
Figure 2: NOMAD analysis of SARS-CoV-2 data.
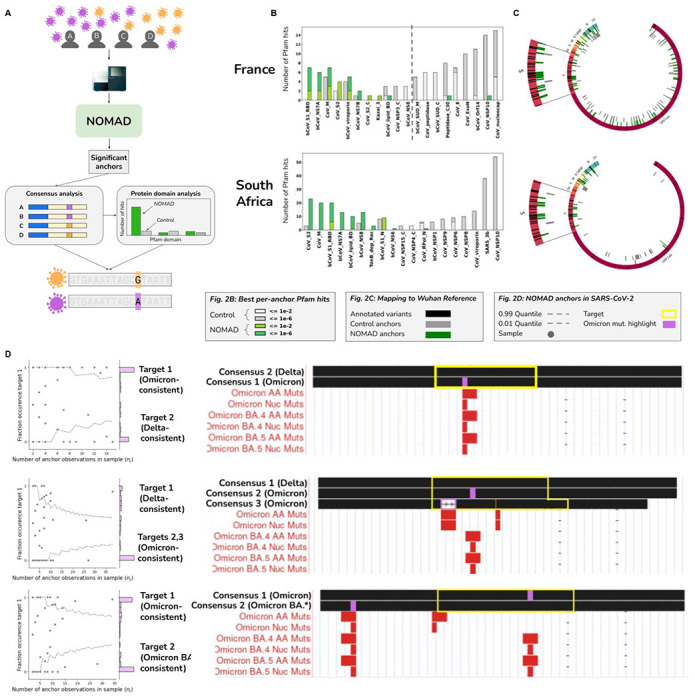
A. Stylized example representing NOMAD workflow for viral data. Patients with varying viral strains are sampled; two representative strains with differentiating mutations are depicted in orange and purple. NOMAD is run on raw FASTQs generated from sequencing patient samples. Significant anchors are called without a reference genome or clinical metadata. Optional post-facto analysis quantifies domain enrichment via in silico translation of consensus sequences derived from NOMAD-called anchors versus controls. Consensuses can also be used to call variants de novo and can be compared to annotated variants e.g. in SARS-CoV-2, Omicron.
B. NOMAD protein profile analysis of SARS-CoV-2. NOMAD SARS-CoV-2 protein profile hits (anchor effect size >.5) to the Pfam database (greens) and control (greys) for France and South Africa datasets; ordered by enrichment in NOMAD hits compared to control showing large distributional differences (chi-squared test p-values France: < 1.1E-12, SA: <2.5E-39). Spike protein domains are highly enriched in the NOMAD versus control. In the France data, the most NOMAD-enriched domain is the betacoronavirus S1 receptor binding domain (hypergeometric p=2.9E-4, corrected) followed by Orf7A (hypergeometric p=1.6E-3, corrected), known to directly interact with the host innate immune defense. In the South Africa data, the most enriched NOMAD profiles are CoV S2 (p=2.9E-6) and the coronavirus membrane protein (p=8.4E-8). Plots were truncated for clarity of presentation as indicated by dashed grey lines (Fig. S2A, B).
C. NOMAD anchors are enriched near annotated variants of concern. NOMAD anchors (effect size >.5) for SARS-CoV2 mapping to the Wuhan reference (NC_045512) show enrichment near variants of concern. SARS-CoV2 genome depicted with annotated ORFs and lines depicting positions of variants of concern (VOC) annotated as Omicron and Delta variants. No control anchor maps to spike or other areas of VOC density except in N (nucleocapsid).
D. NOMAD consensuses identify variants of concern de novo. Examples of NOMAD-detected anchors in SARS-CoV2 (France data). Scatterplots (left) show the fraction of each sample’s observed fraction of target 1 (the most abundant target) for three representative anchors, binomial confidence intervals: (.01,.99), p=empirical fraction occurrence of target 1 (Supplement). y-axis shows histogram of the fraction occurrence of target 1. Mutations (right)found in the targets are highlighted in purple, BLAT shows single nucleotide mutations match known Omicron mutations. Binomial p-values of 6.8E-8, 3.1E-7, and 6.7E-15 respectively (Methods). The anchor in (top) maps to the coronavirus membrane protein; anchors in (middle and bottom) map to the spike protein. One sample (out of 26) depicted in the bottom plot has a consensus mapping perfectly to the Wuhan reference; 3 other consensuses contain annotated Omicron mutations, some designated as VOC in May of 2022, 3 months after these samples were collected.