

Abstract
Modification of the ribose moiety of nucleotides and nucleosides has provided new insights into structural and conformational requirements for ligands at P2Y nucleotide receptors and at adenosine receptors (ARs). Methanocarba derivatives (containing a rigid bicyclic ring system in place of ribose) of adenosine, ATP, ADP, UTP, UDP, and other receptor agonist analogs were synthesized. Biological evaluation led to the conclusion that in general the Northern (N)-conformation was favored over the Southern (S)-conformation of the pseudoribose moiety at A1 and A3 ARs and at P2Y1, P2Y2, P2Y4, or P2Y11 receptors, but not P2Y6 receptors. At the hA3 AR a new full agonist, MRS1898, the (N)-methanocarba equivalent of CI-IB-MECA (2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyladenosine), had a Ki value of 1.9 nM in binding to the hA3 AR expressed in CHO cells. Functional assays confirmed the selectivity of MRS1898, although CI-IB-MECA was even more functionally selective for human A3 vs. hA1 and hA2A ARs. Thirty μM MRS1898 did not induce apoptosis in HL-60 cells, suggesting that some of the proapoptotic effects of CI-IB-MECA may be nonreceptor-mediated. Manipulation of the sequence of A3 ARs through site-directed mutagenesis has led to pharmacologically unique constructs: constitutively active receptors and “neoceptors.” Such engineered receptors may later prove to have potential for cardioprotection through gene transfer. Effects of single amino acid replacement were interpreted using a rhodopsin-based model of ligand-A3 receptor interactions, leading to the proposal that a movement of the conserved W243 in TM6 may be involved in AR activation.
Keywords: G-protein-coupled receptors, nucleosides, nucleotides, structure-activity relationships, purines, ARs
INTRODUCTION
We have adopted an integrated approach to the study of drug-receptor interactions in adenosine receptors (ARs) and P2 nucleotide receptors [Fredholm et al., 2001; Jacobson et al., 2002]. The receptor structure may be studied indirectly through exploring ligand structure-activity relationships (SAR) and directly by using mutagenesis and molecular modeling, based on the structure of rhodopsin. Goals of this structural approach are to generate a testable hypothesis for location of the binding site and subsequently to enable the rational design of new agonists and antagonists. Specifically, this review will describe recent advances both in the design of ligands, in which specific conformations are locked, and the mutation of the receptors, which have resulted in unique constructs that display unique pharmacological properties, such as constitutive activity. Finally, this review will present the concept of neoceptors, which are engineered for activation only by structurally tailored small molecules (“neoligands”) that are selective for the neoceptor vs. the native receptor.
LIGAND DESIGN
Modification of the ribose moiety of nucleotides and nucleosides has provided new insights into structural and conformational requirements for ligands at P2Y nucleotide receptors and ARs.
P2Y Receptor Ligands
In collaboration with the laboratory of T.K. Harden of the University of North Carolina, we studied the medicinal chemistry of nucleotides as agonists, partial agonists, and antagonists at P2Y receptors [Nandanan et al., 1999, 2000; Kim et al., 2001]. Molecular modeling of the docking of adenine nucleotides in the P2Y1 receptor [Moro et al., 1998] has helped us predict that there is no specific interaction of the ribose ring oxygen with the receptor. Consequently, we replaced the ribose of adenine nucleotides with a variety of ribose substitutes lacking this oxygen, i.e., carbocyclics, smaller and larger rings, conformationally constrained rings, and acyclics. Biological evaluation led to the conclusion that affinity at the P2Y1 receptor could be preserved or even enhanced by such substitution.
The ribose ring of pentose derivatives, such as adenosine and ATP, can adopt a range of conformations as described by a “pseudorotational cycle” [Marquez et al., 1996]. One approach to “freezing” the ribose-like moiety of nucleoside/nucleotide receptor ligands in what may be a receptor-preferred conformation is to use a carbocyclic modification in which the cyclopentane ring is sterically constrained through the inclusion of a second, fused ring (Fig. 1) [Marquez et al., 1996]. The “methanocarba” ring system, in which a fused cyclopropane moiety at either of two positions, has two conformational variations, depending on the position of fusion: Northern (N) or Southern (S) envelope conformations.
Fig. 1.
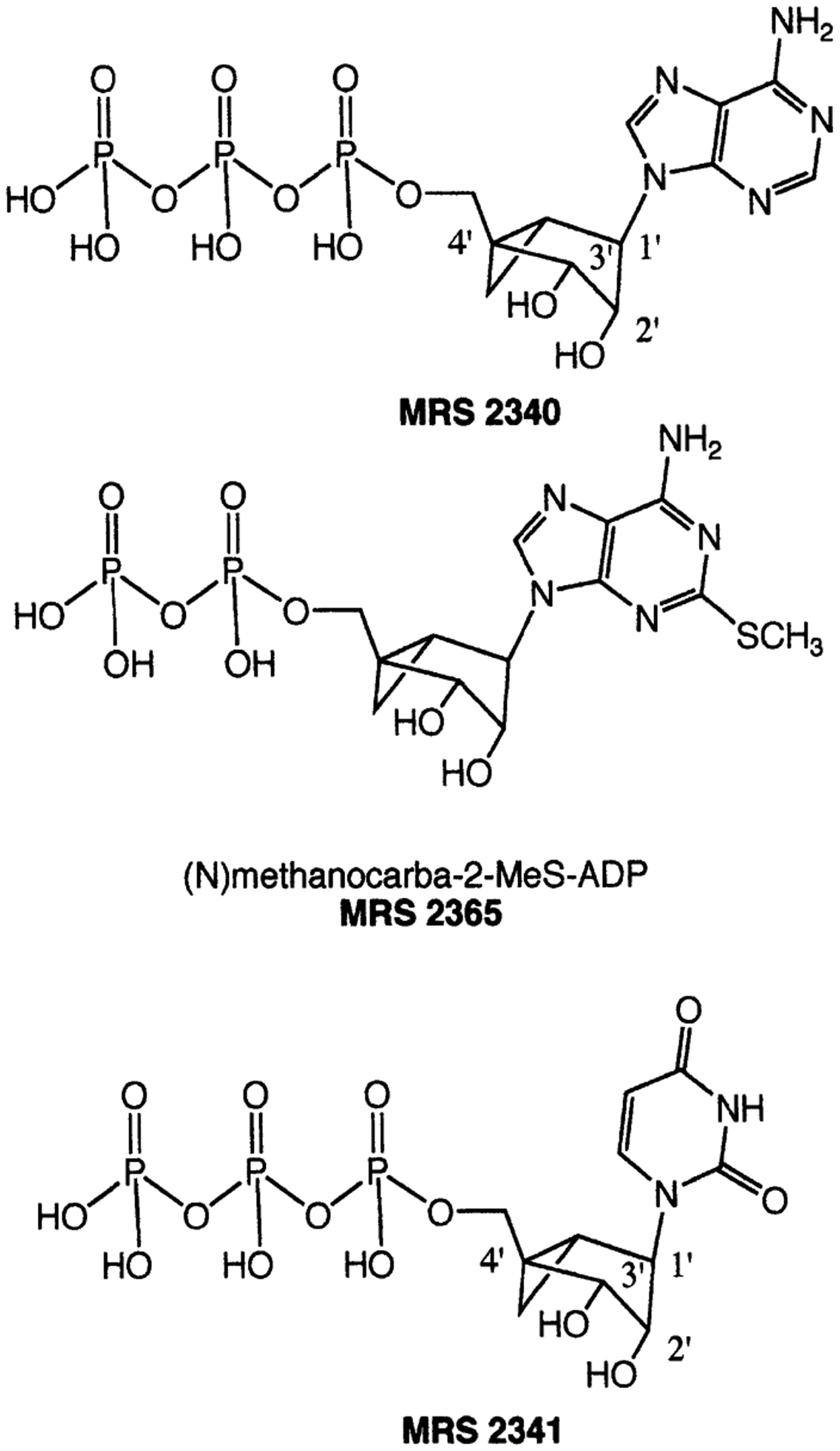
Structures of selected (N)-methanocarba analogs of nucleotides described in this review.
