
Figure 1: Overview of sample-dependent sequence diversity and NOMAD model and pipeline.
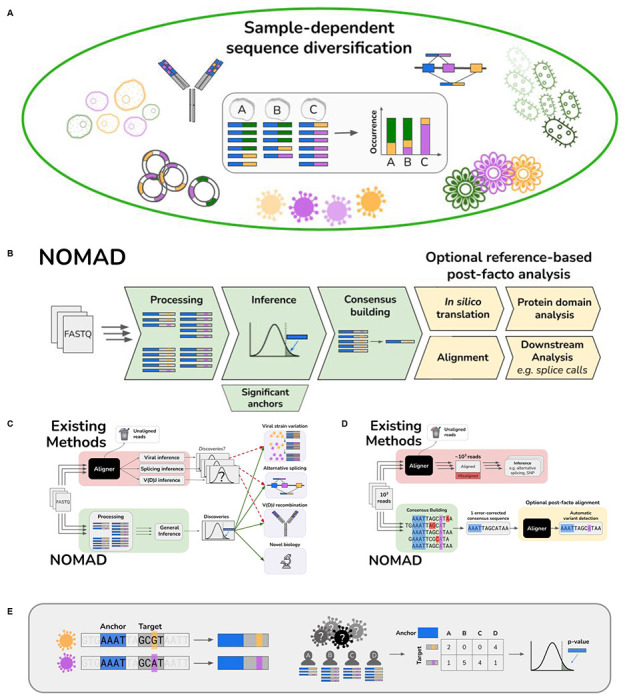
A. Biological generality of sample-dependent sequence diversification. The study of sample-dependent sequence diversification unifies problems in disparate areas of genomics which are currently studied with application-specific models and algorithms. Viral genome mutations, alternative splicing, and V(D)J recombination all fit under this framework, where sequence diversification depends on the sample (through cell-type or infection strain type). Myriad problems in plant genomics, metagenomics and biological adaptation are subsumed by this framework.
B. Overview of NOMAD pipeline. NOMAD takes as input raw FASTQ files for any number of samples >1 and processes them in parallel, counting (anchor, target) pairs per sample. NOMAD performs inference on these aggregated counts, outputting statistically significant anchors. For each significant anchor, a denoised per-sample consensus sequence is built (Fig. 1D). NOMAD also enables optional reference-based post-facto analysis. If a reference genome is available, NOMAD can align the consensus sequences to the reference, enabling denoised downstream analysis (e.g. SNPs, indels, or splice calls). In silico translation of consensuses can optionally be used to study relationships of anchors to protein domains by mapping to databases such as Pfam (Methods).
C. Overview of NOMAD versus existing workflows. Existing workflows (red) discard low-quality reads during FASTQ processing and alignment, only then performing statistical testing after algorithmic bias is introduced; p-values are then not unconditionally valid. Further, for every desired inferential task, a different inference pipeline must be used. NOMAD (green) performs direct statistical inference on raw FASTQ reads, bypassing alignment and enabling data-scientifically driven discovery. Due to its generality, NOMAD can simultaneously detect myriad biological examples of sample-dependent sequence diversification.
D. NOMAD consensus building. NOMAD constructs a per-sample consensus sequence for every significant anchor by taking all reads in which the anchor (blue) appears, and recording plurality votes for each nucleotide, denoising reads while preserving the true variant; sequencing errors in red and biological mutations in purple. Existing approaches require alignment of all reads to a reference prior to error correction, requiring orders of magnitude more computation, discarding reads in both processing and alignment, and potentially making erroneous alignments due to sequencing error. They further require inferential steps, e.g. to detect if there is a SNP or alternatively spliced variant.
E. Example construction of NOMAD anchor, target pairs. A stylized expository example of viral surveillance: 4 individuals A-D are infected with one of two variants (orange and purple), differing by a single basepair (orange and purple). NOMAD anchor k-mers are blue (k=4), followed by a lookahead distance of L=2, and the corresponding k-mer targets. Given sequencing reads from the 4 individuals as shown, NOMAD generates a target by sample contingency table for this blue anchor, and computes a p-value to test if this anchor has sample-dependent sequence diversity.