms. Brain Res. 2012;1441:27–37. doi: 10.1016/j.brainres.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 15.Jiang B, Shen RF, Bi J, Tian XS, Hinchliffe T, Xia Y. Catalpol: a potential therapeutic for neurodegenerative diseases. Curr Med Chem. 2015;22:1278–91. doi: 10.2174/0929867322666150114151720. [DOI] [PubMed] [Google Scholar]
- 16.Zhang RX, Li MX, Jia ZP. Rehmannia glutinosa: review of botany, chemistry and pharmacology. J Ethnopharmacol. 2008;117:199–214. doi: 10.1016/j.jep.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Wan D, Wan G, Wang J, Zhang J, Zhu H. Catalpol induces cell activity to promote axonal regeneration via the PI3K/AKT/mTOR pathway in vivo and in vitro stroke model. Ann Transl Med. 2019;7:756–70. doi: 10.21037/atm.2019.11.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li DQ, Duan YL, Bao YM, Liu CP, Liu Y, An LJ. Neuroprotection of catalpol in transient global ischemia in gerbils. Neurosci Res. 2004;50:169–77. doi: 10.1016/j.neures.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Cai QY, Chen XS, Zhan XL, Yao ZX. Protective effects of catalpol on oligodendrocyte death and myelin breakdown in a rat model of chronic cerebral hypoperfusion. Neurosci Lett. 2011;497:22–6. doi: 10.1016/j.neulet.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Zhu HF, Wan D, Luo Y, Zhou JL, Chen L, Xu XY. Catalpol increases brain angiogenesis and up-regulates VEGF and EPO in the rat after permanent middle cerebral artery occlusion. Int J Biol Sci. 2010;6:443–53. doi: 10.7150/ijbs.6.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Xu X, Yin Y, Yu S, Ren H, Xue Q, et al. Catalpol protects vascular structure and promotes angiogenesis in cerebral ischemic rats by targeting HIF-1α/VEGF. Phytomedicine. 2020;78:153300. doi: 10.1016/j.phymed.2020.153300. [DOI] [PubMed] [Google Scholar]
- 22.Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26:943–54. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- 23.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 24.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 25.Yasuhara T, Shingo T, Kobayashi K, Takeuchi A, Yano A, Muraoka K, et al. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;19:1494–504. doi: 10.1111/j.1460-9568.2004.03254.x. [DOI] [PubMed] [Google Scholar]
- 26.Rosenstein JM, Krum JM. New roles for VEGF in nervous tissue–beyond blood vessels. Exp Neurol. 2004;187:246–53. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 27.Rosenstein JM, Krum JM, Ruhrberg C. VEGF in the nervous system. Organogenesis. 2010;6:107–14. doi: 10.4161/org.6.2.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumpich M, Theiss C. VEGF in the nervous system: an important target for research in neurodevelopmental and regenerative medicine. Neural Regen Res. 2015;10:1725–6. doi: 10.4103/1673-5374.170287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Svensson B, Peters M, König HG, Poppe M, Levkau B, Rothermundt M, et al. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J Cereb Blood Flow Metab. 2002;22:1170–5. doi: 10.1097/01.wcb.0000037988.07114.98. [DOI] [PubMed] [Google Scholar]
- 30.Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–7. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Yang H, Shi Y, Xiao Y, Yin Y, Jiang B, et al. Reconstituting neurovascular unit with primary neural stem cells and brain microvascular endothelial cells in three-dimensional matrix. Brain Pathol. 2021;31:e12940. doi: 10.1111/bpa.12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H, Hu Y, Zhang X, Na W, Peng X. Improved electrocoagulation method for establishing rat cerebral apoplexy model. Third Milit Med Univ. 2011;33:1798–802. [Google Scholar]
- 33.Llovera G, Roth S, Plesnila N, Veltkamp R, Liesz A. Modeling stroke in mice: permanent coagulation of the distal middle cerebral artery. J Vis Exp. 2014;31:e51729. doi: 10.3791/51729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J Neurotrauma. 1996;13:557–68. doi: 10.1089/neu.1996.13.557. [DOI] [PubMed] [Google Scholar]
- 35.Shen LH, Li Y, Chen J, Zhang J, Vanguri P, Borneman J, et al. Intracarotid transplantation of bone marrow stromal cells increases axon-myelin remodeling after stroke. Neuroscience. 2006;137:393–9. doi: 10.1016/j.neuroscience.2005.08.092. [DOI] [PubMed] [Google Scholar]
- 36.Su WS, Wu CH, Chen SF, Yang FY. Low-intensity pulsed ultrasound improves behavioral and histological outcomes after experimental traumatic brain injury. Sci Rep. 2017;7:15524. doi: 10.1038/s41598-017-15916-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–8. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods. 2019;16:1169–75. doi: 10.1038/s41592-019-0586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bond AM, Ming GL, Song H. Adult mammalian neural stem cells and neurogenesis: five decades later. Cell Stem Cell. 2015;17:385–95. doi: 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–6. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- 42.Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–44. doi: 10.1161/01.STR.28.10.2039. [DOI] [PubMed] [Google Scholar]
- 43.Abedi H, Zachary I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem. 1997;272:15442–51. doi: 10.1074/jbc.272.24.15442. [DOI] [PubMed] [Google Scholar]
- 44.Le Boeuf F, Houle F, Huot J. Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J Biol Chem. 2004;279:39175–85. doi: 10.1074/jbc.M405493200. [DOI] [PubMed] [Google Scholar]
- 45.Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, et al. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J. 2001;15:1218–20. doi: 10.1096/fj.00-0495fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.