Methanocarba derivatives of ATP, UTP, UDP, and other nucleotide analogs (Fig. 1) were synthesized [Kim et al., 2002a; Ravi et al., 2002] by methods requiring some adaptation of standard procedures for nucleotide synthesis [Burgess and Cook, 2000]. The routine phosphorylation method using phosphorous oxychloride followed by a diphosphate (the “one pot” synthesis of nucleoside 5′-triphosphates) could not be used in the (N)-methanocarba series due to steric hindrance. When attempted, that method produced principally 3′,5′-cyclic monophosphate analogs [Kim et al., 2002a]. Instead, reaction of the 5′-hydroxyl group with a phosphoramidite reagent, followed by oxidation and deprotection, provided the 5′-monophosphates, which then were treated with 1,1′-carbonyldiimidazole for condensation with additional phosphate groups.
Professor H. Zimmermann and co-workers at the University of Frankfurt [Ravi et al., 2002] studied the stability of the methanocarba analogs of ATP and AMP toward the action of ectonucleotidases and concluded that in general cleavage occurs less readily with methanocarba nucleotides than with the native nucleotides. (N)- and (S)-methanocarba-ATP analogs were exposed to recombinant NTPDases 1 and 2 and methanocarba-AMP derivatives to recombinant 5′-nucleotidases. Phosphate cleavage by these ectonucleotidases occurring progressively closer to the 5′-position was increasingly impeded by the methanocarba modification (Table 1). The monophosphates were poor substrates for 5′-nucleotidase, suggesting that the activity of these compounds in pharmacological assays is less likely to result in receptor activation by their metabolites as compared to the unmodified, native nucleotides.
TABLE 1.
Rates of Hydrolysis of Nucleotide Analogs by Recombinant Ecto-Nucleotidases [Ravi et al., 2002]
| Enzyme | NTPDase1 | NTPDase2 | 5′-NT | |
|---|---|---|---|---|
| ATP | 100% | 100% | AMP | 100% |
| (N)-mcATP | 52% | 4% | (N)-mcAMP | 0.14% |
| (S)-mcATP | 57% | 34% | (S)-mcAMP | 0% |
| 100%= | 42.1 | 13.5 | 5.6 | |
| nmol/min/106cells | nmol/min/μg prot |
Harden and co-workers [Kim et al., 2002a] carried out functional assays of phospholipase C activated by the methanocarba nucleotide analogs through various P2Y receptors. There was a preference for the (N)- vs. the (S)-conformation of nucleotides acting at P2Y1, P2Y2, P2Y4, and P2Y11 receptors (Fig. 2), but not at P2Y6 receptors. At recombinant human P2Y1 and P2Y2 receptors, (N)-methanocarba-ATP, MRS2340 (Figs. 1, 2A) was 138- and 41-fold, respectively, more potent than racemic (S)-methanocarba-ATP as an agonist (note that the enantiomerically pure (S)-methanocarba-ATP was not prepared for comparison). MRS2340 activated P2Y11 receptors with a potency similar to ATP. A (N)-methanocarba-2-methylthio-ADP analog, MRS2365 (Fig. 1), displayed an EC50 at the hP2Y1 receptor of 0.40 nM and was 55-fold more potent than the corresponding triphosphate and 16-fold more potent than the riboside 5′-diphosphate, i.e., 2-MeSADP. (N)-Methanocarba-UTP, MRS2341 (Figs. 1, 2B), was equipotent to UTP as an agonist at human P2Y2 receptors and also activated P2Y4 receptors with an EC50 of 85 nM. (N)-Methanocarba-UDP was inactive at hP2Y6 receptors.
Fig. 2.
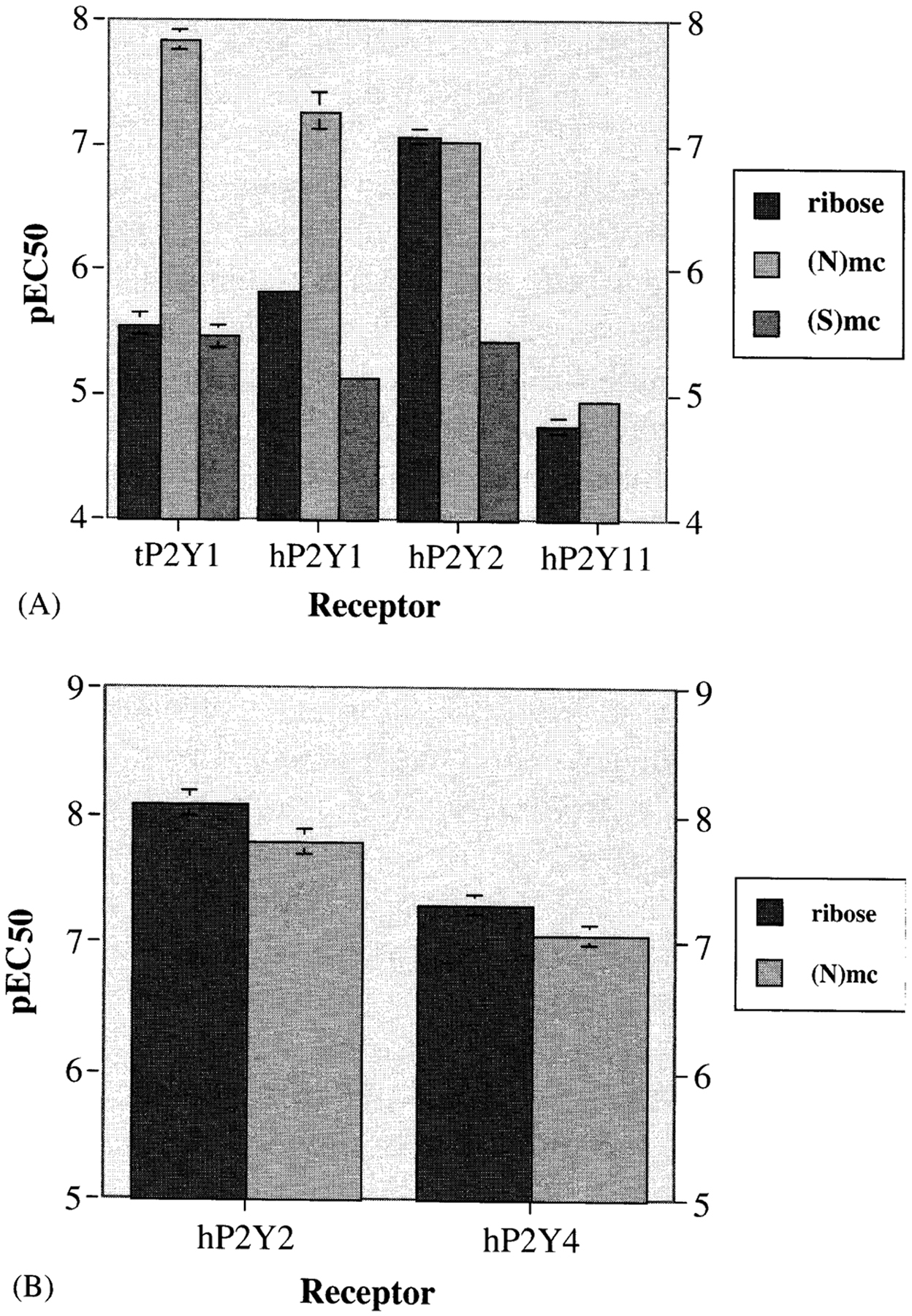
Effects of ribose substitution of nucleoside 5′-triphosphates with (N) or (S)-methanocarba ring systems. Potency at turkey P2Y1 and recombinant human P2Y receptors was determined [Kim et al., 2002a]. A: ATP and its analogs. B: UTP and its analog.
The vascular effects of (N)-methanocarba-UTP and (N)-methanocarba-UDP were studied in a model of the rat mesenteric artery by David Erlinge and co-workers (Univ. of Lund, Sweden) [Kim et al., 2002a]. MRS2341 was more potent than UTP in inducing a dilatory P2Y4 response (pEC50 = 6.1 ± 0.2). Consistent with its inactivity at recombinant P2Y6 receptors, the corresponding diphosphate was inactive as either an agonist or an antagonist in a P2Y6 receptor-mediated contractile response.
We have extensively explored the SAR of nucleotide bisphosphate analogs as P2Y1 receptor antagonists [Nandanan et al., 2000], based on the initial discovery of receptor antagonism by adenosine-3′-phosphate-5′-phosphate (A3P5P) [Boyer et al., 1996]. The ability to constrain such analogs in a receptor-preferring conformation using the (N)-methanocarba ring system has aided the development of such antagonists as well as P2 receptor agonists. The (N)-methanocarba analog MRS2279 ((1R,2S,4S,5S)-1-[(phosphato)methyl]-4-(2-chloro-6-aminopurin-9-yl) bicyclo [3.1.0]-hexane-2-phosphate) proved to be a selective P2Y1 receptor antagonist, with a KB of 12 nM [Boyer et al., 2002]. The pharmacological profile of MRS2279 was characterized at recombinant P2Y receptors and in human platelets, in which it impedes the proaggregatory effects of ADP. As described by Harden and co-workers [Waldo et al., 2003], [3H]MRS2279 was synthesized and shown to be the first generally applicable radioligand for the P2Y1 receptor.
In the antiviral field, acyclic analogs of nucleotides have proven exceptionally useful as inhibitors. We applied the acyclic approach to P2Y receptors. A series of acyclic nucleotide derivatives (all bisphosphates) were moderately potent P2Y1 antagonists, demonstrating that the ribose moiety, or a cyclic substitute, was not required for antagonism of this receptor [Kim et al., 2001]. In fact, at least with respect to antagonists, the ribose appears to be nothing more than a spacer group, maintaining the preferred geometry of phosphate groups with respect to the adenine moiety. We evaluated the SAR of this series of acyclic antagonists in chain length and the terminal (phosphate or phosphate substitutes) groups. The optimal chain length occurred with MRS2298, which contains a single methylene group adjacent to the adenine moiety (Fig. 3A). MRS2298 displayed high selectivity for the P2Y1 receptor [Brown et al., 2000]. The substitution of one of the phosphate groups with an uncharged ester group greatly reduced potency at the P2Y1 receptor, indicating that both phosphates are required (Fig. 3B). An attempt to reduce the conformational degrees of freedom in the acyclic series by introducing a cyclopropyl ring in the alkyl chain failed to enhance potency [Kim et al., 2001].
Fig. 3.
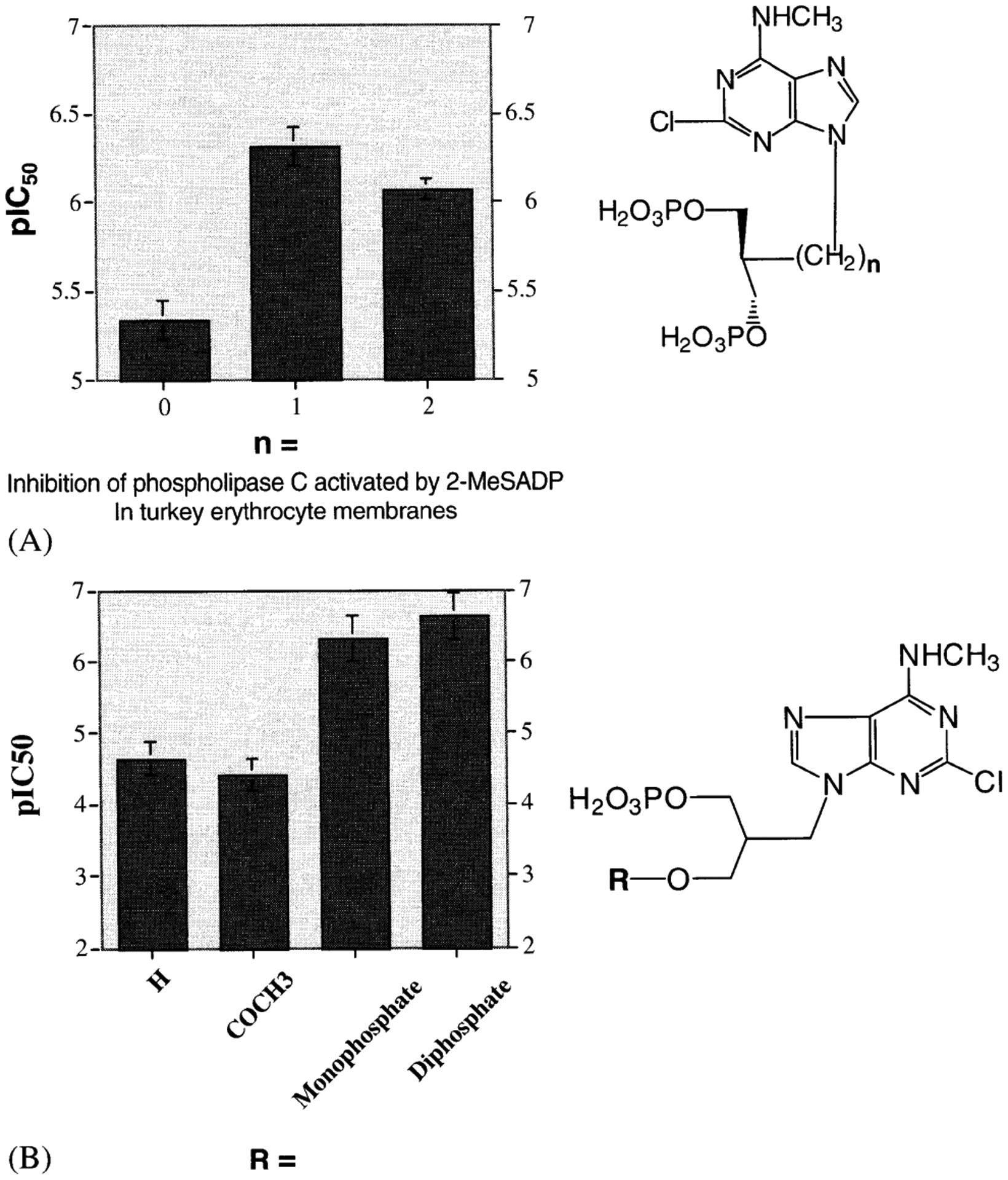
Effects of (A) alkyl chain spacer length substitution and (B) terminal groups substitution on the antagonistic potency of bisphosphate P2Y1 receptor antagonists. The optimal chain length antagonistic potency was found to be n = 1 (MRS 2298). plC50 refers to inhibition of phospholipase C activated by 30 nM 2-MeSADP in turkey erythrocyte membranes [Kim et al., 2001].
AR Ligands
The analysis of ribose or ribose-like ring conformation also proved useful for studying preferred conformations at the ARs. As with certain P2Y receptors, ARs demonstrated a preference for the (N) conformation at A1 and A3 ARs. As demonstrated in binding assays, the affinity preserving or enhancing effects of the (N)-methocarba ring system were especially evident at the A3 AR. (N)- and (S)-Methanocarba derivatives of adenosine and adenosine analogs substituted at the N6-, 2-, and/or 5′-positions were synthesized [Jacobson et al., 2000a; Lee et al., 2001].
Specific partial agonists selective at the A3 AR receptor have been synthesized [Jacobson et al., 2000a] and characterized in CHO (Chinese hamster ovary) cells transfected with the hA3 AR [Gao et al., 2002a]. A 2-H derivative (N)-Methanocarba N6-(3-iodobenzyl) adenosine, MRS1743 (Fig. 1) and its 2-chloro derivative, MRS1760, displayed Ki values of 9.2 and 1.9 nM, respectively, in binding to the hA3 ARs and were highly selective partial agonists with high functional potency (EC50 < 1 nM) but low efficacy (Table 2). These two partial agonists displayed cardioprotective properties (see below).
TABLE 2.
Affinity and Efficacy of Adenosine Derivatives in CHO Cells Expressing hA3AR
| Derivative | Ki (nM)a | EC50(nM)a | Efficacymaxc |
|---|---|---|---|
| Riboside | |||
| CI-IB-MECA | 2.8 | 1.9 | 100% |
| (N)-Methanocarba | |||
| MRS 1743 | 9.2 | 0.70b | 13%b |
| MRS 1760 | 1.9 | 0.67b | 3%b |
| MRS 1898 | 1.9 | 2.1 | 100% |
Ki values refer to inhibition of the binding of [125I]I-AB-MECA and EC50 refers to inhibition of cyclic AMP production, unless noted.
In a functional assay based on guanine nucleotide binding [Jacobson et al., 2000], the efficacy of MRS 1743 and MRS 1760 was determined to be 45% and 22%, respectively.
cAMP effects.
MRS1898, the (N)-methanocarba equivalent of Cl-IB-MECA (2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyladenosine), had a Ki value of 1.9 nM in binding to the hA3 AR expressed in CHO cells and was a full agonist [Lee et al., 2001; Gao et al., 2002a]. Binding assays confirmed the selectivity of MRS1898 (Fig. 4), although Cl-IB-MECA was even more selective (Table 2) for the hA3 AR vs. hA1 and hA2A ARs.
Fig. 4.
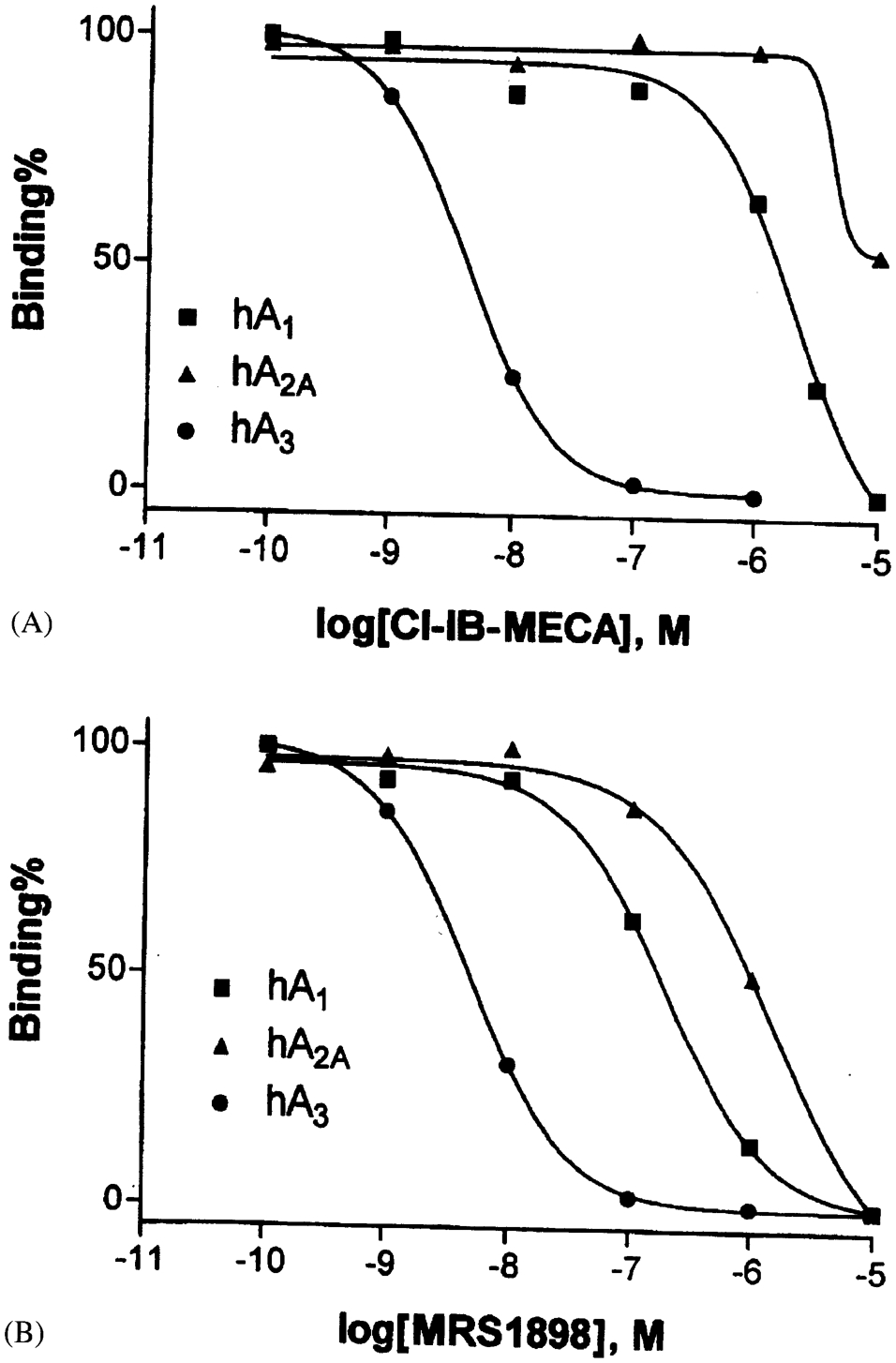
Binding displacement curves for Cl-IB-MECA (A) and MRS1898 (B) at human ARs expressed in CHO cells. A1 (n), A2A(s), and A3 (I). K1 values (Cl-IB-MECA; MRS 1989) at the following human ARs are (nM): A1 (1,240 ± 320; 136 ± 22), A2A(5,360 ± 2,470; 784 ± 97), and A3 (1.4 ± 0.3; 1.9 ± 0.4).
Effects of A3 AR Agonists on Apoptosis
The prototypical selective A3 AR agonists, IB-MECA and Cl-IB-MECA, at high (≥20 μM) concentrations have been noted to induce apoptosis in various cell lines [Kim et al., 2002b; Gessi et al., 2002]. The apoptotic effect of IB-MECA is distinct from the noted anticancer (cytostatic) properties of the compound [Fishman et al., 2002], which occur at lower concentrations. The synthesis of this novel, selective A3 agonist MRS1898 allowed the further examination of mechanism of this cell death phenomenon [Kim et al., 2002b]. First, the effects of Cl-IB-MECA on apoptosis in human leukemia cell lines, HL-60 and MOLT-4, were investigated. Cl-IB-MECA (30 μM) increased the apoptotic fractions as determined using FACS analysis and activated caspase 3 and poly-ADP-ribose-polymerase (PARP). Known messengers coupled to A3 AR (phospholipase C and intracellular calcium) did not seem to play a role in the induction of apoptosis. Selective A3 AR antagonists did not prevent the induction of apoptosis during a 48-h exposure of the cells to Cl-IB-MECA. Furthermore, micromolar concentrations of the A3 AR antagonist MRS1220, which would also antagonize A1 and A2A receptors, were also ineffective in preventing apoptosis. Although Cl-IB-MECA has been shown in other systems [Shneyvais et al., 2000; Gessi et al., 2002] to cause apoptosis through an A3 AR-mediated mechanism, in these cells it appeared to be an AR-independent effect that required prolonged incubation. Unlike its ribose analog, 30 μM MRS1898 did not induce apoptosis in HL-60 cells (Fig. 5), supporting the conclusion that at least under certain conditions the proapoptotic effects of Cl-IB-MECA may be nonreceptor-mediated.
Fig. 5.
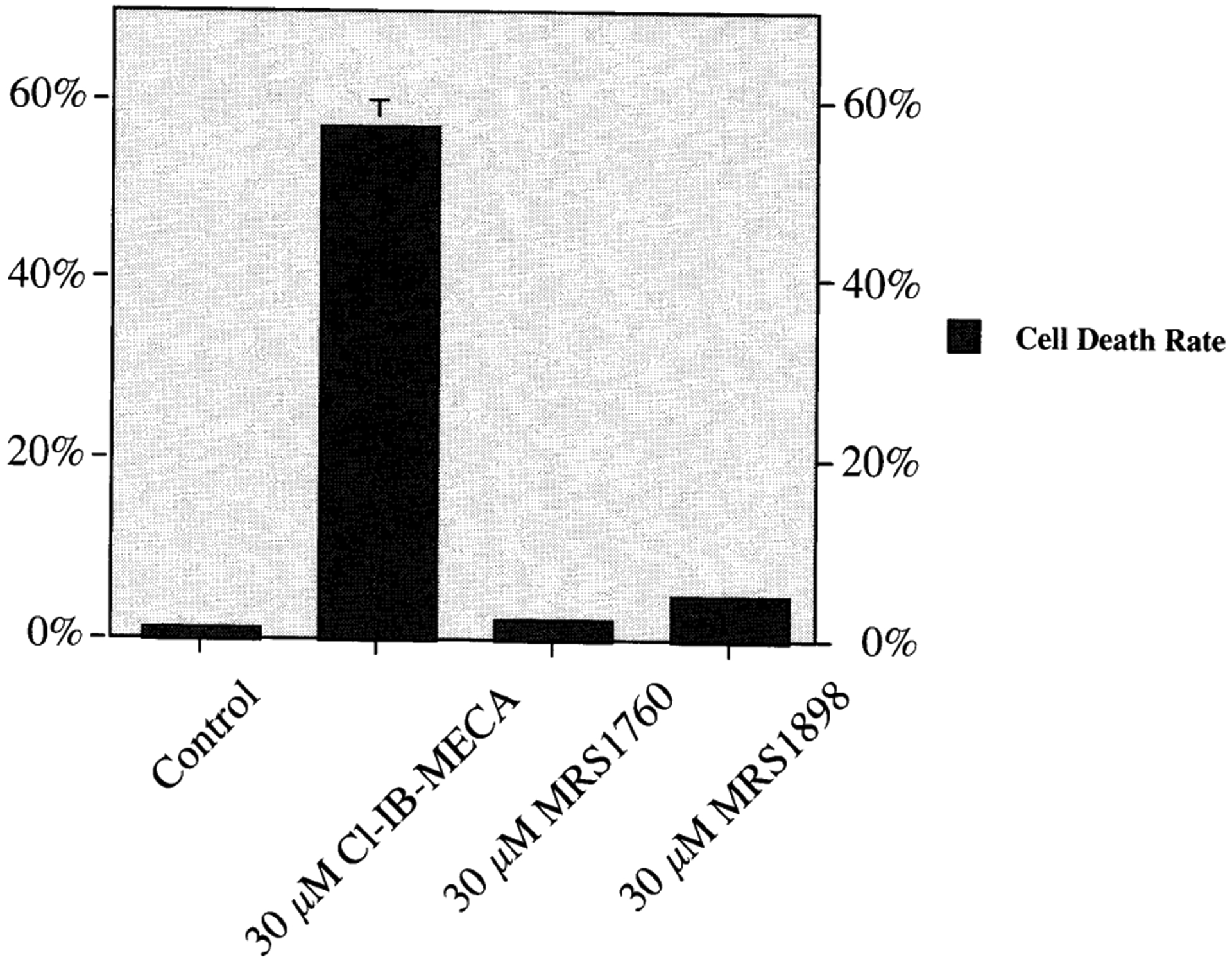
Induction of apoptosis in HL-60 (human leukemia) cells upon 48-h exposure to nucleoside analogs selective for the A3 AR [Kim et al., 2002b]. These included the full A3 AR agonist Cl-IB-MECA, low efficacy A3 AR partial agonist MRS1760, and full A3 AR agonist MRS1898 (each 30 μM). Evidence indicates that in this system, CL-IB-MECA at this high concentration is not acting through an AR-mediated mechanism.
Nevertheless, the induction of apoptosis by Cl-IB-MECA in leukemic cells displayed unique mechanistic features, e.g., dependence on the expression of Fas, a death receptor, which appeared not to depend on p53 [Kim et al., 2002b]. Cl-IB-MECA-induced apoptosis in HL-60 cells was augmented by the immunological agonism of Fas and was suppressed by antagonism of Fas. Therefore, 30 μM Cl-IB-MECA induced apoptosis in HL-60 and MOLT-4 cells via a novel, p53-independent up regulation of Fas.
SAR of Efficacy of Nucleoside Derivatives at A3 ARs
In the typical pharmacological characterization of a series of receptor agonists, the binding affinity or potency are the criteria which give rise to a pattern of SAR [Müller, 2000]. The effects of structural modification of adenosine derivatives on the efficacy SAR at the A3 AR have been studied recently in a systematic fashion by our and other laboratories [van Tilburg et al., 2002; Gao and Jacobsen, 2002; Gao et al., 2002a]. In general, the efficacy of adenosine derivatives at the A3 AR appears to be readily reduced by certain adenine or ribose modifications, such as 2-chloro or N6-iodobenzyl substituents. Therefore, agonist derivatives that are used widely as pharmacological tools at other AR subtypes, such as A1 ARs (see below), must not be assumed automatically to be agonists (albeit weaker) at the other subtypes.
The functional effects of the potent adenosine A1 receptor agonists N6-cyclopentyladenosine (CPA) and 2-chloro-N6-cyclopentyladenosine (CCPA) were compared [Gao and Jacobsen, 2002]. In CHO cells expressing the hA3 AR, CPA, but not CCPA, induced phosphoinositide turnover, indicating greatly reduced efficacy of CCPA at this subtype. CPA inhibited forskolin-stimulated cyclic AMP production (EC50 value of 242 ± 47 nM). CCPA competitively antagonized the effects of the agonist Cl-IB-MECA with a KB value of 5.0 nM. CPA competition curves vs. the selective A3 AR antagonist radioligand [3H]PSB-11(8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2.1-i]purin-5-one) [Müller et al., 2002] were right-shifted 4-fold by 100 μM GTP, whereas the competition by CCPA or the A3 AR antagonist MRS1220 (N-[9-chloro-2-(2-furanyl) [1,2,4]triazolo[1,5-c] quinazolin-5-yl]benzene-acetamide) was not affected by GTP. Thus, CCPA proved to be a moderately potent antagonist (Ki = 38 nM) of the hA3 AR.
The 2-chloro substitution of adenosine derivatives, in combination with certain N6-substitution (such as 3-iodobenzyl), caused a selective decrease in efficacy at both human and rat A3 ARs [Gao et al., 2002a]. In addition to the 2-chloro group, other structural factors influencing the degree of efficacy at the A3 AR were flexibility of the ribose ring and the presence of a uronamide group (such as in NECA and IB-MECA) at the 5′-position. Rigidity of the ribose-like moiety (such as in MRS1743) reduced efficacy; however, the addition of a 5′-uronamide group restored full efficacy in the cases examined. The efficacy-enhancement by the uronamide dominated all other modifications that were found to reduce efficacy.
A3 RECEPTOR STRUCTURAL CHARACTERIZATION AND MANIPULATION
Molecular Modeling of ARs
We are exploring the steric and electronic requirements of the A3 AR binding site using molecular modeling [Gao et al., 2002a,b]. The docking of several ligands into the putative binding site of the A3 AR model was accomplished.
Mutagenesis of ARs
To provide new insights into ligand–receptor interactions, site-directed mutagenesis of the A3 AR was carried out [Gao et al., 2002b]. While mutagenesis of A1 and A2a ARs has been performed [Tucker et al., 1994; IJzerman et al., 1996; Rivkees et al., 1999], only recently were single amino acids replaced in the A3 AR. A number of amino acid residues in the transmembrane (TM) domains III, VI, and the second extracellular loop were individually replaced with Ala and other amino acids. These residues are homologous to those that have been predicted in previous molecular modeling studies of ARs [IJzerman et al., 1996, and references therein] to be involved in the ligand recognition, including: His95, Trp243, Ser247, Asn250, and Lys152. The N250A mutant receptor lost the ability to bind both radiolabeled agonist ([125I]I-AB-MECA) and antagonist ([3H]PSB-11). The H95A (3.37) mutation significantly reduced affinity of both agonists and antagonists. In contrast, the K152A (EL2), W243A (6.48), and W243F (6.48) mutations did not significantly affect the agonist binding but decreased antagonist affinity approximately 3–38-fold, suggesting that these residues were required for the high affinity of A3 AR antagonists. Activation of phospholipase C by wild-type (WT) and mutant receptors was measured. The A3 agonist Cl-IB-MECA stimulated phosphoinositide turnover in the WT, but at concentrations as high as 100 μM failed to evoke a response in cells expressing W243A and W243F mutant receptors. Curiously, the binding affinity of Cl-IB-MECA (Ki = 2–3 nM) was unchanged in this functionally impaired W243 mutant. Thus, the binding modes of nucleoside agonists and nonnucleoside antagonists at the hA3 AR are different. Residues needed for antagonist but not agonist binding include Trp243 and Lys152. The residues proposed to be in proximity to the bound selective agonist Cl-IB-MECA and to the bound nonselective antagonist CGS15943 are shown (Fig. 6). Residues that are needed for both antagonist and agonist binding include His95 (3.37), His272 (7.43), and Asn250 (6.55). Furthermore, it was possible to separate the structural bases for binding and activation processes. Trp243 was required for activation of the receptor but not for binding of agonists.
Fig. 6.
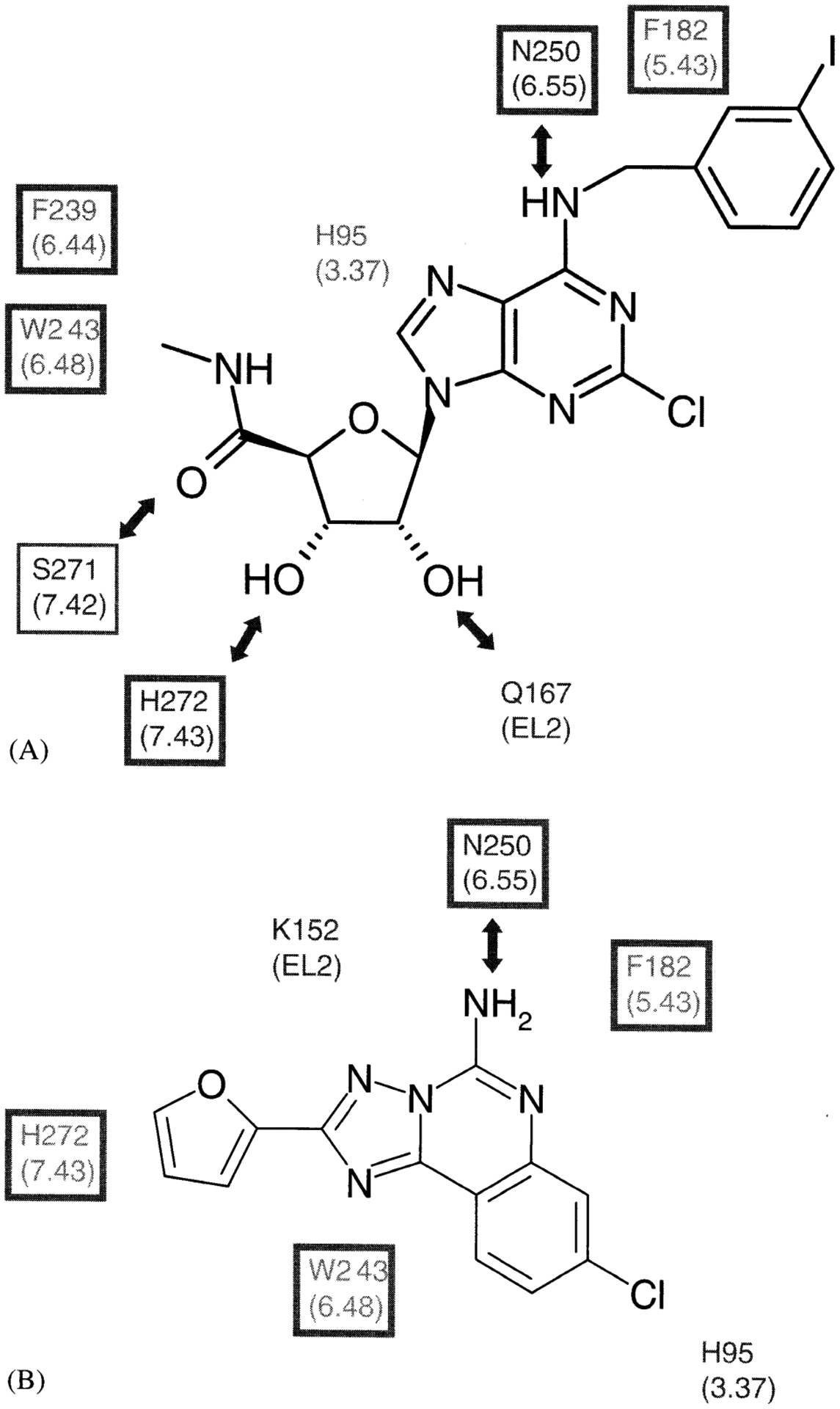
Summary of the results of human A3 AR docking of a selective A3 AR agonist, CI-IB-MECA (A), a nonselective, nonpurine AR antagonist, CGS15943 (B). Residues surrounding the putative binding site are shown [Gao et al., 2002a]. The residues in the double-squared box indicate identity among four subtypes of ARs and those in the single-squared box are conserved amino acids similarity among ARs. lntermolecular H-bonds are shown with arrows.
Hypothesis for Conformational Changes Involved in Activation of ARs
The results were interpreted using a rhodopsin-based model of ligand-A3 AR interactions. We have proposed that a movement of the conserved W243 may be involved in A3 AR activation [Gao et al., 2002a,b]. In the docked A3 AR complexes, a major difference between agonist and antagonist binding was that agonist binding required a rotation of the indole sidechain of W6.48, which was important for constraining the intramolecular TM network through both hydrophobic and H-bonding interactions. This rearrangement upon agonist binding suggested that the destabilization of the ground-state structure through the disruption of the intramolecular TM network facilitated the conformational change from inactive to active form. The conserved P6.50 above W6.48 may act as a flexible hinge for the straightening of TM6 and subsequently TM7 to rearrange IL3 and helix 8 important for signaling to the G-protein, as proposed for the activation of rhodopsin [Ballesteros et al., 2001].
Cardioprotective Effects of AR Activation Suggest Gene Therapy Using Engineered A3 Receptors
Full activation of A3 ARs or co-activation of A1 and A3 ARs can induce potent anti-ischemic effects. Adenosine is released in large amounts during myocardial ischemia and is capable of exerting potent protective effects in heart muscle cells [Strickier et al., 1996]. Thus, a synthetic AR agonist, selective for either the A1 or A3 subtype, might be beneficial to the survival of the ischemic heart [Liang and Jacobson, 1998]. The activation of both receptors exerted a cardioprotective effect greater than the activation of either receptor alone.
We have characterized the cardioprotective effects of novel ligands, including: binary drugs and amino acid conjugates that bind to both A1 and A3 ARs (and less potently to A2A ARs), and selective, partial A3 agonists. The binary agonist MRS1741, consisting of covalently fused moieties derived from the A3 agonist IB-MECA and the A1 agonist ADAC, coactivated A1 and A3 ARs, and full cardioprotection (EC50 ~0.1 nM) was dependent on expression of both subtypes [Jacobson et al., 2000b].
Partial A3 agonists bearing the (N)-methanocarba ring system, e.g., MRS1760 (Fig. 1) and its 2-H analog, MRS1743, were further characterized at the level of native A3 ARs in isolated chick cardiac myocytes. Both agonists were able to induce a marked protective effect against ischemia. However, the maximal protective effect of MRS1760 was less than the maximal effect caused by Cl-IB-MECA (P < 0.05, t-test). Consistent with the property of partial agonism, both MRS1760 and MRS1743 behaved as antagonists in the presence of Cl-IB-MECA by inhibiting the protection induced by a full agonist (Cl-IB-MECA). The underlying signaling mechanism for this protection was via phospholipase D, identical to that of the full A3 agonist Cl-IB-MECA. The cardioprotective effect of MRS1760 was also demonstrated in isolated mouse hearts.
Manipulation of the sequence of A3 ARs through site-directed mutagenesis has led to pharmacologically unique constructs that may later prove to have potential for cardioprotection through gene transfer.
Constitutively active mutant human A3 ARs were created using single amino acid replacements, based on findings from other G-protein-coupled receptors [Chen et al., 2001]. Three mutant receptors, A229E in TM6 and R108A and R108K in the DRY motif of TM3, were found to be constitutively active in functional assays of phospholipase C and adenylyl cyclase following expression in COS-7 cells (Fig. 7A).
Fig. 7.
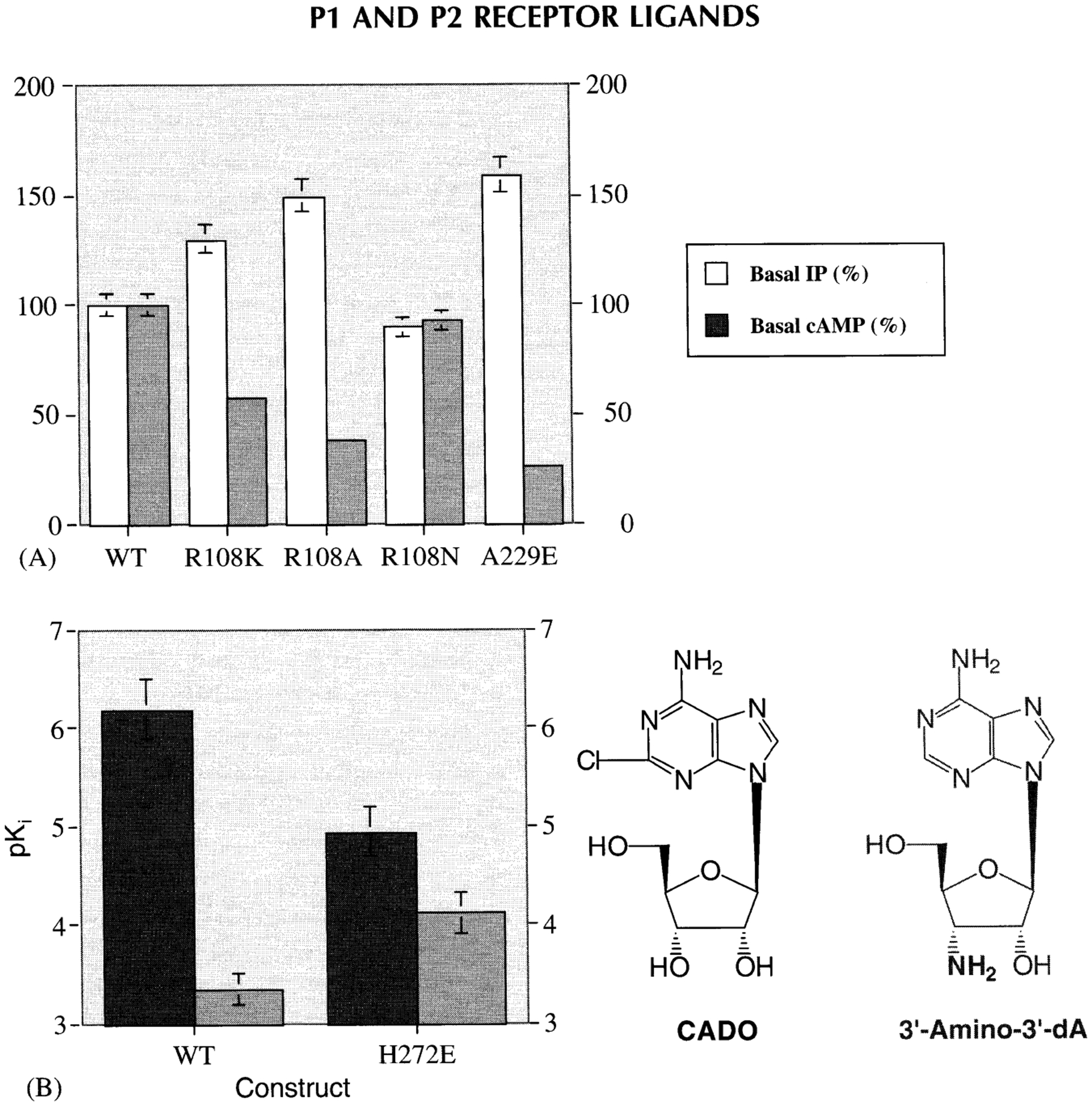
Functional effects of constitutively active mutant human A3 ARs [Chen et al 2001] (A) and enhancement of binding affinity of a human neoceptor derived from the A3 AR, upon of a complementary functional group in the neoligand [Jacobson et al., 2001] (B).
A general concept of engineered “neoceptors” has been demonstrated for the hA3 AR [Jacobson et al., 2001]. Such mutant receptors are designed to be activated only by “neoligands”, e.g., structurally modified adenosine derivatives of reduced potency at wild-type ARs. Molecular modeling suggested His272 in TM7 as a site for A3 AR mutation, leading to selective activation of a receptor mutant (H272E) by adenosine derivatives that were modified in a chemically complementary fashion (Fig. 7B).
CONCLUSIONS
The design of novel ligands for purine and pyrimidine receptors, in some cases based on a rational, structural approach, has resulted in selective receptor probes that promise to be useful in pharmacological studies. These probes display enhanced potency and/or selective, and in some cases stability, over the previously used probes. The ribose ring has been a recent focus of this medicinal chemical effort in terms of effects of modification on both potency and efficacy of the nucleoside/nucleotide derivatives. In general the (N)-conformation was favored over the (S)-conformation for achieving selectivity for P2Y1, P2Y2, P2Y4, or P2Y11 receptors, but not P2Y6 receptors, leading to the new P2Y1 agonist MRS 2365. Also, the P2Y2,4 agonist MRS 2341 and the P2Y1 antagonists MRS 2297 and MRS 2298 have been characterized. At A3 ARs, the new, potent agonist MRS 1898, containing a rigid (N)-methanocarba ring system that is locked in a receptor-preferred conformation, has been introduced. MRS 1898 has been shown to be free of the apoptosis-inducing effects previously observed with Cl-IB-MECA in human leukemic cells. Also, partial agonists of the A3 AR MRS 1743 and MRS 1760 and A1/A3 AR binary agonists MRS 1740 and MRS 1741 have been reported and shown to be cardioprotective.
Advances in the general methods of molecular modeling and the resolution of the template structures of rhodopsin have brought this integrated perspective to a stage of practicality as a medicinal chemical approach to studying GPCRs in general and purine/pyrimidine receptors, specifically. Molecular models of the A3 AR and P2Y1 receptors have provided insights into the putative binding sites and recognition elements. Identification of microscopic complementarity between residues of the putative receptor binding site and the docked high-affinity ligands, i.e., energetically stabilizing elements in ligand recognition, has aided in the design process. The design of mutant A3 ARs having unique pharmacological features has been accomplished, with the envisioned application in gene therapy. Constitutively active A3 ARs and a neoceptor, which is activated selectively by tailored synthetic neoligands, have been designed.
Footnotes
| Strategy, Management and Health Policy | ||||
|---|---|---|---|---|
| Venture Capital Enabling Technology | Preclinical Research | Preclinical Development Toxicology, Formulation Drug Delivery, Pharmacokinetics | Clinical Development Phases I-III Regulatory, Quality, Manufacturing | Postmarketing Phase IV |
REFERENCES
- Ballesteros JA, Shi L, Javitch JA. 2001. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol 60:1–19. [PubMed] [Google Scholar]
- Boyer JL, Romero-Avila T, Schachter JB, Harden TK. 1996. Identification of competitive antagonists of the P2Y1 receptor. Mol Pharmacol 50:1323–1329. [PubMed] [Google Scholar]
- Boyer JL, Adams M, Ravi RG, Jacobson KA, Harden TK. 2002. 2-Chloro-N6-methyl-(N)-methanocarba-2’-deoxyadenosine-35-bisphosphate is a selective high affinity P2Y1 receptor antagonist. Br J Pharmacol 135:2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SG, King BF, Kim YC, Burnstock G, Jacobson KA. 2000. Activity of novel adenine nucleotide derivatives as agonists and antagonists at recombinant rat P2X receptors. Drug Dev Res 49:253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess K, Cook D. 2000. Synthesis of nucleoside triphosphates. Chem Rev 100:2047–2059. [DOI] [PubMed] [Google Scholar]
- Chen A, Gao ZG, Barak D, Liang BT, Jacobson KA. 2001. Constitutive activation of A3 adenosine receptors by site-directed mutagenesis. Biochem Biophys Res Commun 284:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P, Madi L, Bar-Yehuda S, Barer F, Del Valle L, Khalili K. 2002. Evidence for involvement of Wnt signaling pathway in IB-MECA mediated suppression of melanoma cells. Oncogene 21:4060–4064. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. 2001. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53: 527–552. [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Jacobson KA. 2002. 2-Chloro-N6-cyclopentyladenosine, adenosine A1 receptor agonist, antagonizes the adenosine A3 receptor. Eur J Pharmacol 443:39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. 2002a. Structural determinants of A3 adenosine receptor activation: nucleoside ligands at the agonist/antagonist boundary. J Med Chem 45:4471–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Chen A, Barak D, Kim S-K, Müller CE, Jacobson KA. 2002b. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J Biol Chem 277:19056–19063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessi S, Varani K, Merighi S, Cattabriga E, Iannotta V, Leung E, Baraldi PG, Borea PA. 2002. A3 ARs in human neutrophils and promyelocytic HL60 cells: a pharmacological and biochemical study. Mol Pharmacol 61:415–424. [DOI] [PubMed] [Google Scholar]
- IJzerman AP, von Frijtag Drabbe Künzel JK, Kim J, Jiang Q, Jacobson KA. 1996. Site-directed mutagenesis of the human A2a adenosine receptor. Critical involvement of Glu13 in agonist recognition. Eur J Pharmacol 310:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Ji X-D, Li AH, Melman N, Siddiqi MA, Shin KJ, Marquez V, Ravi RG. 2000a. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem 43:2196–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Xie R, Young L, Chang L, Liang BT. 2000b. A novel pharmacological approach to treating cardiac ischemia: binary conjugates of A1 and A3 adenosine receptor agonists. J Biol Chem 275:30272–30279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, van Rompaey P, van Calenbergh S, Liang BT. 2001. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J Med Chem 44:4125–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Jarvis MF, Williams M. 2002. Perspective: purine and pyrimidine (P2) receptors as drug targets. J Med Chem 45: 4057–4093, [DOI] [PubMed] [Google Scholar]
- Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA. 2001. Acyclic and cyclopropyl anlaogues of adenosine bisphosphate antagonists of the P2Y1 receptor: structure activity relationships and receptor docking. J Med Chem 44:3092–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. 2002a. Methanocarba modification of uracil and adenine nucleotides: high potency of Northern ring conformation at P2Y1 P2Y2 or P2Y4 and P2Y11 but not P2Y6 receptors. J Med Chem 45: 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Ravi RG, Hoffmann CA, Jung YJ, Kim M, Chen A, Jacobson KA. 2002b. p53-Independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative Cl-IB-MECA. Biochem Pharmacol 63:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ravi RG, Ji XD, Marquez VE, Jacobson KA. 2001. Ring-constrained (N)methanocarba-nucleosides as adenosine receptor agonists: independent 5’-uronamide and 2’-deoxy modifications. Biorg Med Chem Lett 11:1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang BT, Jacobson KA. 1998. A physiological role of the adenosine A3 receptor: sustained cardioprotection. Proc Natl Acad Sci USA 95:6995–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. 1996. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem 39:3739–3747. [DOI] [PubMed] [Google Scholar]
- Moro S, Jacobson KA. 2002. Molecular modeling as a tool to investigate molecular recognition in P2Y receptors. Curr Pharmaceut Design 8:99–110. [DOI] [PubMed] [Google Scholar]
- Moro S, Guo DP, Camaioni E, Boyer JL, Harden TK, Jacobson KA. 1998. Human P2Y1 receptor: molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J Med Chem 41:1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CE. 2000. Adenosine receptor ligands—recent developments. Curr Med Chem 7:1269–1288. [DOI] [PubMed] [Google Scholar]
- Müller CE, Diekmann M, Thorand M, Ozola V. 2002. [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-lH-imidazo[2,1-i]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 ARs. Bioorg Med Chem Lett 12:501–503. [DOI] [PubMed] [Google Scholar]
- Nandanan E, Jang SY, Moro S, Kim H, Siddiqi MA, Russ P, Marquez V, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. 2000. Synthesis, biological activity, and molecular modeling of ribose-modified adenosine bisphosphate analogues as P2Y1 receptor ligands. J Med Chem 43:829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Young L, Lee JE, Jacobson KA, Liang BT. 2000. Distinct cardioprotective effects of adenosine mediated by differential coupling of receptor subtypes to phospholipases C and D. FASEB J 14:1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA. 2002. Adenine nucleotides analogues locked in a Northern methanocarba conformation: enhanced stability and potency as P2Y1 receptor agonists. J Med Chem 45:2090–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkees SA, Barbhaiya H, IJzerman A. 1999. Identification of the adenine binding site of the human A1 adenosine receptor. J Biol Chem 274:3617–3621. [DOI] [PubMed] [Google Scholar]
- Shneyvais V, Jacobson KA, Li AH, Nawrath H, Zinman T, Isaac A, Shainberg A. 2000. Induction of apoptosis in rat cardiac myocytes by A3 adenosine receptor activation and its suppression by isoproterenol. Exp Cell Res 257:111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickler J, Jacobson KA, Liang BT. 1996. Direct preconditioning of cultured chick ventricular myocytes: novel functions of cardiac adenosine A2A and A3 receptors. J Clin Invest 98:1773–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AL, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, Linden J. 1994. A1 adenosine receptors. Two amino acids are responsible for species differences in ligand recognition. J Biol Chem 269:27900–27906. [PubMed] [Google Scholar]
- van Tilburg EW, von Frijtag Drabbe Kunzel J, de Groote M, IJzerman AP. 2002. 2,5’-Disubstituted adenosine derivatives: evaluation of selectivity and efficacy for the adenosine A1, A2A, and A3 receptor. J Med Chem 45:420–429. [DOI] [PubMed] [Google Scholar]
- Waldo GL, Corbitt J, Boyer JL, Ravi G, Kim HS, Ji X-D, Lacy J, Jacobson KA, Harden TK. 2002. Quantitation of the P2Y1 receptor with a high affinity radiolabeled antagonist. Mol Pharmacol 202:1249–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]